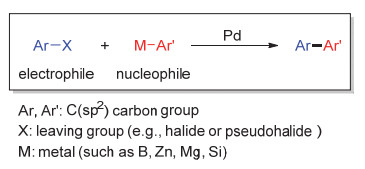
图 1.
钯催化交叉偶联反应
Figure 1.
Pd-catalyzed cross-coupling reactions

碳—碳键(C—C键)是有机化合物结构中最常见也是最重要的组成部分.构筑C—C键的反应研究贯穿有机化学发展历史, 例如格氏反应、Diels-Alder反应、Ziegler-Natta聚合反应、Wittig反应以及烯烃复分解反应等.在构筑C—C键的反应中, 过渡金属催化的偶联反应是最为高效的方法之一,代表性的偶联反应包括钯催化Heck反应、Negishi偶联与Suzuki偶联反应等(图 1).这些反应为C(sp2)—C(sp2)键构筑提供了新方法, 在有机合成、药物合成以及材料合成中得到了广泛应用, 并于2010年被授予诺贝尔化学奖.
与形成C(sp2)—C(sp2)键的芳基或烯基交叉偶联反应相比, 形成C(sp3)—C(sp3)键的烷基交叉偶联反应一直是合成化学家面临的一个挑战.例如, 利用钯催化剂进行烷基偶联时, 反应往往难以实现.主要难点在于钯催化剂难以对大位阻烷基卤代物等底物进行氧化加成, 以及烷基钯中间体容易发生β-H消除等副反应.
同为第十族元素的镍有着不同于钯的物理和化学性质, 这些性质使得镍催化剂在催化烷基交叉偶联反应中展现出较好的催化活性.具体而言, 低价态的镍比钯更容易失去外层价电子发生氧化加成反应, 其可逆过程还原消除反应则相对困难; 由于镍原子半径比钯小, 发生β-H消除反应时,相应烷基镍物种需要比烷基钯物种克服更大的旋转能垒才能到达合适构象, 因此在偶联反应中C(sp3)—Ni更难发生β-H消除反应; 相对于钯而言, 镍的价态变化更加丰富, 目前已经分离鉴定的镍化合物氧化态包括Ni(0)、Ni(I)、Ni(II)、Ni(III)、Ni(IV), 而钯的常见价态为Pd(0)、Pd(II), 少数情况下为Pd(IV); 在催化交叉偶联反应中, 钯通常经历双电子转移过程, 即Pd(0)/Pd(II)循环, 而镍既有Ni(0)/Ni(II)双电子转移过程, 也有Ni(I)/Ni(II)/Ni(III)单电子转移过程; 另外, 镍金属在地壳中的含量丰富, 而钯非常稀少(图 2)[1].所有这些特点使镍金属催化剂成为了烷基交叉偶联反应中的研究热点.
在近二十年里, 镍催化烷基交叉偶联反应发展迅速, 已经成为构筑C(sp3)—C(sp3)键的有效方法, 并且在天然产物、药物合成中得到了应用.本文将从镍催化烷基亲电试剂与金属有机试剂交叉偶联反应、导向基参与的C(sp3)—H键活化的偶联反应、镍-光反应催化剂协同催化偶联反应、烷基亲电试剂之间的还原偶联反应和烯烃加成反应等方面介绍镍催化烷基-烷基偶联构筑C(sp3)—C(sp3)键的研究进展.
1995年, Knochel等利用Ni(acac)2催化剂实现了远端带有烯基[2]或者羰基[3]等配位基团的一级烷基溴化物与烷基锌试剂的交叉偶联反应.控制实验表明, 远端带有烯基官能团时, 烷基溴代物1可以以较高收率得到产物2, 当采用远端无烯基的溴代物3时, 得到的仅仅是锌卤交换产物4 (图 3).作者认为烯烃双键会对镍原子配位形成中间体Int-1, 由于中心金属镍达到配位饱和, 有效抑制β-H消除副反应.

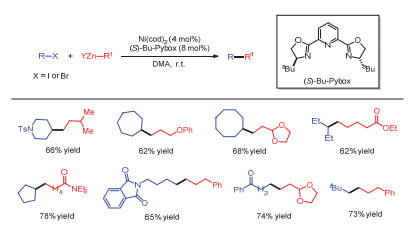
在随后的研究中, Knochel[3]以及Kambe[4]等通过外加苯乙酮或者烯烃达到同样的配位作用, 避免了在底物中预先引入特定官能团, 进一步拓展了该类交叉偶联反应的一级烷基卤代物底物范围(图 4).对于二级和三级烷基卤化物该偶联反应难以发生.有研究者认为金属催化剂对烷基卤化物的氧化加成是SN2过程, 二级或者三级烷基卤化物相对于一级烷基卤化物位阻增大, 金属催化剂对其氧化加成难以发生[5].

2003年, Fu等[6]利用Ni(cod)2为催化剂、PyBOX为配体,在室温下首次实现了二级烷基卤化物与烷基溴化锌试剂的交叉偶联反应, 反应体系温和, 不同类型官能团例如杂环、羰基等都能较好兼容(图 5).实验证明三齿螯合配体PyBOX对反应的高效进行至关重要.作者推测这是由于多齿配体能有效占据金属的配位点, 阻碍β-H消除副反应的发生.这一研究工作开启了多取代烷基卤化物与金属有机试剂交叉偶联反应的序幕, 为叔碳或者季碳手性中心的不对称构筑提供了可能性.
紧接着, 多个课题组对这一反应的底物适用范围进行了研究, 发现亲电试剂可以拓展到氯代物、氟代物等; 烷基可以是二级和三级烷基; 金属试剂可以是烷基硼、镁、锌、硅化物等[7].
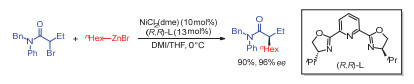
2005年, Fu等[8]首次报道了二价镍作催化剂、(iPr)-PyBOX为手性配体条件下, 消旋α-溴代酰胺与烷基溴化锌的不对称交叉偶联反应, 反应以较高收率和对映选择性得到手性酰胺化合物(图 6).作者发现底物中酰胺基团对于反应的对映选择性控制至关重要.反应回收的溴化物仍然为消旋体, 因此排除了反应经过动力学拆分历程的可能性.
随后, 一系列镍催化烷基卤代物的不对称交叉偶联反应被报道.在这些反应中, 使用最多的是含氮手性配体(例如手性噁唑啉配体), 且不同反应只有采用合适的手性含氮配体才能取得高对映选择性[9] (图 7).

与此同时, 镍催化不对称烷基卤代物交叉偶联反应对亲电性底物具有一定的要求, 往往采用活化的烷基卤化物[10a-10e](如苄基溴和烯丙基溴等)或者带有导向基团(如羰基、酯基、氨基等)的底物[10f-10i]类型时, 反应才可取得较优异的对映选择性结果(图 8).

2016年, Fu等[11]利用二价镍作催化剂, 手性二胺作配体, THF/DMA作混合溶剂, 在0 ℃下实现了α-卤代硼酸酯与有机锌试剂的不对称Negishi偶联, 得到手性烷基硼酸酯类产物.在最优反应条件下, 不同取代的烷基锌试剂都可以获得很高的收率和对映选择性.反应对醚、氰基、缩醛、酯基、卤素等官能团都可以很好地兼容.手性的烷基硼酸酯类产物可以应用于迭代增碳反应中, 实现连续手性中心的构筑(图 9).

2017年, Fu等[12]以二价镍作催化剂、(iBu)-PyBOX作手性配体, 实现了α-溴代硅烷与烷基锌试剂的不对称Negishi交叉偶联反应, 为手性硅化合物的合成提供了新方法(图 10).

Hu等[7c]利用镍-三齿配体螯合物实现了一级和二级烷基卤化物与烷基格氏试剂的Kumada偶联反应.在低温条件下, 反应对酮羰基或者酯基都能有较好的官能团耐受性(图 11a).基于该催化体系, Hu等[13]进行了动力学实验以及密度泛函理论(DFT)计算, 首次提出了自由基参与的双金属氧化加成机理:二价镍催化剂1首先与格氏试剂作用得到配合物2, 随后发生转金属化得到中间体3, 中间体3与烷基卤化物发生单电子还原作用得到烷基自由基以及三价镍物种4; 烷基自由基与二价镍中间体3进行自由基加成反应得到三价镍中间体5, 随后还原消除得到Kumada偶联产物以及一价镍配合物6; 活泼的一价镍6与三价镍4发生逆歧化反应得到二价溴化镍1′和烷基镍3′, 完成催化循环(图 11b).反应过程中镍催化剂的富集状态是二价镍物种, 而一价镍以及三价镍均为瞬态出现, 相应的顺磁性中间体也无法通过EPR手段检测到.在催化循环中, 二价镍中间体3与3′之间处于动态平衡, 分别参与到烷基自由基的生成以及氧化加成过程中, 这与Fu等[14]提出的双金属机理较为相似.采用不同催化体系反应机理会有所不同, 但总体而言, 该类型反应目前被认为是经由Ni(I)/Ni(II)/Ni(III)多价态镍中间体的自由基过程[15].

低价镍金属催化剂易于对苄位C—O键进行氧化加成, 苄基醚、酯甚至活性较低的苄醇化合物都可以在低价镍催化下断裂C—O键参与到偶联反应中[16].
2008年, 施章杰等[17a]利用镍催化剂实现苄基烷基醚与烷基格氏试剂的偶联反应.反应具有较高的化学选择性, 当底物中同时存在C(sp2)—OMe与苄位C(sp3)—OMe键时, 可通过使用双膦配体调控反应的选择性, 使之专一性地发生在苄位(图 12). 2012年, 该课题组[17b]又实现了镍催化苄醇与甲基格氏试剂的偶联反应.

2011年, Jarvo等[18]报道了光学活性苄基甲基醚与烷基格氏试剂的交叉偶联反应.反应底物在向产物的转化过程中光学纯度得以保持, 但是构型发生了翻转.利用该方法可以方便地合成1, 1′-双芳基乙烷类抗癌活性化合物(图 13).

脂肪羧酸在自然界中广泛存在, 具有化学性质稳定、廉价易得等特点.受Barton酯在自由基引发条件下脱羧形成烷基自由基的启发, Baran等[19]在2016年报道了镍催化氧化还原活化酯与烷基锌试剂的交叉偶联反应.反应底物可以是含一、二、或三级烷基的羧酸酯、杂环羧酸酯等, 反应可用于固相肽的合成和官能团化.关于反应机理, 作者认为脂肪羧酸衍生物的反应路径与烷基卤代烷相似, 一价镍首先和锌试剂发生转金属化反应形成烷基镍中间体, 再和羧酸酯经单电子转移(SET)过程形成二价镍中间体; 羧酸酯得到电子并脱羧形成烷基自由基, 烷基自由基与二价镍再结合形成三价镍中间体; 三价镍中间体发生还原消除得到偶联产物, 同时生成一价镍完成催化循环(图 14).

伯胺基团广泛存在于有机化合物中, 从简单的小分子到合成中间体、大分子、药物以及天然产物, 都可见到它的身影.使用胺替代烷基卤化物作为亲电试剂可以拓展烷基-烷基C—C键偶联反应的应用范围.
2019年, Watson等[20]将脂肪胺与2, 4, 6-三甲基吡喃鎓四氟硼酸盐经过缩合反应得到Katritzky吡啶盐, 以二价镍作为催化剂、取代三联吡啶或者二吡唑基吡啶作为配体, 实现了这种吡啶盐与烷基锌试剂的偶联反应.常见的一级胺、二级胺甚至天然胺类化合物都能发生衍生化并进行偶联反应.反应具有较广的底物范围、优良的官能团兼容性(图 15).

在机理研究方面, 作者进行了自由基捕获以及自由基探针实验, 实验结果均证明反应中存在自由基中间体.结合以往镍催化烷基卤化物偶联反应机理, 作者提出了胺的偶联反应的可能机理:吡啶盐底物首先与一价镍发生SET反应, 接下来吡啶基离去形成烷基自由基以及二价镍中间体, 烷基自由基与二价镍结合后经还原消除过程得到最终产物.
在近年来, 导向基参与的C—H键活化被广泛应用于过渡金属催化反应.这些反应采用导向基策略对惰性C—H键、C=C键的反应活性和反应位点进行精准调控[21].
2014年, Ge等[22]利用氨基喹啉作为导向基实现了一级烷基C—H键与烷基卤化物的交叉偶联反应(图 16).作者发现, 膦配体的选择对反应至关重要, 当使用碘化铯时, 烷基溴化物以及氯化物都可以作为偶联组分.在机理研究中, 作者进行了自由基捕获实验, TEMPO的加入使反应收率降低, 同时能分离到烷基自由基与TEMPO的加合物.基于此, 作者提出了Ni(II)/Ni(III)/Ni(I)催化循环机理.但是, 也不能完全排除Ni(II)/Ni(IV)催化循环机理.

2014年, MacMillan、Doyle以及Molander等[23]同时提出镍-光氧化还原协同催化概念, 在光照条件下利用镍催化剂和光反应催化剂协同实现羧酸或者氟硼酸钾化合物与芳基卤化物的脱羧或者脱硼交叉偶联反应构筑C(sp3)—C(sp2)键, 这种协同催化策略拓展了偶联反应底物范围[24].
2016年, MacMillan等[25]采用脂肪羧酸和卤代烷作为偶联底物, 在光反应催化剂和镍催化剂的协同作用下, 实现了更为困难的烷基-烷基偶联反应.该反应条件温和, 避免使用当量金属试剂, 操作简便, 底物适用性广, 适用于许多一级和二级脂肪羧酸.作者还用该反应合成了药物分子Triofiban-HCl (图 17).

作者推测了镍-光反应催化剂协同催化反应机理: (1)光反应催化剂被光激发到高氧化性的激发态, 随后对羧酸底物进行单电子氧化, 光反应催化剂变成还原性的低价态.与此同时, 失去电子之后的羧酸脱羧脱质子得到烷基自由基; (2)零价镍催化剂捕获烷基自由基得到一价烷基镍中间体; (3)一价镍中间体与烷基卤化物经过连续单电子转移过程生成三价镍中间体, 三价镍中间体发生还原消除反应得到目标产物和一价镍中间体; (4)一价镍中间体经低价态光反应催化剂还原生成零价镍, 完成催化循环.
有机分子一般含有大量的C(sp3)—H键, 如果能在过渡金属催化的交叉偶联反应中直接使用它们作为亲核试剂, 将可以避免底物官能团化过程, 能在很大程度上简化合成步骤.但是, 有机分子中含有各种C(sp3)— H键, 如何控制反应的区域选择性就成为了巨大的挑战.自然界中, 酶通过识别有机分子中C—H键的各种热力学和动力学特征完成C—H键的选择性官能化.受生物化学反应途径的启发, MacMillan等推测, 有机小分子催化剂也可能利用键能和键极化度差别区分不同的C—H键.基于此, 他们发展了镍-光反应-氢原子转移三重催化C(sp3)—H键偶联反应.反应中, 氢原子转移催化剂奎宁环在光反应催化剂的协助下形成活性阳离子铵自由基, 随后攫取脂肪胺C(sp3)上的氢原子产生碳自由基, 碳自由基被镍催化剂捕获并发生偶联反应, 生成的一价镍中间体经光反应催化剂还原得到零价镍催化剂, 完成催化循环(图 18).该策略直接利用C(sp3)—H键作为亲核试剂构筑C(sp3)—C(sp3)键, 具有广阔的应用前景.这一策略已经被用于α-氨基、α-烷氧基及α-硫代化合物C(sp3)—H键与烷基卤化物的直接偶联反应[26].

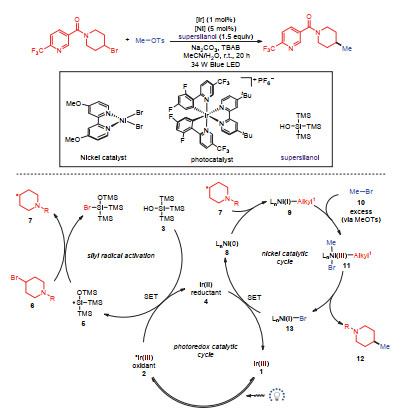
2018年, MacMillan等[27]利用镍-光氧化还原协同催化剂实现双烷基亲电试剂的交叉偶联反应.为了提高反应的化学选择性, 避免底物自身偶联副反应, 需要控制双亲电试剂底物用量物质的量比在2:1~5:1.在建立的标准条件下, 一级或者二级烷基卤代烷均可有效地参与到反应中完成烷基-烷基碳—碳键构筑.作者基于实验结果以及前期工作的报道, 提出机理路径如下: (1)在光照条件下, 光催化剂受到激发生成氧化性更强的激发态催化剂2, 对硅醇进行单电子氧化得到硅自由基, 由于Si—Br键相对于C—Br键具有更高的键解离能, 硅自由基会攫取溴代物的卤原子生成具有亲核性的烷基自由基7; (2)体系中的Ni(0)会捕获烷基自由基得到Ni(I)物种9, 伴随着一价镍物种对另一组分烷基亲电试剂氧化加成得到三价镍中间体11, 还原消除得到目标产物12以及Ni(I) 13; (3)一价镍13经低价态光反应催化剂4还原生成零价镍, 完成催化循环(图 19).
本节中, 还原偶联反应是指在还原剂作用下多组分亲电试剂之间的交叉偶联反应[28].相比于氧化还原中性的交叉偶联反应, 还原偶联反应能避免使用水氧敏感的金属有机试剂, 提高反应的实用性; 还原偶联反应的一大挑战在于如何避免亲电试剂自身偶联, 即如何控制反应的化学选择性.实际上, 烷基亲电试剂之间的还原交叉偶联反应是最为困难的偶联反应之一.
2011年, Gong等[29]采用镍-吡啶双噁唑啉催化剂, 以锌粉作为还原剂, 实现了两种不同烷基卤化物的还原偶联反应, 得到烷基-烷基交叉偶联产物.反应虽然表现出良好的底物适用范围以及官能团耐受性, 但是也难以避免地发生自身偶联, 反应中需要将一种底物的用量增加到三倍(图 20).

为了解决烷基亲电试剂与烷基亲电试剂还原交叉偶联反应中化学选择性问题, Gong等[30]利用[B(pin)]2做还原试剂, 将烷基卤化物用量物质的量比降为1.5:1, 实现了二级或者更高级烷基卤化物与一级烷基卤化物的还原交叉偶联反应, 反应具有较高的收率和选择性.作者通过机理研究排除了反应中现场生成C—Bpin化合物再进行Suzuki偶联的过程(图 21).

多数情况下, 镍催化双烷基亲电试剂交叉偶联反应局限在二级烷基卤代物与一级烷基卤代物或者大位阻烷基卤代物与小位阻烷基卤代物这些活性存在一定差别的组分之间进行. 2019年, Osaka等[31]基于镍、钴协同催化体系成功实现了烷基对甲苯磺酸酯与烷基卤代物之间的还原交叉偶联反应.该催化体系反应条件温和, 底物用量物质的量比为1.5:1.反应不仅可以实现二级和一级烷基亲电试剂交叉偶联, 还可以进行一级和一级亲电试剂的还原偶联.作者认为反应经过了如下过程: (1)在锰还原剂作用下, VB12被还原为亲核性的一价钴物种F, 随后与ROTs经过氧化加成得到三价钴中间体G; (2)中间体G与Ni(0)经过转金属化得到烷基镍物种B, 在锰还原作用下, 生成一价镍C, 接下来与另一组分烷基卤代烷发生氧化加成得到双烷基镍中间体D, 还原消除得到目标产物以及一价镍中间体E; (3)在锰作用下, 中间体E被还原为零价镍A, 完成循环(图 22).

烯烃是一类非常重要的化工原料,具有来源广泛、方便易得等特点.过渡金属催化烯烃加成反应是从简单原料出发合成高附加值化合物的有效途径[32], 是由二维线性分子构筑三维立体分子重要的方式.在本节中, 将介绍近些年来烯烃经过氢烷基化以及双碳官能团化构筑C(sp3)—C(sp3)键的研究进展.
镍催化烯烃氢烷基化反应是在烯烃双键两端分别引入氢原子和烷基, 生成一个C(sp3)—H键和一个C(sp3)—C(sp3)键.烯烃氢烷基化反应近年来受到广泛的关注, 已经发展出多种催化体系, 催化模式各异, 反应选择性也不尽相同[33].
2016年, 傅尧、刘磊等[34]报道在硅烷存在下, 镍-双氮配体催化剂能够催化烷基卤化物与α-烯烃的氢烷基化反应, 得到反马氏加成产物.反应避免了活泼金属有机试剂的使用, 所用烯烃、卤化物底物范围较广, 能够兼容羟基、羰基、含氮杂环等官能团, 可用于天然产物等复杂化合物的后期官能团修饰(图 23).
2018年, 朱少林[35]以及王细胜[36]等分别报道了分子内烯烃与烷基卤代物的还原氢烷基化反应, 所得产物都为烯烃移位到端位再加成产物.他们均认为反应中存在Ni—H物种参与的烯烃链行走过程.除了烷基卤代物[37]外, 烷基羧酸衍生物[38]及烷基胺衍生物[39]均可作为烷基自由基前体参与到镍催化烯烃的氢烷基化反应中.
2018年, Fu等[40]发展了手性镍-双噁唑啉催化烯烃与二级、三级卤化物的对映选择性氢烷基化反应, 产物的收率和对映选择性都较高.反应在水和空气条件下进行时, 对映选择性不受影响, 只是产率稍有降低.从外消旋的三级烷基亲电试剂构建手性季碳中心的反应的对映选择性控制是一个长期的难题.作者发现手性镍-双噁唑啉催化剂可以有效控制α-卤代内酰胺对烯烃的加成反应的对映选择性(图 24).

作者提出了两种可能的反应途径, 第一种是烯烃在镍催化下发生氢硅化得到烷基硅烷, 烷基硅烷作为亲核试剂与亲电试剂发生交叉偶联反应.为了验证该过程的可能性, 作者直接用烷基硅化合物参与反应, 但仅获得少量产物, 由此证明该反应途径的可能性较小.另外一种反应途径是LBrNiII—Br (A)首先与硅烷反应形成镍氢物种LBrNiII—H (B), 随后与烯烃发生迁移插入反应形成烷基镍中间体LBrNiII—CH2CH2R (C), C与烷基自由基结合并发生还原偶联反应, 得到产物(图 25).为了验证这一猜想, 作者制备了镍中间体A和C, 并通过当量实验证明了这两个中间体的存在.此外, 电子顺磁共振光谱也证明了反应中存在自由基中间体.

在Fu等报道以后, 朱少林[41]以及傅尧[42]等也报道了类似的镍催化不对称烷基卤化物与烯烃的不对称还原偶联反应, 卤代物范围拓展到了α-溴代酰胺、α-溴代磷酸酯、α-溴代磺酸酯等化合物. 2020年, Fu课题组[43]在前期工作的基础上, 利用二价镍-手性双噁唑啉催化体系, 进一步实现氧原子α位卤代物参与的烯烃不对称氢烷基化反应, 完成手性双烷基甲醇类化合物的构筑(图 26).

在还原条件下, 镍催化烯烃与烷基卤化物的氢官能团化反应由于其经历单电子自由基过程所得均为反马氏加成产物, 马氏加成产物则难以合成. 2019年, Shenvi等[44]利用双金属协同催化体系巧妙实现了烷基卤化物对烯烃的马氏加成(图 27).作者发现, 同时使用镍催化剂和锰催化剂才能得到马氏加成产物; 而同时使用醇和硅烷对于提高收率也十分必要.需要强调的是, 反应虽然是个还原体系, 但需要在空气下进行才能取得好的结果.各种烷基碘代物都呈现较高的反应活性.反应对于酯基、酰胺、硅醚、环氧等官能团都表现出较好的兼容性.

关于反应机理, 作者认为不同金属催化剂分别参与到两个独立的循环中.首先三价锰化合物与硅烷进行负氢转移得到锰氢化合物, 随后与烯烃发生迁移加成反应得到马氏加成产物, 经过均裂过程得到烷基自由基以及二价锰, 后者经氧气氧化得到三价锰参与下一次循环; 烷基自由基与零价镍作用得到烷基镍中间体, 随后与烷基碘化物发生氧化加成得到三价镍中间体.三价镍中间体发生还原消除得到产物以及一价镍, 一价镍随后被硅烷还原为零价镍完成催化循环(图 28).

在镍催化烯烃与烷基卤化物的氢烷基化反应中负氢来自硅烷, 所以反应往往需要使用当量以上的硅烷试剂, 这降低了反应的原子经济性.因此, 开发出氧化还原中性的催化体系是该领域的重要课题.
2018年, 周其林等[45]发现醇可以作为零价镍催化体系的负氢来源.当用醇作反应溶剂时, 在催化量的碱存在下, 零价镍可以催化酮与共轭二烯的加成反应.反应条件温和, 不同类型的酮如芳香酮、脂肪酮以及醛和酯等均能反应, 并能以高收率和高区域选择性得到相应的氢烷基化产物.在此基础上, 他们使用手性联苯双膦配体实现了反应对映选择性控制.关于反应机理, 他们认为首先是镍与醇以及共轭二烯经过氢转移过程(经过A或者C中间体)得到烯丙基镍中间体, 随后与酮的烯醇负离子发生配体交换, 再经还原消除得到目标产物.与以往硅烷作为氢源的烯烃氢烷基化反应不同, 该催化反应直接用溶剂乙醇作为氢源, 不需要外加负氢试剂, 催化体系呈氧化还原中性, 具有较高原子经济性, 并且对环境友好(图 29).类似的工作还包括邢栋课题组[46]报道的镍催化简单酮与1, 3-丁二烯氢烷基化反应.

2019年, 周其林[47a]和李朝军[47b]等几乎同时报道了腙作为碳亲核试剂与共轭二烯的氢烷基化反应.反应为氧化还原中性, 负氢由醇提供.反应拓展了烷基亲核试剂的类型, 为在烯丙位引入烷基提供了新策略.反应的局限性在于只能使用苯甲醛腙, 因而引入的烷基只能是苄基(图 30).

2020年, 陈庆安等[48]利用烯丙基硼酸酯作为烷基亲核试剂与共轭二烯进行氢烯丙基化反应得到1, 5-二烯化合物(图 31).

镍催化分子间烯烃双碳官能团化反应是在烯烃双键两端分别引入含碳基元, 是由简单化合物一步构建多官能团化合物最有效的方法之一.该反应近年来得到了广泛的关注, 并且取得了出色研究成果[49].
共轭烯烃例如丙烯酸酯等能与自由基发生Giese加成反应, 得到烯烃氢官能团化产物.如果用镍(II)对Giese加成过程中产生的烷基自由基进行捕获就可以实现烯烃的1, 2-双官能团化(图 32a). 2016年, Baran等[50]利用丙烯酸苄酯、N-羟基邻苯二甲酰亚胺酯、苯基氯化锌实现了烯烃的1, 2-双官能团化反应(图 32b).在标准条件下, 一系列烷基羧酸衍生的活化酯如杂环烷基羧酸、张力环烷基羧酸、含杂原子取代的烷基羧酸等均可以实现1, 2-双官能团化反应.在反应机理上, 作者认为首先是羧酸酯经过单电子转移过程脱酸产生烷基自由基, 随后自由基对烯烃双键进行加成得到新的烷基自由基, 二价镍与烷基自由基结合得到三价镍, 再经过还原消除得到产物. 2018年, Giri等[51]报道了苯乙烯类型底物的1, 2-烷基芳基化反应, 作者通过机理研究认为反应经历了自由基加成过程(图 32c).

2019年, Morken等[52]报道了以烯基硼酸酯为底物的不对称1, 2-双烷基化反应.反应使用二价镍作为催化剂, 手性双胺作为配体, 反应中相应的偶联组分是烷基卤化锌、三级烷基卤化物, 烷基硼酸酯产物可以通过氧化得到相应的手性醇. α-硼自由基的稳定性是反应能够控制对映选择性的重要原因(图 32d).
2017年, Nevado等[53]利用四三(二甲胺基)乙烯(TDAE)作为还原试剂, 发展了镍催化烯烃分子间还原1, 2-烷基芳基化反应, 烯烃底物类型为特定位点有螯合基团的α-烯烃(图 33). 2019年, Nevado等[54]基于类似的还原偶联体系实现了非活化脂肪烯烃的1, 2-烷基芳基化反应.烯烃底物不需要引入导向基团或者活化基团, 较以往工作在底物范围上取得了重要突破.
2019年, 基于镍-光氧化还原协同催化策略, 储玲玲[55a]、Nevado[55b]、Molander[55c]等几乎同时报道了分子间烯烃的1, 2-烷基芳基化反应.反应机理也较为类似, 反应之间不同点主要在于反应所使用的烷基自由基前体、光催化剂种类(图 34).以储玲玲的工作为例, 反应的机理如下: (1)在光照条件下, 光催化剂受到激发得到氧化性更强的激发态催化剂2, 对草酸盐进行单电子氧化脱除二氧化碳得到烷基自由基, 随后与烯烃双键发生自由基转移反应得到二级烷基自由基物种6; (2)体系中的Ni(0)会捕获烷基自由基得到Ni(I)物种8, 伴随着一价镍物种对另一组分芳基亲电试剂氧化加成得到三价镍中间体10, 还原消除得到目标产物11以及Ni(I)中间体12; (3)中间体12经低价态光反应催化剂4还原生成零价镍, 完成催化循环(图 34d).

非活化的脂肪烯烃是加成反应的重要底物类型, 但是这类底物由于缺少稳定自由基的基团, 1, 2-双官能团化非常具有挑战.为了解决脂肪烯烃底物的官能团化反应性低和选择性差的问题, 多个课题组进行了有益探索, 其中导向基策略成为最主要的解决方案.
2017年, Engle等[56]利用可脱除的8-氨基喹啉作为导向基团, 实现了β, γ-不饱和羰基化合物的区域选择性的1, 2-芳基烷基化反应.端烯或者内烯都可以与烷基锌试剂、芳基碘试剂发生三组分偶联反应得到产物.机理上, 首先是镍与芳基碘代物发生氧化加成得到二价镍物种, 随后与底物的导向基进行配位, 烯烃双键对芳基镍物种进行分子内的顺式迁移插入形成五元镍杂环.由于镍杂环非常稳定, 使得β-H消除过程被抑制.随后与锌试剂发生转金属化和还原消除得到1, 2-双官能团化产物, 完成催化循环(图 35).

基于同样的导向基策略, Engle等[57]利用烷基锌、烷基卤化物实现了非共轭烯烃的1, 2-双烷基化反应.反应中使用的导向基仍然是氨基喹啉, 但是反应却给出与上述芳基化反应不同的区域选择性.具体而言, 烷基亲核试剂加成到γ-位.在标准反应条件下, 许多烯烃底物如三取代烯烃、γ, δ-不饱和烯烃等都能反应, 得到顺式加成产物(图 36).

过去几十年见证了镍催化偶联反应的快速发展, 镍催化剂也被寄托了更多的希望, 它已不仅仅被用作贵金属钯催化剂的替代物, 更是展现出与其它金属催化剂的互补性与独特性, 镍催化剂在烷基-烷基碳—碳键构筑反应中的表现更是如此.例如:在烷基亲电试剂与烷基金属试剂的交叉偶联反应中镍催化剂表现出比其它金属催化剂更高的催化活性和选择性, 烷基亲电试剂已经从溴化物、碘化物拓展到氯化物、氟化物以及羧酸衍生物和胺类化合物等; 镍催化反应与光反应结合实现了不同烷基亲核试剂与烷基卤化物的交叉偶联反应, 反应用光驱动更加符合绿色合成化学的要求, 反应使用羧酸、惰性C—H键等作为亲核试剂前体更进一步丰富了烷基-烷基碳—碳键构筑方式; 烯烃是一类来源广泛的大宗化工原料, 镍催化烯烃参与的官能团化反应是由简单原料出发合成高附加值化合物的重要方法, 具有高原子经济性和步骤经济性, 有效地提高了合成效率.
尽管目前镍催化烷基-烷基碳—碳键构筑反应研究已经取得了很大进展, 但是还有很多挑战, 例如: (1)目前很多烷基-烷基偶联反应使用零价镍催化剂, 其前体是20世纪60年代由Wilke发展的Ni(cod)2, 该催化剂前体对水氧较为敏感, 操作和保存要求十分严格, 使用不方便.今后需要发展更加稳定的、易操作的高活性零价镍催化剂[58]. (2)目前镍催化不对称烷基-烷基偶联反应种类非常有限, 底物中有苯基、烯丙基、吸电子基团或者导向基等稳定自由基中间体时才能取得高对映选择性.非活化的烷基自由基前体参与的不对称偶联反应尚未得到有效解决; (3)随着镍催化烷基-烷基碳—碳键构筑方法多样化, 反应机理的认识越发成熟, 今后将会在天然产物和药物分子合成中有更多的应用.
For reviews on nickel catalysis: (a) Tasker, S. Z.; Standley, E. A.; Jamison, T. F. Nature, 2014, 509, 299. (b) Ananikov, V. P. ACS Catal. 2015, 5, 1964. (c) Clevenger, A. L.; Stolley, R. M.; Aderibigbe, J.; Louie, J. Chem. Rev. 2020, 120, 6124. (d) Choi, J.; Fu, G. C. Science 2017, 356, eaaf7230. (e) Modern Organonickel Chemistry, Eds.: Tamaru, Y., Wiley-VCH, Weinheim, 2005. (f) Nickel Catalysis in Organic Synthesis: Methods and Reactions, Eds.: Ogoshi, S., Wiley-VCH, Weinheim, 2020.
Devasagayaraj, A.; Stüdemann, T.; Knochel, P. Angew. Chem. Int. Ed. 1996, 34, 2723. doi: 10.1002/anie.199527231
(a) Giovannini, R.; Stüdemann, T.; Dussin, G.; Knochel, P. Angew. Chem. Int. Ed. 1998, 37, 2387. (b) Giovannini, R.; Stüdemann, T.; Devasagayaraj, A.; Dussin, G.; Knochel, P. J. Org. Chem. 1999, 64, 3544. (c) Piber, M.; Jensen, A. E.; Rottl nder, M.; Knochel, P. Org. Lett. 1999, 1, 1323.
Terao, J.; Watanabe, H.; Ikumi, A.; Kuniyasu, H.; Kambe, N. J. Am. Chem. Soc. 2002, 124, 4222. doi: 10.1021/ja025828v
Hills, I. D.; Netherton, M. R.; Fu, G. C. Angew. Chem. Int. Ed. 2003, 42, 5749. doi: 10.1002/anie.200352858
Zhou, J.; Fu, G. C. J. Am. Chem. Soc. 2003, 125, 14726. doi: 10.1021/ja0389366
(a) Saito, B.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 9602. (b) Smith, S. W.; Fu, G. C. Angew. Chem. Int. Ed. 2008, 47, 9334. (c) Vechorkin, O.; Hu, X. Angew. Chem. Int. Ed. 2009, 48, 2937.
Fisher, C.; Fu, G. C. J. Am. Chem. Soc. 2005, 127, 4594. doi: 10.1021/ja0506509
Binder, J. T.; Cordier, C. J.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 17003. doi: 10.1021/ja308460z
(a) Arp, F. O.; Fu, G. C. J. Am. Chem. Soc. 2005, 127, 10482. (b) Son, S.; Fu, G. C. J. Am. Chem. Soc. 2008, 130, 2756. (c) Lundin, P. M.; Esquivias, J.; Fu, G. C. Angew. Chem. Int. Ed. 2009, 48, 154. (d) Smith, S. W.; Fu, G. C. J. Am. Chem. Soc. 2008, 130, 12645. (e) Liang, Y.; Fu, G. C. J. Am. Chem. Soc. 2014, 136, 5520. (f) Owston, N. A.; Fu, G. C. J. Am. Chem. Soc. 2010, 132, 11908. (g) Lu, Z.; Wilsily, A.; Fu, G. C. J. Am. Chem. Soc. 2011, 133, 8154. (h) Wilsily, A.; Tramutola, F.; Owston, N. A.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 5794. (i) Huo, H.; Gorsline, B. J.; Fu, G. C. Science 2020, 367, 559.
Schmidt, J.; Choi, J.; Liu, A. T.; Slusarczyk, M.; Fu, G. C. Science, 2016, 354, 1265. doi: 10.1126/science.aai8611
Schwarzwalder, G. M.; Matier, C. D.; Fu, G. C. Angew. Chem. Int. Ed. 2019, 58, 3571. doi: 10.1002/anie.201814208
Breitenfeld, J.; Ruiz, J.; Wodrich, M. D.; Hu, X. J. Am. Chem. Soc. 2013, 135, 12004. doi: 10.1021/ja4051923
Schley, N. D.; Fu, G. C. J. Am. Chem. Soc. 2014, 136, 16588. doi: 10.1021/ja508718m
Hu, X. Chem. Sci. 2011, 2, 1867.
(a) Tollefson, E. J.; Hanna, L. E.; Jarvo, E. R. Acc. Chem. Res. 2015, 48, 2344. (b) Su, B.; Cao, Z.-C.; Shi, Z.-J. Acc. Chem. Res. 2015, 48, 886.
(a) Guan, B.-T.; X, S.-K.; Wang, B.-Q.; Sun, Z.-P.; Wang, Y.; Zhao, K.-Q.; Shi, Z.-J. J. Am. Chem. Soc. 2008, 130, 3268. (b) Yu, D.-G.; Wang, X.; Zhu, R.-L.; Luo, S.; Wang, B.-Q.; Wang, L.; Shi, Z.-J. J. Am. Chem. Soc. 2012, 134, 14638.
Taylor, B. L. H.; Swift, E. C.; Waetzig, J. D.; Jarvo, E. R. J. Am. Chem. Soc. 2011, 133, 389. doi: 10.1021/ja108547u
Qin, T.; Cornella, J.; Li, C.; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell, B. D.; Eastgate, M. D.; Baran, P. S. Science 2016, 352, 801. doi: 10.1126/science.aaf6123
Plunkett, S.; Basch, C. H.; Santana, S. O.; Watson, M. P. J. Am. Chem. Soc. 2019, 141, 2257. doi: 10.1021/jacs.9b00111
占贝贝, 刘斌, 胡芳, 史炳锋, 中国科学-化学, 2015, 60, 2097.Zhan, B.-B.; Liu, B.; Hu, F.; Shi, B.-F. Sci. Chin. Chem. 2015, 60, 2097(in Chinese).
Wu, X.; Zhao, Y.; Ge, H. J. Am. Chem. Soc. 2014, 136, 1789. doi: 10.1021/ja413131m
(a) Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C. Science 2014, 345, 437. (b) Tellis, J. C.; Primer, D. N.; Molander, G. A. Science 2014, 345, 433.
(a) Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, 0052. (b) Tells, J. C.; Kelly, C. B.; Primer, D.V.; Jouffroy, M.; Patel, N. R.; Molander, G. A. Acc. Chem. Res. 2016, 49, 1429. (c) Milligan, J. A.; Phelan, J. P.; Badir, S. O.; Molander, G. A. Angew. Chem. Int. Ed. 2019, 58, 6152.
Johnston, C. P.; Smith, R. T.; Allmendinger, S.; MacMillan, D. W. C. Nature 2016, 536, 322. doi: 10.1038/nature19056
Le, C.; Liang, Y.; Evans, R. W.; Li, X.; MacMillan, D. W. C. Nature 2017, 547, 79. doi: 10.1038/nature22813
Smith, R. T.; Zhang, X.; Rincón, J. A.; Agejas, J.; Mateos, C.; Barberis, M.; García-Cerrada, S.; Frutos, O. D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2018, 140, 17433. doi: 10.1021/jacs.8b12025
For selected reviews of reductive cross-couplings: (a) Knappke, C. E. I.; Grupe, S.; G rtner, D.; Corpet, M.; Gosmini, C.; Jacobi von Wangelin, A. Chem. Eur. J. 2014, 20, 6828. (b) Gu, J.; Wang, X.; Xue, W.; Gong, H. Org. Chem. Front. 2015, 2, 1411. (c) Wang, X.; Dai, Y.; Gong, H. Top. Curr. Chem. 2016, 374, 43. (d) Lucas, E. L.; Jarvo, E. R. Nat. Rev. Chem. 2017, 1, No. 0065. (e) Poremba, K. E.; Dibrell, S. E.; Reisman, S. E. ACS Catal. 2020, 10, 8237.
Yu, X.; Yang, T.; Wang, S.; Xu, H.; Gong, H. Org. Lett. 2011, 13, 2138. doi: 10.1021/ol200617f
Xu, H.; Zhao, C.; Qian, Q.; Deng, W.; Gong, H. Chem. Sci. 2013, 4, 4022. doi: 10.1039/c3sc51098k
Komeyama, K.; Michiyuki, T.; Osaka, I. ACS Catal. 2019, 9, 9285. doi: 10.1021/acscatal.9b03352
(a) Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307. (b) Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. (c) McDonald, R. I.; Liu, G.; Stahl, S. S. Chem. Rev. 2011, 111, 2981. (d) Dong, Z.; Ren, Z.; Thompson, S. J.; Xu, Y.; Dong, G. Chem. Rev. 2017, 117, 9333. (e) Yan, T.; Guironnet, D. Sci. Chin. Chem. 2020, 63, 755.
Wang, X.-X.; Lu, X.; Li. Y.; Wang, J.-W.; Fu, Y. Sci Chin. Chem.10.1007/s11426-020-9838-x. doi: 10.1007/s11426-020-9838-x
Lu, X.; Xiao, B.; Zhang, Z.-Q.; Gong, T.-J.; Su, W.; Yi, J.; Fu, Y.; Liu, L. Nat. Commun. 2016, 7, 11129. doi: 10.1038/ncomms11129
Zhou, F.; Zhu, J.; Zhang, Y.; Zhu, S. Angew. Chem. Int Ed. 2018, 57, 4058. doi: 10.1002/anie.201712731
Wang, Z.-Y.; Wan, J.-H.; Wang, G.-Y.; Wang, R.; Jin, R.-X.; Lan, Q.; Wang, X.-S. Tetrahedron Lett. 2018, 59, 2302. doi: 10.1016/j.tetlet.2018.05.008
(a) Sun, S.-Z.; Borjesson, M.; Martin-Montero, R.; Martin, R. J. Am. Chem. Soc. 2018, 140, 12765. (b) Qian, D.; Hu, X. Angew. Chem. Int. Ed. 2019, 58, 18519.
Lu, X.; Xiao, B.; Liu, L.; Fu, Y. Chem. Eur. J. 2016, 22, 11161. doi: 10.1002/chem.201602486
Sun, S.-Z.; Romano, C.; Martin, R. J. Am. Chem. Soc. 2019, 141, 16197. doi: 10.1021/jacs.9b07489
Wang, Z.; Yin, H.; Fu, G. C. Nature 2018, 563, 379. doi: 10.1038/s41586-018-0669-y
Zhou, F.; Zhang, Y.; Xu, X.; Zhu, S. Angew. Chem. Int. Ed. 2019, 58, 1754. doi: 10.1002/anie.201813222
He, S.-J.; Wang, J.-W.; Li, Y.; Xu, Z.-Y.; Wang, X.-X.; Lu, X.; Fu, Y. J. Am. Chem. Soc. 2020, 142, 214. doi: 10.1021/jacs.9b09415
Yang, Z.-P.; Fu, G. C. J. Am. Chem. Soc. 2020, 142, 5870. doi: 10.1021/jacs.0c01324
Green, S. A.; Huffman, T. R.; McCourt, R. O.; van der Puyl, V.; Shenvi, R. A. J. Am. Chem. Soc. 2019, 141, 7709. doi: 10.1021/jacs.9b02844
Cheng, L.; Li, M.-M.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. J. Am. Chem. Soc. 2018, 140, 11627. doi: 10.1021/jacs.8b09346
Chen, T.; Yang, H.; Yang, Y.; Dong, G.; Xing, D. ACS Catal. 2020, 10, 4238. doi: 10.1021/acscatal.0c00019
(a) Cheng, L.; Li, M.-M.; Wang, B.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. Chem. Sci. 2019, 10, 10417. (b) Lv, L.; Zhu, D.; Qiu, Z.; Li, J.; Li, C.-J. ACS Catal. 2019, 9, 9199.
Ji, D.-W.; He, G.-C.; Zhang, W.-S.; Zhao, C.-Y.; Hu, Y.-C.; Chen, Q.-A. Chem. Commun. 2020, 56, 7431. doi: 10.1039/D0CC02697B
(a) Dhungana, R. K.; KC, S.; Basnet, P.; Giri, R. Chem. Rec. 2018, 18, 1314. (b) Giri, R.; KC, S. J. Org. Chem. 2018, 83, 3013. (c) Derosa, J.; Apolinar, O.; Kang, T.; Tran, V. T.; Engle, K. M. Chem. Sci. 2020, 11, 4287. (d) Luo, Y.-C.; Xu, C.; Zhang, X. Chin. J. Chem. 2020, 38, 1371. (e) Qi, X.; Diao, T. ACS Catal. 2020, 10, 8542.
Qin, T.; Cornella, J.; Li, C.; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell, B. D.; Eastage, M. D.; Baran, P. S. Science, 2016, 352, 801. doi: 10.1126/science.aaf6123
KC, S.; Dhungana, R. K.; Shrestha, B.; Thapa, S.; Khanal, N.; Basnet, P.; Lebrun, R. W.; Giri, R. J. Am. Chem. Soc. 2018, 140, 9801. doi: 10.1021/jacs.8b05374
Chierchia, M.; Xu, P.; Lovinger, G. J.; Morken, J. P. Angew. Chem. Int. Ed. 2019, 58, 14245. doi: 10.1002/anie.201908029
García-Domínguez, A.; Li, Z.; Nevado, C. J. Am. Chem. Soc. 2017, 139, 6835. doi: 10.1021/jacs.7b03195
Shu, W.; García-Domínguez, A.; Quirós, M. T.; Mondal, R.; Cárdenas, D. J.; Nevado, C. J. Am. Chem. Soc. 2019, 141, 13812. doi: 10.1021/jacs.9b02973
(a) Guo, L.; Tu, H.-Y.; Zhu, S.; Chu, L. Org. Lett. 2019, 21, 4771. (b) García-Domínguez, A.; Mondal, R.; Nevado, C. Angew. Chem. Int. Ed. 2019, 58, 12286. (c) Campbell, M. W.; Compton, J. S.; Kelly, C. B.; Molander, G. A. J. Am. Chem. Soc. 2019, 141, 20069.
Derosa, J.; Tran, V. T.; Boulous, M. N.; Chen, J. S.; Engle, K. M. J. Am. Chem. Soc. 2017, 139, 10657. doi: 10.1021/jacs.7b06567
Derosa, J.; van der Puyl, V. A.; Tran, V. T.; Liu, M.; Engle, K. M. Chem. Sci. 2018, 9, 5278. doi: 10.1039/C8SC01735B
(a) Nattmann, L.; Saeb, R.; N thling, N.; Cornella, J. Nat. Catal. 2020, 3, 6. (b) Tran, V. T.; Li, Z.-Q.; Apolinar, O.; Derosa, J.; Joannou, . W. V.; Wisniewski, S. R.; Eastgate, M. D.; Engle, K. M. Angew. Chem. Int. Ed. 2020, 59, 7409.

图 3 镍催化一级烷基卤代物与烷基锌的Negishi偶联
Figure 3 Ni-catalyzed Negishi coupling of primary alkyl halide

图 4 添加剂辅助的镍催化一级烷基卤化物与金属试剂的交叉偶联反应
Figure 4 Additive-promoted Ni-catalyzed cross-coupling reactions of primary alkyl halide

图 5 镍催化二级烷基卤化物的C(sp3)—C(sp3)交叉偶联
Figure 5 Ni-catalyzed C(sp3)—C(sp3) cross-coupling reaction using a secondary halides

图 6 镍催化α-溴代酰胺与烷基溴化锌的不对称偶联反应
Figure 6 Ni-catalyzed asymmetric cross-coupling between α-bromo- amide and aliphatic organozinc reagent

图 7 手性噁唑啉配体对二级烷基溴化物不对称偶联反应的影响
Figure 7 Effects of chiral oxazoline ligands on asymmetric coupling reactions of secondary alkyl bromide

图 8 镍催化的活化烷基卤代物或者带有导向基团的底物不对称交叉偶联反应
Figure 8 Ni-catalyzed asymmetric cross-coupling using activated alkyl halides or substrates with directing groups

图 9 镍催化不对称合成烷基硼酸酯
Figure 9 Ni-catalyzed asymmetric synthesis of enantioenriched alkylboronates

图 10 镍催化α-溴代硅烷与烷基锌试剂不对称交叉偶联反应
Figure 10 Ni-catalyzed asymmetric cross-coupling of a-halosilanes and alkylzinc reagents

图 11 镍催化烷基溴代物与烷基格氏试剂的Kumada偶联反应及反应机理
Figure 11 Ni-catalyzed Kumada coupling of primary alkyl bromides with Grignard reagents and mechanism

图 12 镍催化苄基甲基醚与烷基格氏试剂的偶联反应
Figure 12 Ni-catalyzed cross-coupling of benzyl methyl ether with alkyl Grignard reagents

图 13 镍催化苄基甲基醚与格氏试剂的立体选择性偶联反应
Figure 13 Ni-catalyzed coupling of benzyl methyl ethers and Grignard reagent

图 14 镍催化脂肪羧酸酯与金属试剂的交叉偶联反应
Figure 14 Ni-catalyzed cross-coupling of activated esters and alkylzinc reagents

图 15 镍催化脂肪胺与烷基锌试剂的偶联反应
Figure 15 Ni-catalyzed cross-coupling of aliphatic amines and alkylzinc reagents

图 16 镍催化非活化烷基C—H键与卤代烷的交叉偶联反应
Figure 16 Nickel-catalyzed alkylation of inactivated C(sp3)—H bonds

图 17 镍-光反应催化剂协同催化羧酸与烷基溴化物的偶联反应
Figure 17 Dual nickel-photoredox catalyzed cross-coupling of carboxylic acids with alkyl halides

图 18 镍-光催化剂-有机催化剂催化的C(sp3)—H键烷基化偶联反应及机理
Figure 18 Nickel-, photoredox- and organocatalysis for C(sp3)—H alkylation and mechanism

图 19 镍-光反应催化剂协同催化烷基卤代烷交叉偶联反应及机理
Figure 19 Dual nickel-photoredox catalyzed cross-coupling of alkyl halides and mechanism

图 20 镍催化烷基亲电试剂的还原交叉偶联反应
Figure 20 Ni-catalyzed reductive cross-coupling of inactivated alkyl halides

图 21 [B(pin)]2还原剂存在下的Ni催化卤化物的还原交叉偶联反应
Figure 21 Ni-catalyzed cross-coupling of unactivated alkyl halides using [B(pin)]2 as reductant

图 22 镍钴共催化烷基卤代物与烷基对甲苯磺酸酯交叉偶联反应
Figure 22 Ni/Co-catalyzed cross coupling of alkyl halides with alkyl tosylates

图 24 镍催化二级或三级烷基卤化物与烯烃的不对称还原偶联反应
Figure 24 Ni-catalyzed enantioselective coupling of secondary and tertiary halides with olefins

图 25 镍催化烷基卤化物与烯烃的偶联机理
Figure 25 Mechanism for Ni-catalyzed coupling of alkyl halides and olefins

图 26 镍催化α-酯基烷基卤化物与烯烃的不对称还原偶联反应
Figure 26 Ni-catalyzed enantioselective reductive hydroalkylation of α-ester alkyl bromides and oleifins

图 27 镍锰共催化非活化烯烃的氢烷基化构筑季碳化合物
Figure 27 Hydroalkylation of inactivated olefins to form quaternary carbon centers using Mn/Ni dual catalysis

图 28 镍锰协同催化烯烃氢烷基化反应机理
Figure 28 Mechanism of Ni/Mn-catalyzed hydroalkylation of olefins

图 29 镍催化共轭二烯与简单酮的不对称氢烷基化反应及反应机理
Figure 29 Ni-catalyzed asymmetric hydroalkylation of 1, 3-dienes and catalytic cycle

图 30 镍催化共轭二烯与腙的氢烷基化反应
Figure 30 Ni-catalyzed hydroalkylation of 1, 3-dienes with hydrazones

图 31 镍催化共轭二烯与烯丙基硼酸酯的氢烷基化反应
Figure 31 Ni-catalyzed allyl-allyl coupling reactions between 1, 3-dienes and allylboronates

图 32 镍催化共轭烯烃、芳基烯烃或者杂原子取代烯烃双官能团化反应
Figure 32 Ni-catalyzed difunctionalization of conjugated alkenes, styrenes and heteroatom-substituted alkenes

图 34 镍-光反应催化剂协同催化分子间烯烃1, 2-烷基芳基化及反应机理
Figure 34 Dual nickel-photoredox catalyzed intermolecular 1, 2-alkylarylation of alkenes and mechanism

图 35 镍催化导向基策略的烯烃1, 2-芳基烷基化反应及机理
Figure 35 Directed Ni-catalyzed 1, 2-arylalkylation of alkenes and mechanism

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




 下载:
下载:




 下载:
下载: