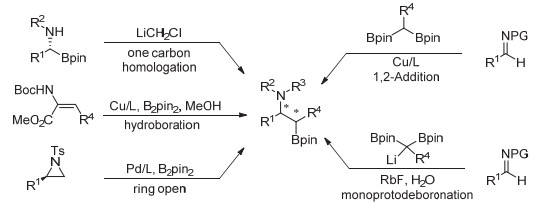
图 1.
手性β-胺基硼酯合成途径
Figure 1.
Approaches for the synthesis of enantioenriched β-aminoboronate

有机硼化合物是有机化学中一类重要中间体, 常用于构建各类碳-碳(C—C)键或碳-杂(C—X)键[1].其中, 手性β-氨基硼酯类化合物因其在药物分子、天然产物合成中的广泛应用而得到极大关注[2].因此, 对该类化合物的合成研究在过去也取得了长足发展.手性α-氨基硼化合物的增碳反应常用于合成手性β-氨基硼化合物[3a]; 此外, 烯胺的不对称硼氢化[3b]、手性2-芳基氮丙啶的开环硼化反应[3c]以及偕二硼类化合物对手性亚胺或非手性亚胺的1, 2-加成[3d-3i]均可以高效地构建手性β-氨基硼类化合物(图 1).铜催化烯烃的1, 2-硼化偶联[4]反应, 可一步实现烯烃的双官能化, 构建多官能化有机硼化合物.通过铜催化烯烃的1, 2-硼胺反应[5]构建手性β-氨基硼酯化合物是较为理想的方法之一. Mirua课题组在该领域做了大量的研究工作, 他们相继实现了苯乙烯类化合物[5a]、双环烯烃[5e]、烯基硅烷[5f]等的不对成硼胺化反应; 施敏课题组实现了含环丙烷烯烃化合物的不对称硼胺化反应[5h]; 我们课题组在2017年利用自己设计的手性亚砜膦配体实现了简单苯乙烯类化合物的不对称硼胺化反应[8d].到目前为止, 除王建波课题组利用芳基重氮为亲电氮源以外[5i], 针对各类烯烃的1, 2-硼胺化反应, 均以联硼酸酯作为硼源, 以羟胺类衍生物作为亲电氮源, 实现手性β-氨基硼酯化合物的构建, 但该类化合物只能在特定底物条件下才能实现硼酯保留而氨基脱保护合成手性的β-硼酯伯胺[5f], 较大限制β-氨基硼酯化合物的进一步转化应用. 1, 2-苯基异噁唑作为一种新型亲电氮源[6], 可用于苯乙烯化合物的不对称氢胺化反应[6a], 目标产物可以在温和条件下脱去氮保护基, 实现手性伯胺化合物的合成.需要指出的是, 1, 2-苯基异噁唑在碱性条件下极易发生Kemp消除反应[7], 对反应体系兼容性有一定限制(因为基于铜硼的烯烃双官能化一般在碱性条件下进行).近些年来, 我们课题组在手性亚砜膦(SOP)/铜络合物催化的苯乙烯、1, 3-炔烯硼化双官能化取得一定进展[8].在这些前期研究基础上, 我们利用手性亚砜膦/铜络合物为催化剂, 以1, 2-苯基异噁唑为亲电氮源, 对苯乙烯类底物的不对称1, 2-硼胺化反应进行了研究, 高效地合成了系列手性β-氨基硼酯化合物, 获得较高的收率和对映选择性.该类化合物在温和条件下可以实现手性β-硼酯伯胺的转化, 为各类结构丰富的手性氨基化合物提供了一条较为高效的合成路径(图 2).
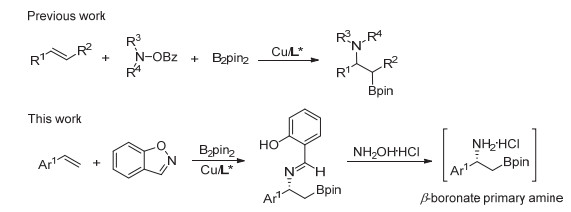
我们选取苯乙烯(1a)为模板底物, 1, 2-苯基异噁唑为亲电胺试剂(2a)和联硼酸频哪醇酯(B2pin2)为亲核试剂, 手性亚砜膦配体(SOP) L1和CuCl原位络合生成活性催化剂, 以叔丁醇锂(LiOtBu)为碱, 四氢呋喃(THF)为溶剂, 设定反应温度为20 ℃, 对反应进行了初步尝试(表 1, Entry 1).反应12 h后, 以中等收率(50%)和优异的对映选择性(95% ee)得到了苯乙烯的硼胺化产物.为了进一步提高反应的收率和对映选择性, 我们对反应条件进行了系统性优化.
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | Ligand | Cu salt | Base | Yieldb/% | eec/% |
| 1 | L1 | CuCl | LiOtBu | 50 | 95 |
| 2 | L1 | CuCl | LiOH | 51 | 97 |
| 3 | L1 | CuCl | LiOMe | 67 | 96 |
| 4 | L1 | CuCl | NaOtBu | 6 | n.d.d |
| 5 | L1 | CuCl | KOtBu | 10 | n.d.d |
| 6e | L1 | CuCl | LiOMe | 81 | 96 |
| 7e | L1 | CuBr | LiOMe | 75 | 97 |
| 8e | L1 | CuI | LiOMe | 84 | 98 |
| 9e | L1 | CuCl2 | LiOMe | trace | n.d.d |
| 10e | L1 | Cu(OTf)2 | LiOMe | 10 | n.d.d |
| 11e | L2 | CuI | LiOMe | 71 | 91 |
| 12e | L3 | CuI | LiOMe | 45 | 89 |
| 13e | L4 | CuI | LiOMe | 24 | -53 |
| 14e | L5 | CuI | LiOMe | 16 | n.d.d |
| 15e | L6 | CuI | LiOMe | 24 | -73 |
| a Conditions: The reaction was performed with 1a (0.5 mmol), 2a (0.75mmol), B2pin2 (0.75 mmol), Base (0.75 mmol), CuCl (10 mol%), L1 (12 mol%) in THF (4 mL) at 20 ℃ for 12 h. b The yield was determined by 1H NMR using dimethyl terephthalate as an internal standard. c Determined by chiral HPLC analysis. d n.d.=not determined. e 1.25 mmol LiOMe was used. | |||||
首先对碱进行了考察(详细信息见SI).我们发现大部分无机碱在此反应中效果较差, 可能与其溶解性较差有关.当碱换作碱性更强的叔丁醇钠、叔丁醇钾时, 仅能得到少量的目标产物, 可能是因为1, 2-苯基异噁唑在强碱性条件下发生竞争反应Kemp消除生成了副产物(表 1, Entries 4~5).几种含锂的碱均表现出了较好的反应结果, 综合考虑, 甲醇锂效果最佳(表 1, Entry 3, 67% yield, 96% ee).当碱的用量增加到1.25 mmol时, 收率可以得到进一步提高(表 1, Entry 6, 81% yield, 96% ee).随后, 我们考察了铜盐对反应的影响(表 1, Entries 7~10).当铜源由CuCl变为CuI时, 收率和对映选择性均有进一步提升(表 1, Entry 8, 84% yield, 98% ee).最后, 我们对手性配体, 包括我们发展的其它手性亚砜膦配体(L2~L3)和商业可得的双膦配体(L4~L6)进行了考察.不管是活性还是对映选择性均没有表现出更好的结果(表 1, Entries 11~15).综合考虑上述实验结果, 我们确定了反应的最优条件:以10 mol% CuI为金属铜源, 12 mol% L1为手性配体, 以四氢呋喃为溶剂, 甲醇锂作为碱, 20 ℃条件下反应12 h.
在最优条件下, 我们对该反应的底物普适性进行了考察(表 2).由于该产物在硅胶上不稳定, 易发生分解, 影响其分离纯化, 因此我们分别测定了产物的核磁收率与分离收率, 其中括号内为分离收率.针对空间效应以及电子效应, 我们对芳环上不同取代的芳基烯烃以及其他类型的烯烃进行了考察.通过我们发展的三组分硼胺化方法, 绝大部分芳基烯烃都能以中等到优秀的收率(47%~84% yield)和优秀的对映选择性(81%~99% ee)转化为相应的目标产物.卤素取代(F, Cl, Br)苯乙烯在该反应中耐受, 可取得较好结果.值得注意的是, 通过具体分析我们发现, 缺电子烯烃和富电子烯烃在空间位阻方面表现出了不一样的趋势:缺电子烯烃的位阻效应对反应的收率和对映选择性均没有明显影响(3c, 3h, 3n), 富电子烯烃具有不同空间位阻时, 均能获得较高对映选择性, 但在收率方面表现不一, 其中邻位取代的苯乙烯收率较高, 间位次之, 对位较差(3f, 3k, 3s).强吸电子基团(CF3)取代苯乙烯也可以在该条件下发生反应获得较好的收率和优秀的对映选择性(3l, 3u).芳基烯烃底物相同位置取代基位阻对反应的对映选择性也有影响, 空间位阻较大的对苯苯乙烯在最优条件下可获得最高99%的对映选择性(3v). 2-噻吩乙烯和β-萘乙烯两个底物通过核磁谱图分析有对应的产物生成, 但产物不稳定且副反应明显, 未能分离纯化得到目标产物.其他芳基烯烃, 如α-取代苯乙烯, β-取代苯乙烯, 可能由于位阻原因不能发生反应.此外, 特殊结构烷基烯烃, 如降冰片二烯可能因其特殊二烯结构和骨架具有较强的张力, 在该反应体系下也能顺利地以中等收率和较好对映选择性获得目标产物(3w), 其他烷基烯烃底物, 如环己烯、4-苯基-1-丁烯则无活性.
下载:
导出CSV
 |
 |
| a Conditions: The reaction was performed with 1a (0.5 mmol), 2a (0.75 mmol), B2pin2 (0.75 mmol), LiOMe (1.25 mmol), CuI (10 mol%), L1 (12 mol%) in THF (4 mL) at 20 ℃ for 12 h. b The yield was determined by 1H NMR spectroscopy. cIsolated yield. dDetermined by chiral HPLC analysis. en.r=no reaction. |
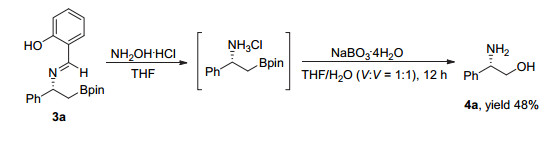
为了证明该反应的实用性, 我们对反应产物进行了转化.手性β-氨基硼酯3a未经分离纯化, 在盐酸羟胺的甲醇溶液条件下可高效地脱除氮保护基得到手性β-硼酯伯胺盐酸盐.较为遗憾的是, 我们尝试了多种分离提纯方法, 均不能获得纯的伯胺产物.该粗产品通过氧化可以以三步反应总收率48%获得手性β-氨基醇化合物4a.化合物4a通过与文献已报道产物的比旋光度[9]进行对比, 确定了产物3a绝对构型为R (图 3).
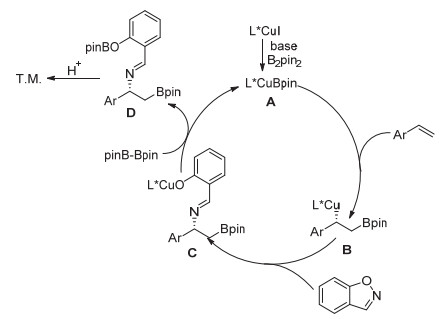
根据我们的前期相关研究[8, 10b]及文献报道[10], 在这里提出一个可能的反应机理(图 4).首先, 手性亚砜膦/碘化亚铜络合物和联硼酸频哪醇酯在碱的作用下生成手性铜硼物种A; 铜硼物种A对烯烃进行1, 2-加成生成β-硼苄基铜中间体B; 1, 2-苯基异噁唑捕捉该苄基铜中间体B而得到中间体C; 中间体C和联硼酸频哪醇酯反应得到中间体D以及再生活性的铜硼物种A.最后, 中间体D在反应后处理过程中质子化得到目标产物.
我们以铜/手性亚砜膦络合物为催化剂, 以商业可得的1, 2-苯基异噁唑为亲电氮源, 发展了一种较为高效的苯乙烯1, 2-硼胺化反应, 取得了较高的收率和最高达99%的对映选择性.目标产物可以在温和条件下保留硼酯脱去氮保护基合成手性β-硼酯伯胺化合物, 为该类化合物进一步转化应用提供更多可能.最后我们通过硼酯氧化得到手性β-氨基醇化合物, 为手性氨基醇化合物合成提供了一种有效的路径.
(a) Pelter, A.; Smith, K.; Brown, H. C. Borane Reagents, Academic Press: London, 1988. (b) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. (c) Davison, M.; Hughes, A. K.; Marder, T. B.; Wade, K. Contemporary Boron Chemistry; RSC: Cambridge, U.K., 2000. (d) Boronic Acids, 2nd ed.; Hall, D. G., Ed.; Wiley-VCH: Weinheim, Germany, 2011. (e) Yang, J.; Deng, M.; Yu, T. Chin. J. Org. Chem. 2013, 33, 693(in Chinese). (杨军, 邓敏智, 于涛, 有机化学, 2013, 33, 693). (f) Wu, Y.; Dou, Z.; Wu, C.; Chen, H.; Zhang, Z.; Wang, X.; Yin, Z.; Song, X.; He, C.; Yue, G. Chin. J. Org. Chem. 2018, 38, 2896(in Chinese). (吴瑶, 豆正杰, 吴彩梅, 陈华保, 张祖民, 王显祥, 殷中琼, 宋旭, 贺常亮, 乐贵洲, 有机化学, 2018, 38, 2896.) (g) Zhu, D.; Xu, M.; Chin. J. Org. Chem. 2020, 40, 255(in Chinese). (祝东星, 徐明华, 有机化学, 2020, 40, 255.) (h) Guan, H.; Chen, L.; Liu L. Acta Chim. Sinica 2018, 76, 440(in Chinese). (关弘浩, 陈磊, 刘磊, 化学学报, 2018, 76, 440.) (i) He, S.; Pi, J.; Li, Y.; Lu, X.; Fu, Y. Acta Chim. Sinica 2018, 76, 956(in Chinese). (何世江, 皮静静, 李炎, 陆熹, 傅尧, 化学学报, 2018, 76, 956.) (j) Wang, D.; He, Y.; Dai, H.; Huang, C.; Yuan, X.-A.; Xie, J. Chin. J. Chem. 2020, 38, 1497. (k) Bai, Y.; Cui, C. Acta Chim. Sinica 2020, 78, 763(in Chinese). (白云平, 崔春明, 化学学报, 2020, 78, 763.)
(a) Gorovoy, A. S.; Gozhina, O.; Svendsen, J.-S.; Tetz, G. V.; Domorad, A.; Tetz, V. V.; Lejon, T. J. Pept. Sci. 2013, 19, 613. (b) Gorovoy, A. S.; Gozhina, O. V.; Svendsen, J. S.; Domorad, A. A.; Tetz, G. V.; Tetz, V. V.; Lejon, T. Chem. Biol. Drug Des. 2013, 81, 408.
(a) Solé, G.; Gulyás, H.; Fernández, E. Chem. Commun. 2012, 48, 3769. (b) He, Z. T.; Zhao, Y. S.; Tian, P.; Wang, C. C.; Dong, H. Q.; Lin, G. Q. Org. Lett. 2014, 16, 1426. (c) Takeda, Y.; Kuroda, A.; Sameera, W. M. C.; Morokuma, K.; Minakata, S. Chem. Sci. 2016, 7, 6141. (d) Park, J.; Lee, Y.; Kim, J.; Cho, S. H. Org. Lett. 2016, 18, 1210. (e) Kim, J.; Ko, K.; Cho, S. H. Angew. Chem., Int. Ed. 2017, 56, 11584. (f) Li, X.; Hall, D. G. Angew. Chem., Int. Ed. 2018, 57, 10304. (g) Kim, J.; Hwang, C.; Kim, Y.; Cho, S. H. Org. Process Res. Dev. 2019, 23, 1663. (h) Kim, J.; Shin, M.; Cho, S. H. ACS Catal. 2019, 9, 8503. (i) Li, X.; Hall, D. G. J. Am. Chem. Soc. 2020, 142, 9063.
Reviews: (a) Semba, K.; Fujihara, T.; Terao, J.; Tsuji, Y. Tetrahedron 2015, 71, 2183. (b) Liu, Y.; Zhang, W. Chin. J. Org. Chem. 2016, 36, 2249(in Chinese). (刘媛媛, 张万斌, 有机化学, 2016, 36, 2249.) (c) Whyte, A.; Torelli, A.; Mirabi, B.; Zhang, A.; Lautens, M. ACS Catal. 2020, 10, 11578.
(a) Matsuda, N.; Hirano, K.; Satoh, T.; Miura, M. J. Am. Chem. Soc. 2013, 135, 4934. (b) Sakae, R.; Matsuda, N.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 1228. (c) Parra, A.; Amenos, L.; Guisan-Ceinos, M.; Lopez, A.; Garcia Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833. (d) Sakae, R.; Hirano, K.; Miura, M. J. Am. Chem. Soc. 2015, 137, 6460. (e) Sakae, R.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2015, 54, 613. (f) Kato, K.; Hirano, K.; Miura, M. Angew. Chem., Int. Ed. 2016, 55, 14400. (g) Nishikawa, D.; Hirano, K.; Miura, M. Org. Lett. 2016, 18, 4856. (h) Jiang, H. C.; Tang, X. Y.; Shi, M. Chem. Commun. 2016, 52, 5273. (i) Huo, J.; Xue, Y.; Wang, J. Chem. Commun. 2018, 54, 12266. (j) Kato, K.; Hirano, K.; Miura, M. Chem. Eur. J. 2018, 24, 5775.
(a) Guo, S.; Yang, J. C.; Buchwald, S. L. J. Am. Chem. Soc. 2018, 140, 15976. (b) Feng, S.; Hao, H.; Liu, P.; Buchwald, S. L. ACS Catal. 2019, 10, 282. (c) Guo, S.; Zhu, J.; Buchwald, S. L. Angew. Chem., Int. Ed. 2020, 59, 20841.
(a) Casey, M. L.; Kemp, D. S.; Paul, K. G.; Cox, D. J. Org. Chem. 1973, 38, 2294.
(a) Chen, B.; Cao, P.; Yin, X.; Liao, Y.; Jiang, L.; Ye, J.; Wang, M.; Liao, J. ACS Catal. 2017, 7, 2425. (b) Jia, T.; Cao, P.; Wang, B.; Lou, Y.; Yin, X.; Wang, M.; Liao, J. J. Am. Chem. Soc. 2015, 137, 13760. (c) Jia, T.; Cao, P.; Wang, D.; Lou, Y.; Liao, J. Chem. Eur. J. 2015, 21, 4918. (d) Zhang, Y.; Wang, M.; Cao, P.; Liao, J. Acta Chim. Sinica 2017, 75, 794(in Chinese). (张涌灵, 王敏, 曹鹏, 廖建, 化学学报, 2017, 75, 794.) (e) Chen, B.; Cao, P.; Liao, Y.; Wang, M.; Liao, J. Org. Lett. 2018, 20, 1346. (f) Wang, B.; Wang, X.; Yin, X.; Yu, W.; Liao, Y.; Ye, J.; Wang, M.; Liao, J. Org. Lett. 2019, 21, 3913. (g) Liao, Y.; Yin, X.; Wang, X.; Yu, W.; Fang, D.; Hu, L.; Wang, M.; Liao, J. Angew. Chem., Int. Ed. 2020, 59, 1176.
(a) Noshita, M.; Shimizu, Y.; Morimoto, H.; Ohshima, T. Org. Lett. 2016, 18, 6062.
(a) Laitar, D. S.; Tsui, E. Y.; Sadighi, J. P. Organometallics 2006, 25, 2405. (b) Jiang, L.; Cao, P.; Wang, M.; Chen, B.; Wang, B.; Liao, J. Angew. Chem., Int. Ed. 2016, 55, 13854. (c) Tobisch, S. Chem. Eur. J. 2017, 23, 17800.

图 1 手性β-胺基硼酯合成途径
Figure 1 Approaches for the synthesis of enantioenriched β-aminoboronate

图 2 铜催化烯烃不对称硼胺化反应
Figure 2 Copper-catalyzed enantioselective aminoboration of styrenes
表 1 反应条件优化a
Table 1. Reaction conditions screening
|
|
|||||
| Entry | Ligand | Cu salt | Base | Yieldb/% | eec/% |
| 1 | L1 | CuCl | LiOtBu | 50 | 95 |
| 2 | L1 | CuCl | LiOH | 51 | 97 |
| 3 | L1 | CuCl | LiOMe | 67 | 96 |
| 4 | L1 | CuCl | NaOtBu | 6 | n.d.d |
| 5 | L1 | CuCl | KOtBu | 10 | n.d.d |
| 6e | L1 | CuCl | LiOMe | 81 | 96 |
| 7e | L1 | CuBr | LiOMe | 75 | 97 |
| 8e | L1 | CuI | LiOMe | 84 | 98 |
| 9e | L1 | CuCl2 | LiOMe | trace | n.d.d |
| 10e | L1 | Cu(OTf)2 | LiOMe | 10 | n.d.d |
| 11e | L2 | CuI | LiOMe | 71 | 91 |
| 12e | L3 | CuI | LiOMe | 45 | 89 |
| 13e | L4 | CuI | LiOMe | 24 | -53 |
| 14e | L5 | CuI | LiOMe | 16 | n.d.d |
| 15e | L6 | CuI | LiOMe | 24 | -73 |
| a Conditions: The reaction was performed with 1a (0.5 mmol), 2a (0.75mmol), B2pin2 (0.75 mmol), Base (0.75 mmol), CuCl (10 mol%), L1 (12 mol%) in THF (4 mL) at 20 ℃ for 12 h. b The yield was determined by 1H NMR using dimethyl terephthalate as an internal standard. c Determined by chiral HPLC analysis. d n.d.=not determined. e 1.25 mmol LiOMe was used. | |||||
 下载: 导出CSV
下载: 导出CSV
表 2 硼胺化底物范围考察a
Table 2. Substrate scope of aminoboration
|
|
|
| a Conditions: The reaction was performed with 1a (0.5 mmol), 2a (0.75 mmol), B2pin2 (0.75 mmol), LiOMe (1.25 mmol), CuI (10 mol%), L1 (12 mol%) in THF (4 mL) at 20 ℃ for 12 h. b The yield was determined by 1H NMR spectroscopy. cIsolated yield. dDetermined by chiral HPLC analysis. en.r=no reaction. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: