Received Date:
28 July 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the Mistry of Science and Technology of the People's Republic of China (No.206YFA0202503) and the National Natural ScienceFoundation of China (Nos. 21925503, 21835004)
Abstract:
Lithium-rich manganese-based oxides (LRMO) are promising cathode materials to build next generation lithium-ion batteries because of high capacity and low cost. However, the severe capacity fade and voltage decay, which originate from surface oxygen loss, side reactions and irreversible phase transformation, restrict their practical application. Proposed approaches to address these issues include electrolyte modification, synthesis condition optimization, tuning elemental composition, bulk doping and surface coating. Surface coating has been proved to be an effective method to stabilize the interface between LRMO and electrolyte. Herein, we report a facile approach to synthesize Li3PO4-coated LRMO (LRMO@LPO) by in-situ carbonate-phosphate precipitate conversion reaction. The formation of Li3PO4 layer and its contribution to enhanced electrochemical performance are investigated in detail. Transmission electron microscopy (TEM) reveals that the surface of carbonate precursor converts to Ni3(PO4)2 after reacting with Na2HPO4 solution, which finally transforms to Li3PO4 coating layer with thickness below 30 nm during calcination process. Quinoline phosphomolybdate gravimetric method gives the optimal Li3PO4 coating content of 0.56%. The modified LRMO@LPO sample exhibits improved cycling stability (191.1 mAh·g-1 after 175 cycles at 0.5C between 2.0~4.8 V and 81.8% capacity retention) and suppressed voltage decay (1.09 mV per cycle), compared with bare LRMO material (72.9% capacity retention, 1.78 mV per cycle). The electrodes are studied by galvanostatic intermittent titration technique, electrochemical impedance spectroscopy, TEM and inductively coupled plasma atomic emission spectrometry. The results suggest efficient mitigation of phase transformation and dissolution of transition metal in LRMO@LPO. As a coating material with lithium-ion conductivity, Li3PO4 not only acts as a physical barrier to inhibit side reaction between the electrolyte and LRMO, but also promotes lithium ion transport at the surface region of cathode. The in-situ surface modification approach simplifies the traditional post coating process, and may provide new insight to build stable and low cost Li-rich cathode for lithium-ion batteries.
Table 1.
Solubility product constant (Ksp) of TM carbonate and phosphate, minimal concentration of PO43- for precipitation-conversion reaction, and the reaction equilibrium constant
Figure 5.
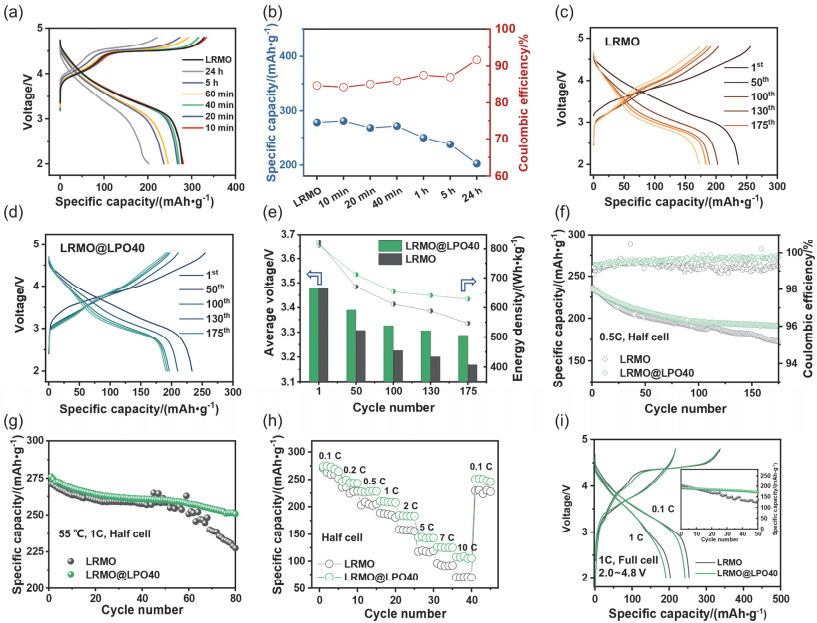
(a) Initial charge-discharge curves (0.1C) of bare and Li3PO4-coated LRMO cathode materials with different treatment time. (b) Specific discharge capacity and initial coulombic efficiency for different samples. (c, d) Charge-discharge curves at selected cycles and (e) corresponding average discharge voltage and energy density. Cycling performance of LRMO and LRMO@LPO40 at (f) 25 ℃ and (g) 55 ℃. (h) Rate performance. (i) Cycling performance of CR2032 full cell
Figure 6.
(a) GITT voltage curves and (b) apparent Li+ diffusion coefficient of LRMO and LRMO@LPO40. (c) Nyquist plot of the electrodes after activation for 3 cycles. Inset is the equivalent circuit
House, R. A.; Maitra, U.; Perez-Osorio, M. A.; Lozano, J. G.; Jin, L.; Somerville, J. W.; Duda, L. C.; Nag, A.; Walters, A.; Zhou, K. J.; Roberts, M. R.; Bruce, P. G. Nature2020, 577, 502.
[10]
Li, Q.; Yao, Z.; Lee, E.; Xu, Y.; Thackeray, M. M.; Wolverton, C.; Dravid, V. P.; Wu, J. Nat. Commun.2019, 10, 1692.
[11]
Lee, S.; Jin, W.; Kim, S. H.; Joo, S. H.; Nam, G.; Oh, P.; Kim, Y. K.; Kwak, S. K.; Cho, J. Angew. Chem. Int. Ed.2019, 58, 10478.
Liu, W.; Oh, P.; Liu, X.; Myeong, S.; Cho, W.; Cho, J. Adv. Energy Mater.2015, 5, 1500274.
[32]
Haynes, W. M. CRC Handbook of Chemistry and Physics, 97th ed., Vol. 5, CRC Press Taylor & Francis Group, Boca Raton, Florida, 2016, pp. 177-178.
[33]
Xiao, Z. B.; Chen, S.; Guo, M. M. Trans. Nonferrous Met. Soc. China2011, 21, 2454.
[34]
Zhang, Y.; Hou, P.; Zhou, E.; Shi, X.; Wang, X.; Song, D.; Guo, J.; Zhang, L. J. Power Sources2015, 292, 58.
[35]
Radin, M. D.; Hy, S.; Sina, M.; Fang, C.; Liu, H.; Vinckeviciute, J.; Zhang, M.; Whittingham, M. S.; Meng, Y. S.; Van der Ven, A. Adv. Energy Mater.2017, 7, 1602888.
[36]
Toby, B. H.; Von Dreele, R. B. J. Appl. Crystallogr.2013, 46, 544.
Figure 5
(a) Initial charge-discharge curves (0.1C) of bare and Li3PO4-coated LRMO cathode materials with different treatment time. (b) Specific discharge capacity and initial coulombic efficiency for different samples. (c, d) Charge-discharge curves at selected cycles and (e) corresponding average discharge voltage and energy density. Cycling performance of LRMO and LRMO@LPO40 at (f) 25 ℃ and (g) 55 ℃. (h) Rate performance. (i) Cycling performance of CR2032 full cell
Figure 6
(a) GITT voltage curves and (b) apparent Li+ diffusion coefficient of LRMO and LRMO@LPO40. (c) Nyquist plot of the electrodes after activation for 3 cycles. Inset is the equivalent circuit
Table 1.
Solubility product constant (Ksp) of TM carbonate and phosphate, minimal concentration of PO43- for precipitation-conversion reaction, and the reaction equilibrium constant


 下载:
下载:






 下载:
下载:








 下载:
下载:
