
图 1.
IT-450, IT-500和IT-550的XRD图
Figure 1.
XRD patterns of IT-450, IT-500 and IT-550

自从1972年Fujishima与Honda[1]发表了TiO2作为光催化剂实现光电催化分解水产氢/氧的工作以来, 光催化制氢一直被认为是制备氢气这种清洁能源最有潜力的方法之一[2].由于TiO2具有出色的生物、化学稳定性与光化学稳定性以及合适的电势电位, 因此受到了科研人员的广泛关注, 其在光催化降解有机污染物[3-5]、光催化制氢[6-9]、有害气体的探测与净化领域[10-11]都有潜在的应用前景.然而由于TiO2本征带隙较宽(Eg=3.1~3.2 eV), 只对紫外光有响应, 而对可见光部分几乎毫无响应, 这限制了其在实际应用中对太阳能的高效利用[12]; 同时, TiO2吸收光后激发产生的载流子—电子和空穴容易发生复合, 使得可以传输到催化剂表面参与反应的载流子数目减少, 降低催化效率[13].因此, 缩短TiO2基光催化剂的带隙, 使其对光谱的吸收拓展至可见光范围内, 以及解决大量电子-空穴对发生复合的问题, 是TiO2基光催化剂走向应用的关键.研究人员发现, 通过调控TiO2的微观形貌与结构, 可以提高半导体的光吸收率, 例如暴露特定的晶面[14], 制备多孔结构使得照射光在材料表面产生多次反射-吸收现象[15].此外, 通过元素掺杂的方式可以有效调节TiO2的能带结构, 如掺杂Fe, Co, Cu, Ni等金属元素[16-18], 掺杂N[19], B[20-22], C[23-24], F[25-27], S[28]等非金属元素.其中, 非金属元素的掺杂主要是通过将掺杂元素的p轨道与TiO2中O 2p轨道混合, 将价带上移以缩短带隙宽度, 同时降低生成复合中心的概率, 因此非金属元素的掺杂可以有效地拓宽TiO2对于太阳光特别是可见光波段的吸收范围.构建异质结结构可以显著提高电子空穴对的分离效率, 使两种光生载流子分别在不同的反应位点发生氧化还原反应[29], 多种非金属元素共掺杂生成的键合结构也有助于载流子的分离[21].
大多数TiO2光催化剂是基于粉末、颗粒状悬浊液来参与光催化反应[30-34], 一方面粉末颗粒有聚集趋势, 另一方面在后续处理、分离回收时有诸多不便, 不利于降低成本.将TiO2粉末负载在其他载体上, 如玻璃、活性炭、钢丝网或高分子材料上[35-36], 由于借助载体成型, 催化剂在使用后仍能维持形态, 便于分离回收与重复利用.但负载型催化剂也存在一定缺陷[37]:由于载体的“阴影效应”, 光的穿透被抑制, 这影响了光的利用效率; 同时, 比表面积的减小以及污染物和牺牲剂在整个基材上的传质减少会降低TiO2在载体上的光催化效率; 另外, 在液体的流动环境中, 负载型催化剂面临着催化剂脱落的问题, 使得其在长期使用过程中催化效率逐渐降低.
为了解决上述问题, 聚合物前驱体法是一种有效的手段, 聚合物前驱体法是制备陶瓷材料和改性催化剂的一种新型方法[3, 38], 相比传统方法, 该方法具有如下显著优点: (1)结构和组成可调, 可以与多种改性方式兼容; (2)合成条件温和, 合成周期短; (3)容易与软模板结合, 煅烧后可形成介孔结构, 增大比表面积; (4)液相前驱体易于加工成型, 方便制备涂层或者纤维.因此, 采用聚合物前驱体法制备纳米纤维有望解决粉体催化剂分离回收困难以及负载型催化剂的脱落等问题.
在本工作中, 我们利用聚合物前驱体法, 结合非金属元素掺杂、构建异质结、引入致孔剂等多种改性手段, 来协同改性TiO2, 成功制备出一种高效稳定的B, N共掺杂的In2O3/TiO2光催化剂, 并且利用气纺丝装置制备了该体系的纤维棉光催化剂.在可见光照射下, 改性催化剂粉体和纤维棉状催化剂均表现出优异的光催化产氢活性, 且纤维棉状催化剂可以极大程度简化分离回收的过程, 表现出了较大的应用潜力.
样品通过聚合物前驱体法制备后煅烧获得.以钛酸异丙酯为钛源, 硝酸铟为铟源, 硼酸为硼源, 乙酰胺为氮源, PEG为致孔剂, 经可控水解聚合生成B, N共掺杂In2O3/TiO2聚合物前驱体, 将前驱体于不同温度(450、500、550 ℃)下裂解处理30 min, 得到B, N共掺杂In2O3/TiO2催化剂, 根据裂解温度不同, 所得催化剂分别命名为IT-450、IT-500、IT-550.利用X-射线衍射(XRD)表征了样品的晶体结构, 如图 1所示.由图可得, 不同温度所得的IT-TiO2样品的晶相均为锐钛矿相, 在25.4°、37.9°、48.1°、54.0°、55.1°和62.8°处的衍射峰, 分别对应于锐钛矿相TiO2的(101), (004), (200), (105), (211)和(204)晶面(PDF No.21-1272, JCPDS), 此外, 掺杂样品的XRD图在2θ=30.3°处均有一个相对较小的峰, 根据体系的元素组成判断该峰应为B2O3和In2O3布拉格衍射峰的重叠峰, 另外B2O3的峰比标准谱(PDF No.06-0297, JCPDS)向小角度偏移, B2O3衍射峰的半峰宽较大, 强度较弱, 因此结晶性较弱, 晶体结构并不明显.
通过扫描电子显微镜(SEM)与高分辨透射电子显微镜(HRTEM)表征了样品IT-500的微观形貌, 由于各样品的形貌差别不大, 这里仅给出IT-500的形貌图.由SEM(图 2A)可以看出, 所制备的B, N共掺杂的In2O3/TiO2催化剂由粒径均一的纳米颗粒组成, 呈葡萄状堆积, 粒径在10~20 nm左右, 与商品化TiO2 P25微观尺寸基本一致[39]. 图 2B~2D为样品的高分辨透射电镜图, 由HRTEM(图 2D)可以清楚地观察到两种尺寸的晶格条纹, 其中0.41 nm的晶格条纹, 为立方In2O3 (211)晶面[40], 而0.35 nm的晶格条纹间距归属于锐钛矿相TiO2的(101)晶面[41], 两种晶格条纹区域紧密接触(图 2D与图S1, 见Supporting Information), 证明成功构建了预期的In2O3/TiO2异质结结构[4].各元素在催化剂上分布均匀, 粉体催化剂的光电子能谱分析(EDS)结果见图S2.

利用BET来表征样品的比表面积以及孔径信息. 表 1列出了三种样品的比表面积以及孔的结构特性, IT-450, IT-500, IT-550的比表面积分别为169.9118, 173.4842, 140.0745 m2/g, 而相同条件下测定的P25的比表面积为60.8553 m2/g.根据N2吸附脱附曲线(图 3A)可知, 三种样品均呈现Ⅳ型等温线, 说明均为介孔材料. 图 3B显示三种样品的孔径主要分布在4~7 nm之间.可见添加致孔剂PEG, 在煅烧裂解过程中能形成介孔结构, 根据文献报道, 介孔允许光在其孔道内散射, 大幅提高了样品的比表面积, 从而提高了光吸收能力[25], 比表面积的增大有利于提高光催化活性[42].
 下载:
导出CSV
下载:
导出CSV
| 样品名称 | BET比表面积/ (m2•g-1) | 孔径尺寸/nm | 孔体积/(cm3•g-1) |
| IT-450 | 169.9118 | 4.7412 | 0.2133 |
| IT-500 | 173.4842 | 5.5640 | 0.2413 |
| IT-550 | 140.0745 | 6.6110 | 0.2315 |

利用光电子能谱XPS来表征IT-500的组成元素以及元素所处的化学环境, 样品的XPS全谱图见图S3.如图 4A所示, In 3d的高分辨XPS谱图(图 4A)去卷积后, 可以得到属于In 3d5/2(444.80 eV)和In 3d3/2(452.55 eV)的两个特征峰, 特征峰的位置与In2O3一致, 未出现偏移, 表明体系中不存在In-B, In-N或者In-Ti结构.谱图中未出现金属铟的In3d5/2信号(结合能443.6 eV), 表明体系中不存在金属铟, 铟仅以In2O3形态存在于体系中[43]. Ti 2p高分辨XPS谱图(图 4B)中, Ti 2p1/2的结合能位于464.51 eV, Ti 2p3/2的结合能位于458.81 eV, 两个特征峰的结合能差值为5.70 eV, 表明In2O3/TiO2异质结体系中Ti为Ti4+.文献所报道的In2O3/TiO2异质结体系中Ti 2p1/2和Ti 2p3/2的特征峰分别位于464.2 eV和458.5 eV[44], 相比之下, 本工作中样品Ti 2p1/2和Ti 2p3/2均降低了0.31 eV, 结合能的移动表明Ti与掺杂元素发生相互作用, 掺杂元素进入到TiO2的晶格内, 提高了Ti的电子密度[45].如图 4C所示, N 1s的高分辨XPS光谱中, 在399.52 eV处有一个突出的特征峰, 对应于TiO2晶格中的Ti-N-B[46]和以填隙方式掺杂的N结构[47]. 图 4D显示了B 1s的特征峰位于192.01 eV和193.79 eV, 分别对应于Ti-O-B结构[48]和B2O3[49], B2O3峰信号的存在与XRD的结果一致.高分辨XPS谱图表明, B和N的掺杂在一定程度上影响了In2O3/TiO2中Ti的化学环境, 体系中有Ti-N-B和Ti-O-B结构的形成, 此外也存在以填隙方式掺杂的N结构, 该结构有助于提高TiO2的光吸收能力[50].

通过紫外可见吸收光谱图可以对比不同样品对于光子的吸收范围, 如图 5A所示, 不同煅烧温度所得的B, N共掺杂In2O3/TiO2催化剂在波长大于380 nm的可见光区域吸收范围获得了不同程度的拓展, 样品IT-500可见光范围吸收红移最多.

利用Tauc方程(1)计算了不同样品的带隙宽度,
|
$ {\left( {ahv} \right)^{1/2}} = {k^*}\left( {hv - {E_{\rm{g}}}} \right) $ |
(1) |
其中α为光吸收系数, h为普朗克常数, ν为光波频率, k为与材料性质有关的常数, Eg为样品的带隙宽度.
如图 5B所示, 商品化P25的带隙为3.09 eV, B, N共掺杂及引入In2O3/TiO2异质结后, TiO2的带隙宽度明显缩短, 尤其是500 ℃煅烧所得的IT-500样品的带隙宽度仅为2.71 eV, 表明500 ℃为制备该体系催化剂的最佳温度.结合XPS表征数据, 可知改性样品中的Ti-N-B结构、Ti-O-B结构与填隙方式掺杂的N对带隙变窄做出了贡献.
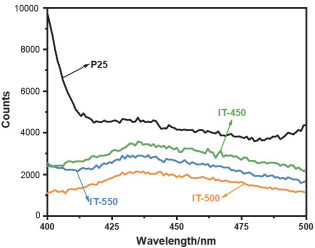
利用光致发光(PL)谱图测量光生载流子的二次结合发光的光强, 来表征不同样品中光生载流子的复合率.如图 6所示, P25的PL光谱强度最高, 表明纯的TiO2中, 电子和空穴比较容易发生复合, 而经过改性的样品的PL光谱强度均低于纯TiO2, 证明In2O3/TiO2异质结的引入, 在两相界面处形成了接触电场, 促进了电子空穴对的分离与转移, 抑制了电荷载流子的复合[44].基于上述表征, 表明我们已经制备了预期的N, B共掺杂的In2O3/TiO2异质结结构.
进一步研究了IT-450, IT-500, IT-550与P25的光催化产氢活性, 使用Pt纳米粒子作为助催化剂, 三乙醇胺(TEOA)作为空穴牺牲剂.如图 7A所示, 负载Pt纳米颗粒的IT-450、IT-500、IT-550在波长大于380 nm的可见光照射下, 光催化产氢速率分别为3839、5961、5709 μmol•g-1•h-1, 而同实验条件下的P25仅有135 μmol• g-1•h-1.通过对比前文的表征结果, 相比其他样品, IT-500有着更大的比表面积、更窄的带隙以及更宽的吸收光谱范围, 并且在IT-500中, 光生载流子发生复合的概率要小于另外两种样品.选择产氢活性最高的IT-500进行循环实验, 如图 7B所示, 在经过5次循环后, 所制备催化剂的光催化产氢活性未发生明显变化.

将B, N共掺杂In2O3/TiO2聚合物前驱体利用气纺丝制备得到B, N掺杂In2O3/TiO2纤维棉.纤维棉的实物图与SEM电镜图如图 8所示, 纤维呈蓬松纤维棉状, 有一定的回弹性.纤维表面较为粗糙, 利于增加其比表面积, 提高光催化效率.各元素在纤维表面均匀分布, 其EDS面扫结果见图S4.

在500 ℃的退火条件下, 该体系纤维的光催化产氢速率可达到1186 μmol•g-1•h-1, 在经过5次循环实验后仍能达到初始产氢速率的97%, 其5次循环数据如图 9所示.由于催化剂整体为一个团状, 在进行光催化反应过程中可维持宏观形态, 可以在实验后轻易地从体系中分离回收, 进行循环重复使用.较文献中TiO2纳米纤维的产氢速率亦有明显提高(见表S4).

根据表征结果分析, 我们推测了N, B掺杂的In2O3/TiO2的反应机理, 如图 10所示.由于非金属元素N和B的掺杂, 在价带顶部生成了局部中间态, 使得TiO2的带隙变窄, 从而将样品对太阳光的吸收范围拓宽至可见光区域, 这提高了样品对于太阳光的吸收利用率; 异质结结构的存在促进了激子的分离, 减少了它们发生复合的概率.具体而言, 当样品在可见光的照射下, 改性TiO2和In2O3吸收光激发后会产生电子-空穴对, 由于TiO2的导带能级比In2O3更负, 因此In2O3所产生的电子会转移至TiO2的导带上, 同时由于较低的价带电势, 在TiO2中产生的空穴也将迁移至In2O3的价带, 促进激子分离为自由电荷, 使得更多电子和空穴分别参与到还原与氧化反应中.另外, 由于助催化剂Pt纳米粒子的负载, 形成界面肖特基势垒, 来自TiO2或In2O3 CB的电子可以通过肖特基势垒迁移到金属中, 并且金属可以储存电子, 形成反应位点, 还原H+生成H2; 金属Pt具有较高的功函数, 转移至金属的光生电子将无法重新回流至受激半导体, 促进了载流子的分离与转移, 同时贵金属助催化剂可以降低产生氢气的过电位[29].

本工作报道了一种通过多种改性手段协同作用来增强TiO2基光催化剂活性的方法. PEG作为致孔剂引入体系, 在煅烧裂解后形成介孔结构, 提高了光催化剂的比表面积. B, N共掺杂可以使TiO2的带隙变窄, 拓宽了TiO2的光谱吸收范围, 使其可见光吸收范围显著拓宽. In2O3/TiO2作为异质结结构可以有效地分离激子为自由电荷, 使电子和空穴分别参与还原和氧化反应, 减少它们复合的概率, 提高了光催化效率.在光催化产氢实验中, 利用光沉积法负载了Pt纳米颗粒, 在大于380 nm的可见光照射下, 粉末状光催化剂IT-500的光催化产氢速率达到了5961 μmol•g-1•h-1, 远优于P25和大部分使用单一改性手段制备的TiO2基光催化剂, 在此基础上利用所合成的聚合物前驱体溶液, 通过气纺丝装置制备成纤维棉状催化剂, 测试得其光催化产氢速率达到1186 μmol•g-1•h-1, 在经过5次循环实验后, 仍能达到初始产氢速率的97%.纤维棉状催化剂在分离回收过程中可以缩短时间周期, 简化操作步骤, 具有经济性与环保性, 在大规模制备、高效稳定使用方面具有巨大潜力.
采用聚合物前驱体法制备了B, N共掺杂的In2O3/TiO2光催化剂.将92.6 g钛酸异丙酯、39 g的PEG (Mn=600)与1.23 g硝酸铟混合, 在搅拌下升温至120 ℃, 保温2 h.降温至90 ℃后, 加入4.82 g硼酸, 升温至120 ℃保温2 h, 降温至90 ℃后加入38.39 g乙酰胺, 升温至120 ℃保温2 h.冷却至85 ℃, 加入9.75 g乙酰丙酮, 随后使用恒压漏斗以1~2滴/秒的速度滴入5.85 g水和23.4 g正丙醇的混合物进行可控水解聚合, 滴加完成后回流1 h, 旋蒸除去溶剂, 得到B, N共掺杂的In2O3/TiO2聚合物前驱体.将该前驱体放置于石英管式炉中, 以3 ℃•min-1升至预定温度(450, 500, 550 ℃), 保温30 min, 随后将产物放入研钵中研磨成粉体, 得到B, N共掺杂的In2O3/TiO2催化剂.
B, N掺杂的In2O3/TiO2纳米纤维的制备:以聚乙烯吡咯烷酮(PVP, Mn=1300000)为纺丝助剂, 以乙醇和乙酸为溶剂, 将前驱体、PVP、乙醇、乙酸按照质量比1:1:3:1均匀混合成纺丝液; 以压缩空气为气源, 采用气纺丝装置将纺丝液从针头中挤出, 利用高流速气体将纺丝液拉伸成纳米纤维形态, 进气气压为0.10 MPa, 进料速度为40 mL•h-1, 接收距离为50 cm, 使用笼状装置收集纳米纤维.随后经过湿气固化、在500 ℃下焙烧30 min, 获得B, N掺杂的In2O3/TiO2纳米纤维.
将10 mg光催化剂, 5 mL水, 1 mL三乙醇胺(TEOA)加入25 mL的试管中, 然后加入30 μL氯铂酸水溶液(氯铂酸浓度为10 mg•mL-1), 通过光沉积法沉积催化剂质量分数3%的Pt纳米粒子作为助催化剂.将体系真空处理几次, 并用氩气鼓泡30 min以除去溶解的空气.光催化产氢反应在300 W氙灯(截止波长380 nm)的可见光照射下开始, 该灯配有冷却系统以保持温度.反应4 h后, 通过气相色谱仪(GC, HP4890D)分析产生氢气的量, 该色谱仪配备了以氦气作为内标的FID和TCD检测器.
Fujishima, A.; Honda, K. Nature 1972, 238, 37.
李鑫, 张太阳, 王甜, 赵一新, 化学学报, 2019, 77, 1075.Li, X.; Zhang, T. Y.; Wang, T.; Zhao, Y. X. Acta Chim. Sinica 2019, 77, 1075(in Chinese).
Yu, H.; Ye, L.; Zhang, T.; Zhou, H.; Zhao, T. RSC Adv. 2017, 7, 15265.
Yu, H.; Chen, F.; Ye, L.; Zhou, H.; Zhao, T. J. Mater. Sci. 2019, 54, 10191.
龙绘锦, 王恩君, 董江舟, 王玲玲, 曹永强, 杨文胜, 曹亚安, 化学学报, 2009, 67, 1533.Long, H. J.; Wang, E. J.; Dong, J. Z.; Wang, L. L.; Cao, Y. Q.; Yang, W. S.; Cao, Y. A. Acta Chim. Sinica 2009, 67, 1533(in Chinese).
郭宇, 李燕瑞, 王成名, 龙冉, 熊宇杰, 化学学报, 2019, 77, 520.Guo, Y.; Li, Y. R.; Wang, C. M.; Long, R.; Xiong, Y. J. Acta Chim. Sinica 2019, 77, 520(in Chinese).
Chen, X. B.; Liu, L.; Yu, P. Y.; Mao, S. S. Science 2011, 331, 746.
Marchal, C.; Cottineau, T.; Mendez-Medrano, M. G.; Colbeau- Justin, C.; Caps, V.; Keller, V. Adv. Energy Mater. 2018, 8, 1702142.
彭正康, 丁慧敏, 陈如凡, 高超, 汪成, 化学学报, 2019, 77, 681.Peng, Z. K.; Ding, H. M.; Chen, R. F.; Gao, C.; Wang, C. Acta Chim. Sinica 2019, 77, 681(in Chinese).
Perillo, P. M.; Rodríguez, D. F. J. Alloys Compd. 2016, 657, 765.
Seo, M.-H.; Yuasa, M.; Kida, T.; Huh, J.-S.; Yamazoe, N.; Shimanoe, K. Sensor. Actuat. B-Chem. 2011, 154, 251.
Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Science 2001, 293, 269.
Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D. W. Chem. Rev. 2014, 114, 9919.
Yang, H. G.; Liu, G.; Qiao, S. Z.; Sun, C. H.; Jin, Y. G.; Smith, S. C.; Zou, J.; Cheng, H. M.; Lu, G. Q. J. Am. Chem. Soc. 2009, 131, 4078.
Erwin, W. R.; Zarick, H. F.; Talbert, E. M.; Bardhan, R. Energy Environ. Sci. 2016, 9, 1577.
Kumaravel, V.; Mathew, S.; Bartlett, J.; Pillai, S. C. Appl. Catal. B-Environ. 2019, 244, 1021.
Chai, Z.; Zeng, T. T.; Li, Q.; Lu, L. Q.; Xiao, W. J.; Xu, D. J. Am. Chem. Soc. 2016, 138, 10128.
Huang, H.; Jin, Y.; Chai, Z.; Gu, X.; Liang, Y.; Li, Q.; Liu, H.; Jiang, H.; Xu, D. Appl. Catal. B-Environ. 2019, 257, 117869.
Di Valentin, C.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Giamello, E. J. Phys. Chem. B 2005, 109, 11414.
Geng, H.; Yin, S.; Yang, X.; Shuai, Z.; Liu, B. J. Phys. Condens. Matter 2006, 18, 87.
Liu, G.; Zhao, Y.; Sun, C.; Li, F.; Lu, G. Q.; Cheng, H. M. Angew. Chem. Int. Ed. 2008, 47, 4516.
Finazzi, E.; Di Valentin, C.; Pacchioni, G. J. Phys. Chem. C 2009, 113, 3382.
Sakthivel, S.; Kisch, H. Angew. Chem. Int. Ed. 2003, 42, 4908.
Di Valentin, C.; Pacchioni, G.; Selloni, A. Chem. Mater. 2005, 17, 6656.
Huang, D.-G.; Liao, S.-J.; Liu, J.-M.; Dang, Z.; Petrik, L. J. Photochem. Photobiol., A 2006, 184, 282.
Yu, J. C.; Yu, J. G.; Ho, W. K.; Jiang, Z. T.; Zhang, L. Z. Chem. Mater. 2002, 14, 3808.
Park, H.; Choi, W. J. Phys. Chem. B 2004, 108, 4086.
Bidaye, P. P.; Khushalani, D.; Fernandes, J. B. Catal. Lett. 2009, 134, 169.
Marschall, R. Adv. Funct. Mater. 2014, 24, 2421.
Li, X.; Zhou, X.; Guo, H.; Wang, C.; Liu, J.; Sun, P.; Liu, F.; Lu, G. ACS Appl. Mater. Interfaces 2014, 6, 18661.
Wang, M.; Han, J.; Xiong, H.; Guo, R. Langmuir 2015, 31, 6220.
He, Q.; Sun, H.; Shang, Y.; Tang, Y.; She, P.; Zeng, S.; Xu, K.; Lu, G.; Liang, S.; Yin, S.; Liu, Z. Appl. Surf. Sci. 2018, 441, 458.
Hu, W.; Zhou, W.; Zhang, K.; Zhang, X.; Wang, L.; Jiang, B.; Tian, G.; Zhao, D.; Fu, H. J. Mater. Chem. A 2016, 4, 7495.
Ding, D.; Liu, K.; He, S.; Gao, C.; Yin, Y. Nano Lett. 2014, 14, 6731.
Gao, Y. M.; Shen, H. S.; Dwight, K.; Wold, A. Mater. Res. Bull. 1992, 27, 1023.
Zhang, X.; Lei, L. J. Hazard. Mater. 2008, 153, 827.
Zhang, L.; Wang, L.; Wei, Y.; Zhang, M.; Jiang, H.; Li, J.; Li, S.; Li, J. Eur. J. Inorg. Chem. 2015, 2015, 5039.
Soares, G. B.; Bravin, B.; Vaz, C. M. P.; Ribeiro, C. Appl. Catal. B-Environ. 2011, 106, 287.
Tan, Y.; Shu, Z.; Zhou, J.; Li, T.; Wang, W.; Zhao, Z. Appl. Catal. B-Environ. 2018, 230, 260.
Wang, X.; Zhang, J.; Wang, L.; Li, S.; Liu, L.; Su, C.; Liu, L. J. Mater. Sci. Technol. 2015, 31, 1175.
Yang, Y.; Liang, Y.; Wang, G.; Liu, L.; Yuan, C.; Yu, T.; Li, Q.; Zeng, F.; Gu, G. ACS Appl. Mater. Interfaces 2015, 7, 24902.
Cavalcante, R. P.; Dantas, R. F.; Bayarri, B.; González, O.; Giménez, J.; Esplugas, S.; Machulek, A. Catal. Today 2015, 252, 27.
Pujilaksono, B.; Klement, U.; Nyborg, L.; Jelvestam, U.; Hill, S.; Burgard, D. Mater. Charact. 2005, 54, 1.
Mu, J.; Chen, B.; Zhang, M.; Guo, Z.; Zhang, P.; Zhang, Z.; Sun, Y.; Shao, C.; Liu, Y. ACS Appl. Mater. Interfaces 2012, 4, 424.
Cong, Y.; Zhang, J.; Chen, F.; Anpo, M. J. Phys. Chem. C 2007, 111, 6976.
Xing, M.-Y.; Li, W.-K.; Wu, Y.-M.; Zhang, J.-L.; Gong, X.-Q. J. Phys. Chem. C 2011, 115, 7858.
Patel, N.; Jaiswal, R.; Warang, T.; Scarduelli, G.; Dashora, A.; Ahuja, B. L.; Kothari, D. C.; Miotello, A. Appl. Catal. B-Environ. 2014, 150-151, 74.
Ling, Q.; Sun, J.; Zhou, Q. Appl. Surf. Sci. 2008, 254, 3236.
Feng, N.; Zheng, A.; Wang, Q.; Ren, P.; Gao, X.; Liu, S.-B.; Shen, Z.; Chen, T.; Deng, F. J. Phys. Chem. C 2011, 115, 2709.
Zhang, K.; Wang, X.; He, T.; Guo, X.; Feng, Y. Powder Technol. 2014, 253, 608.

图 2 (A) IT-500的SEM图和(B~D) IT-500的HRTEM图
Figure 2 (A) SEM image of IT-500 and (B~D) HRTEM images of IT-500

图 3 (A) IT-450, IT-500, IT-550的N2吸附脱附曲线(77K); (B) IT-450, IT-500与IT-550的BJH孔径分布图
Figure 3 (A) N2 absorption-desorption isotherms of IT-450, IT-500 and IT-550; (B) BJH pore size distribution plots of IT-450, IT-500 and IT-550

图 4 样品IT-500的高分辨In 3d (A), Ti 2p (B), N 1s (C)和B 1s (D) XPS谱图
Figure 4 Fitted XPS spectra of In 3d (A), Ti 2p (B), N 1s (C) and B 1s (D)

图 5 (A) IT-450, IT-500, IT-550和P25的紫外-可见吸收光谱图; (B) Tauc图用于确定带隙
Figure 5 (A) UV-vis absorption spectra of IT-450, IT-500, IT-550 and P25; (B) Tauc plot for band gap determination.

图 6 IT-450, IT-500和IT-550的光致发光光谱图
Figure 6 Photoluminescence spectra of IT-450, IT-500 and IT-550

图 7 (A) P25, IT-450, IT-500和IT-550的光催化产氢速率; (B) IT-500的光催化产氢循环实验
Figure 7 (A) Photocatalytic hydrogen production rates of P25, IT-450, IT-500 and IT-550; (B) photocatalytic hydrogen production cycle experiments of IT-500

图 8 B, N-In2O3/TiO2纤维实物图(A)和SEM图(B)
Figure 8 Picture of real products (A) and SEM images (B) of B, N-In2O3/TiO2 fibers

图 9 B, N-In2O3/TiO2纳米纤维的光催化产氢循环实验
Figure 9 Photocatalytic hydrogen production cycle experiments of B, N-In2O3/TiO2 nanofibers.

图 10 N, B掺杂的In2O3/TiO2的光催化机理示意图
Figure 10 Schematic illustration of photocatalysis mechanism of N, B co-doped In2O3/TiO2
表 1 不同样品的BET比表面积
Table 1. BET surface areas of different samples
| 样品名称 | BET比表面积/ (m2•g-1) | 孔径尺寸/nm | 孔体积/(cm3•g-1) |
| IT-450 | 169.9118 | 4.7412 | 0.2133 |
| IT-500 | 173.4842 | 5.5640 | 0.2413 |
| IT-550 | 140.0745 | 6.6110 | 0.2315 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: