Department of Applied Chemistry, College of Life Science, Fujian Agriculture and Forestry University, Fuzhou 350002, China
b.
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
Received Date:
12 August 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21520102001), Natural Science Foundation of Fujian Province of China (No. 2020J01549), and Fujian Agriculture and Forestry University (Nos. 118360020, XJQ201616)
Abstract:
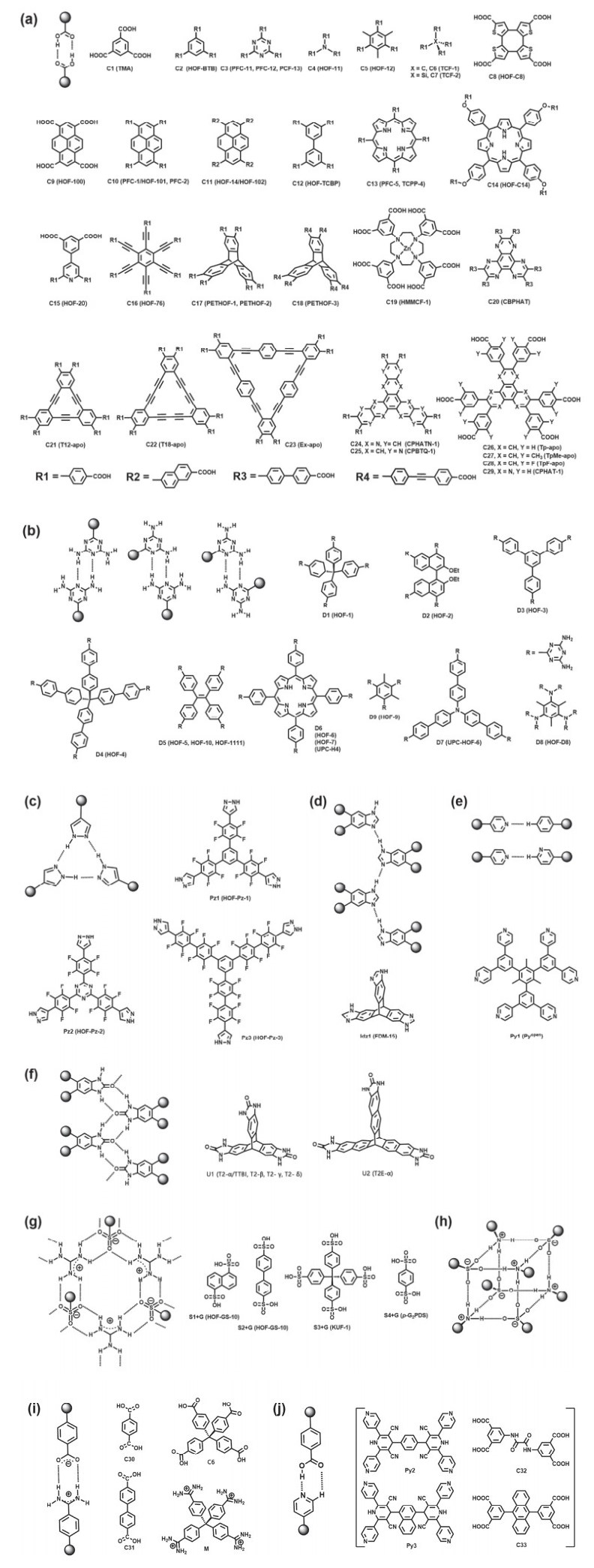
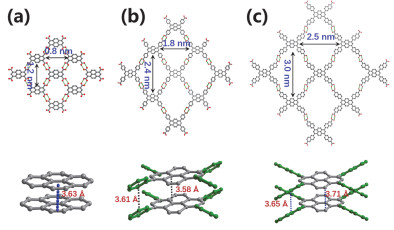
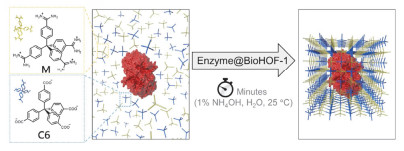
Hydrogen-bonded organic frameworks (HOFs), usually self-assembled by organic or metal-organic building blocks via intermolecular H-bonding interactions, have become a unique type of crystalline porous material. Although the weak and flexible nature of hydrogen bonds makes most HOFs fragile, the high stability and permanent porosity could be realized by the judicious selection of rigid building blocks with special spatial configuration as well as the introduction of framework interpenetration and/or other intermolecular interactions like π-π stacking and electrostatic interactions, etc. Compared with other crystalline porous materials like metal-organic frameworks (MOFs) and covalent-organic frameworks (COFs), HOFs feature mild preparation condition, high crystallinity, permissible solution processability, easy healing and regeneration, etc. These distinguishing merits make HOFs capable to be used as unique multifunctional porous materials. Herein, we first review the basic rules to design and synthesize stable and porous HOFs, and then systematically summarize the representative supramolecular synthons and backbones that have been used to build stable and porous HOFs. Emphasis is put on the potential applications of HOFs in gas adsorption and separation, proton conduction, heterogeneous catalysis, luminescence and sensing, biological applications, enantiomeric resolution and aromatic compounds separation, pollutants removal, and structure determination, etc.
Figure 1.
The construction of sql networks by (a) 4-connected planner SBUs like Cu2(CO2)4 and linear ligands like terephthalic acid in MOFs, and (b) 4-connected planner scaffold (or backbone, blue) and supramolecular synthon (like carboxy dimer, red) in HOFs.
Figure 6.
(a) The connection motifs between M and C30 building block, and (b) M and C31 building block. HOFs with dia topology built by (c) M-C30 and (d) M-C31 building blocks, respectively. Reprinted with permission from ref. [110, 114], Copy right 2017 Royal Society of Chemistry and 2019 Wiley-VCH
Figure 7.
(a) The structure of MPM-1-TIFSIX and its building block; (b) CO2, CH4, and N2 adsorption over MPM-1-TIFSIX at 298 K; (c) The IAST selectivities of CO2/N2 (50:50) and CO2/CH4 (50:50) at 298 K. Reprinted with permission from ref. [126], Copy right 2013 American Chemical Society
Figure 8.
(a) The structure of SOF-7. (b) CO2 and CH4 adsorption over SOF-7 at 273 and 298 K. Reprinted with permission from ref. [119], Copy right 2014 American Chemical Society
Figure 9.
(a) The structure of IISERP-HOF1 and its 1D channels along crystallographic a axis. (b) CO2 and N2 adsorption over IISERP-HOF1; (c) IAST selectivities of CO2/N2 (15:85). Reprinted with permission from ref. [43], Copy right 2016 Royal Society of Chemistry
Figure 10.
(a) The structure of KUF-1. (b) N2, H2 and O2 sorption isotherms over KUF-1. (c) NH3 adsorption over KUF-1. Reprinted with permission from ref. [104], Copy right 2019 Wiley-VCH
Figure 11.
(a) The structure of HOF-4. (b) C2H4 and C2H6 adsorption over HOF-4. Reprinted with permission from ref. [86], Copy right 2014 Royal Society of Chemistry
Figure 12.
(a) The carboxy dimer and π-π stacking in HOF-76. (b) The structure of HOF-76. (c) C2H4 and C2H6 adsorption over HOF-76. The interactions between (d) C2H4, (e) or C2H6 and HOF-76 framework. (f) Experimental breakthrough curves for the C2H6/C2H4 binary mixture at 298 K and 1×105 Pa. Reprinted with permission from ref. [69], Copy right 2020 American Chemical Society
Figure 13.
(a) The 1D channels in HOF-1. (b) The sorption isotherms of C2H2 and C2H4 over HOF-1 at 273 K. Reprinted with permission from ref. [6], Copy right 2011 American Chemical Society
Figure 14.
(a) The structure of HOF-21. (b) Adsorption isotherms of C2H2 (solid) and C2H4 (hollow) on HOF-21 (blue) and MPM-1-TIFSIX at 298 K. (c) Experimental column breakthrough curves for C2H2/C2H4 (50:50) binary mixture at 298 K and 1×105 Pa in an adsorber bed packed with HOF-21a (blue) or MPM-1-TIFSIX (red). (d) The interactions between C2H2 and HOF-21 calculated by DFT. Reprinted with permission from ref. [132], Copy right 2018 American Chemical Society
Figure 15.
(a) The building units and structure of HOF-3. (b) C2H2 and CO2 sorption isotherms over HOF-3. (c) Comparison of the adsorption enthalpies in HOFs and various MOFs. (d) IAST adsorption selectivities of C2H2/CO2 in equimolar mixture in HOF-3a and various MOFs at 296 K. (e) Breakthrough curve for an equimolar C2H2/CO2 mixture at 1×105 Pa and 296 K. Reprinted with permission from ref. [85], Copy right 2015 Wiley-VCH
Figure 16.
(a) The carboxy dimer and π-π stacking in HOF-TCBP; (b) 1D channel in HOF-TCBP; (c~e) The adsorption isotherms of various C4 over HOF-TCBP at 295 K; (f) IAST predicted the adsorption selectivities of C4/C1 for HOF-TCBP at 295 K and 101 kPa. Reprinted with permission from ref. [116], Copy right 2016 Nature Publishing Group
Figure 17.
(a) The structure of HOF-5. (b) The sorption isotherms of CO2 and CH4 over HOF-5. (c) The sorption isotherms of C2H2 and CH4 over HOF-5. (d) IAST selectivities of CO2/CH4 and C2H2/CH4. Reprinted with permission from ref. [87], Copy right 2015 American Chemical Society
Figure 18.
(a) The building unit of UPC-HOF-6. (b) Digital photo of UPC-HOF-6. (c) The structure of UPC-HOF-6. (d) 1D channel in UPC-HOF-6. (e) Top-view SEM of UPC-HOF-6-120 film. (f) Single gas permeation of UPC-HOF-6-120 membrane at 25 ℃. Reprinted with permission from ref. [93], Copy right 2020 Wiley-VCH
Figure 19.
(a) The structure of HOF-GS-10 and HOF-GS-11. (b) Arrhenius plot of HOF-GS-10 and HOF-GS-11. (c) Proton conduction values of HOF-GS-10 and HOF-GS-11 at various humidity and 30 ℃. Reprinted with permission from ref. [103], Copy right 2016 Wiley-VCH
Figure 20.
(a) The construction of homochiral nanoscale metal-organic cages. (b) Asymmetric [3+2] coupling of 3-substituted indoles with quinone monoimine. (c) Asymmetric Friedel-Crafts reaction of N-sulfonyl aldimines with indole. Reprinted with permission from ref. [70], Copy right 2020 Nature Publishing Group
Figure 21.
(a) The cage-based organic building units and the intramolecular hydrogen bonding between building units. (b) Suzuki-Miyaura coupling reactions catalyzed by HOF-19⊃pd(Ⅱ). Reprinted with permission from ref. [131], Copy right 2019 American Chemical Society
Figure 22.
(a) The structure of Tp-1, T12-1, T18-1, and Ex-1. The luminescence (solid line) of (b) Tp-1, (c) T12-1, (d) T18-1, and (e) Ex-1. Reprinted with permission from ref. [50, 52], Copy right 2016 American Chemical Society
Figure 23.
(a) The construction of HOF-10 and HOF-5. The structure of (b) HOF-10 and (c) HOF-5. Luminescent response of (d) HOF-10 and (e) HOF-5 to various concentration of Ag(Ⅰ). Reprinted with permission from ref. [91], Copy right 2017 Royal Society of Chemistry
Figure 24.
(a) The structure of 8PN. (b) Photographs of 8PN taken under UV-light irradiation (365 nm). (c) Emission spectra of 8PN excited at 365 nm. Reprinted with permission from ref. [35], Copy right 2019 Nature Publishing Group
Figure 25.
(a) Carboxy dimer and building unit in HOF-20. (b) The structure of HOF-20. (c) Luminescent response of HOF-20 to various aromatic compounds. Reprinted with permission from ref. [74], Copy right 2020 American Chemical Society
Figure 27.
The structure and synergistic chemo-photodynamic therapy of nr-HOF@Doxo. Reprinted with permission from ref. [121], Copy right 2019 American Chemical Society
Figure 29.
(a) The structure of HOF-2. (b) The topology of HOF-2. (c) The structure of HOF-2 after the adsorption of R-1-phenylethanol. (d) The chiral cavity in HOF-2. The interactions between HOF-2 and (e) R-1-phenylethanol/(f)
S-1-phenylethanol. Reprinted with permission from ref. [84], Copy right 2014 American Chemical Society
Figure 30.
(a) Synthesis of HcOF-1 from 1crystal. (b) A saturated iodine aqueous solution (ca. 1.2 mmol•L-1) upon addition of HCOF-1 (3.0 mg). (c) The iodine desorption of I2@HCOF-1 in DMSO. Reprinted with permission from ref. [150], Copy right 2017 American Chemical Society
Figure 31.
(a) The structure of (G2NDS)⊃(3aR)-(+)-Sclareolide and encapsulated Sclareolide, (b) (G2NDS)⊃(drospirenone)(methanol)0.84- (H2O)0.1 and encapsulated drospirenone, and (c) (G2BDPYDS)⊃ (progesterone) and encapsulated progesterone. Reprinted with permission from ref. [157], Copy right 2019 Nature Publishing Group
Li, Y.-L.; Alexandrov, E. V.; Yin, Q.; Li, L.; Fang, Z.-B.; Yuan, W.; Proserpio, D. M.; Liu, T.-F. J. Am. Chem. Soc. 2020, 142, 7218.
[37]
Pulido, A.; Chen, L.; Kaczorowski, T.; Holden, D.; Little, M. A.; Chong, S. Y.; Slater, B. J.; McMahon, D. P.; Bonillo, B.; Stackhouse, C. J.; Stephenson, A.; Kane, C. M.; Clowes, R.; Hasell, T.; Cooper, A. I.; Day, G. M. Nature 2017, 543, 657.
[38]
Cui, P.; McMahon, D. P.; Spackman, P. R.; Alston, B. M.; Little, M. A.; Day, G. M.; Cooper, A. I. Chem. Sci. 2019, 10, 9988.
[39]
Cui, P.; Svensson Grape, E.; Spackman, P. R.; Wu, Y.; Clowes, R.; Day, G. M.; Inge, A. K.; Little, M. A.; Cooper, A. I. J. Am. Chem. Soc. 2020, 12743.
[40]
Desiraju, G. R. Angew. Chem. Int. Ed. 1995, 34, 2311.
[41]
Herbstein, F. H.; Kapon, M.; Reisner, G. M. J. Incl. Phenom.1987, 5, 211.
[42]
Zentner, C. A.; Lai, H. W. H.; Greenfield, J. T.; Wiscons, R. A.; Zeller, M.; Campana, C. F.; Talu, O.; FitzGerald, S. A.; Rowsell, J. L. C. Chem. Commun. 2015, 51, 11642.
Hashim, M. I.; Le, H. T. M.; Chen, T.-H.; Chen, Y.-S.; Daugulis, O.; Hsu, C.-W.; Jacobson, A. J.; Kaveevivitchai, W.; Liang, X.; Makarenko, T.; Miljanić, O. Š.; Popovs, I.; Tran, H. V.; Wang, X.; Wu, C.-H.; Wu, J. I. J. Am. Chem. Soc. 2018, 140, 6014.
Morshedi, M.; Ward, J. S.; Kruger, P. E.; White, N. G. Dalton Trans. 2018, 47, 783.
[110]
Boer, S. A.; Morshedi, M.; Tarzia, A.; Doonan, C. J.; White, N. G. Chem. Eur. J. 2019, 25, 10006.
[111]
Boer, S. A.; Wang, P.-X.; MacLachlan, M. J.; White, N. G. Cryst. Growth Des. 2019, 19, 4829.
[112]
Cullen, D. A.; Gardiner, M. G.; White, N. G. Chem. Commun. 2019, 55, 12020.
[113]
Liang, W.; Carraro, F.; Solomon, M. B.; Bell, S. G.; Amenitsch, H.; Sumby, C. J.; White, N. G.; Falcaro, P.; Doonan, C. J. J. Am. Chem. Soc. 2019, 141, 14298.
[114]
Morshedi, M.; Thomas, M.; Tarzia, A.; Doonan, C. J.; White, N. G. Chem. Sci. 2017, 8, 3019.
[115]
Morshedi, M.; White, N. G. CrystEngComm 2017, 19, 2367.
陈之尧, 刘捷威, 崔浩, 张利, 苏成勇, 化学学报, 2019, 77, 242.Chen, Z. Y.; Liu, J. W.; Cui, H.; Zhang, L.; Su, C. Y. Acta Chim. Sinica 2019, 77, 242 (in Chinese).
[142]
Liu, F. Q.; Liu, J. W.; Gao, Z.; Wang, L.; Fu, X.-Z.; Yang, L. X.; Tao, Y.; Yin, W. H.; Luo, F. Appl. Catal. B: Environ. 2019, 258, 117973.
[143]
Aitchison, C. M.; Kane, C. M.; McMahon, D. P.; Spackman, P. R.; Pulido, A.; Wang, X.; Wilbraham, L.; Chen, L.; Clowes, R.; Zwijnenburg, M. A.; Sprick, R. S.; Little, M. A.; Day, G. M.; Cooper, A. I. J. Mater. Chem. A 2020, 8, 7158.
Figure 1
The construction of sql networks by (a) 4-connected planner SBUs like Cu2(CO2)4 and linear ligands like terephthalic acid in MOFs, and (b) 4-connected planner scaffold (or backbone, blue) and supramolecular synthon (like carboxy dimer, red) in HOFs.
Figure 6
(a) The connection motifs between M and C30 building block, and (b) M and C31 building block. HOFs with dia topology built by (c) M-C30 and (d) M-C31 building blocks, respectively. Reprinted with permission from ref. [110, 114], Copy right 2017 Royal Society of Chemistry and 2019 Wiley-VCH
Figure 7
(a) The structure of MPM-1-TIFSIX and its building block; (b) CO2, CH4, and N2 adsorption over MPM-1-TIFSIX at 298 K; (c) The IAST selectivities of CO2/N2 (50:50) and CO2/CH4 (50:50) at 298 K. Reprinted with permission from ref. [126], Copy right 2013 American Chemical Society
Figure 8
(a) The structure of SOF-7. (b) CO2 and CH4 adsorption over SOF-7 at 273 and 298 K. Reprinted with permission from ref. [119], Copy right 2014 American Chemical Society
Figure 9
(a) The structure of IISERP-HOF1 and its 1D channels along crystallographic a axis. (b) CO2 and N2 adsorption over IISERP-HOF1; (c) IAST selectivities of CO2/N2 (15:85). Reprinted with permission from ref. [43], Copy right 2016 Royal Society of Chemistry
Figure 10
(a) The structure of KUF-1. (b) N2, H2 and O2 sorption isotherms over KUF-1. (c) NH3 adsorption over KUF-1. Reprinted with permission from ref. [104], Copy right 2019 Wiley-VCH
Figure 11
(a) The structure of HOF-4. (b) C2H4 and C2H6 adsorption over HOF-4. Reprinted with permission from ref. [86], Copy right 2014 Royal Society of Chemistry
Figure 12
(a) The carboxy dimer and π-π stacking in HOF-76. (b) The structure of HOF-76. (c) C2H4 and C2H6 adsorption over HOF-76. The interactions between (d) C2H4, (e) or C2H6 and HOF-76 framework. (f) Experimental breakthrough curves for the C2H6/C2H4 binary mixture at 298 K and 1×105 Pa. Reprinted with permission from ref. [69], Copy right 2020 American Chemical Society
Figure 13
(a) The 1D channels in HOF-1. (b) The sorption isotherms of C2H2 and C2H4 over HOF-1 at 273 K. Reprinted with permission from ref. [6], Copy right 2011 American Chemical Society
Figure 14
(a) The structure of HOF-21. (b) Adsorption isotherms of C2H2 (solid) and C2H4 (hollow) on HOF-21 (blue) and MPM-1-TIFSIX at 298 K. (c) Experimental column breakthrough curves for C2H2/C2H4 (50:50) binary mixture at 298 K and 1×105 Pa in an adsorber bed packed with HOF-21a (blue) or MPM-1-TIFSIX (red). (d) The interactions between C2H2 and HOF-21 calculated by DFT. Reprinted with permission from ref. [132], Copy right 2018 American Chemical Society
Figure 15
(a) The building units and structure of HOF-3. (b) C2H2 and CO2 sorption isotherms over HOF-3. (c) Comparison of the adsorption enthalpies in HOFs and various MOFs. (d) IAST adsorption selectivities of C2H2/CO2 in equimolar mixture in HOF-3a and various MOFs at 296 K. (e) Breakthrough curve for an equimolar C2H2/CO2 mixture at 1×105 Pa and 296 K. Reprinted with permission from ref. [85], Copy right 2015 Wiley-VCH
Figure 16
(a) The carboxy dimer and π-π stacking in HOF-TCBP; (b) 1D channel in HOF-TCBP; (c~e) The adsorption isotherms of various C4 over HOF-TCBP at 295 K; (f) IAST predicted the adsorption selectivities of C4/C1 for HOF-TCBP at 295 K and 101 kPa. Reprinted with permission from ref. [116], Copy right 2016 Nature Publishing Group
Figure 17
(a) The structure of HOF-5. (b) The sorption isotherms of CO2 and CH4 over HOF-5. (c) The sorption isotherms of C2H2 and CH4 over HOF-5. (d) IAST selectivities of CO2/CH4 and C2H2/CH4. Reprinted with permission from ref. [87], Copy right 2015 American Chemical Society
Figure 18
(a) The building unit of UPC-HOF-6. (b) Digital photo of UPC-HOF-6. (c) The structure of UPC-HOF-6. (d) 1D channel in UPC-HOF-6. (e) Top-view SEM of UPC-HOF-6-120 film. (f) Single gas permeation of UPC-HOF-6-120 membrane at 25 ℃. Reprinted with permission from ref. [93], Copy right 2020 Wiley-VCH
Figure 19
(a) The structure of HOF-GS-10 and HOF-GS-11. (b) Arrhenius plot of HOF-GS-10 and HOF-GS-11. (c) Proton conduction values of HOF-GS-10 and HOF-GS-11 at various humidity and 30 ℃. Reprinted with permission from ref. [103], Copy right 2016 Wiley-VCH
Figure 20
(a) The construction of homochiral nanoscale metal-organic cages. (b) Asymmetric [3+2] coupling of 3-substituted indoles with quinone monoimine. (c) Asymmetric Friedel-Crafts reaction of N-sulfonyl aldimines with indole. Reprinted with permission from ref. [70], Copy right 2020 Nature Publishing Group
Figure 21
(a) The cage-based organic building units and the intramolecular hydrogen bonding between building units. (b) Suzuki-Miyaura coupling reactions catalyzed by HOF-19⊃pd(Ⅱ). Reprinted with permission from ref. [131], Copy right 2019 American Chemical Society
Figure 22
(a) The structure of Tp-1, T12-1, T18-1, and Ex-1. The luminescence (solid line) of (b) Tp-1, (c) T12-1, (d) T18-1, and (e) Ex-1. Reprinted with permission from ref. [50, 52], Copy right 2016 American Chemical Society
Figure 23
(a) The construction of HOF-10 and HOF-5. The structure of (b) HOF-10 and (c) HOF-5. Luminescent response of (d) HOF-10 and (e) HOF-5 to various concentration of Ag(Ⅰ). Reprinted with permission from ref. [91], Copy right 2017 Royal Society of Chemistry
Figure 24
(a) The structure of 8PN. (b) Photographs of 8PN taken under UV-light irradiation (365 nm). (c) Emission spectra of 8PN excited at 365 nm. Reprinted with permission from ref. [35], Copy right 2019 Nature Publishing Group
Figure 25
(a) Carboxy dimer and building unit in HOF-20. (b) The structure of HOF-20. (c) Luminescent response of HOF-20 to various aromatic compounds. Reprinted with permission from ref. [74], Copy right 2020 American Chemical Society
Figure 27
The structure and synergistic chemo-photodynamic therapy of nr-HOF@Doxo. Reprinted with permission from ref. [121], Copy right 2019 American Chemical Society
Figure 29
(a) The structure of HOF-2. (b) The topology of HOF-2. (c) The structure of HOF-2 after the adsorption of R-1-phenylethanol. (d) The chiral cavity in HOF-2. The interactions between HOF-2 and (e) R-1-phenylethanol/(f)
S-1-phenylethanol. Reprinted with permission from ref. [84], Copy right 2014 American Chemical Society
Figure 30
(a) Synthesis of HcOF-1 from 1crystal. (b) A saturated iodine aqueous solution (ca. 1.2 mmol•L-1) upon addition of HCOF-1 (3.0 mg). (c) The iodine desorption of I2@HCOF-1 in DMSO. Reprinted with permission from ref. [150], Copy right 2017 American Chemical Society
Figure 31
(a) The structure of (G2NDS)⊃(3aR)-(+)-Sclareolide and encapsulated Sclareolide, (b) (G2NDS)⊃(drospirenone)(methanol)0.84- (H2O)0.1 and encapsulated drospirenone, and (c) (G2BDPYDS)⊃ (progesterone) and encapsulated progesterone. Reprinted with permission from ref. [157], Copy right 2019 Nature Publishing Group


 下载:
下载:




























 下载:
下载:
































 下载:
下载:
