引用本文:
孙九龙, 曹湾湾, 王宁, 顾林, 李伟华. 氮化硼纳米片在重防腐涂层中的应用进展[J]. 化学学报,
2020, 78(11): 1139-1149.
doi:
10.6023/A20060267

Citation: Sun Jiulong, Cao Wanwan, Wang Ning, Gu Lin, Li Weihua. Progress of Boron Nitride Nanosheets Used for Heavy-duty Anti-Corrosive Coatings[J]. Acta Chimica Sinica, 2020, 78(11): 1139-1149. doi: 10.6023/A20060267
Citation: Sun Jiulong, Cao Wanwan, Wang Ning, Gu Lin, Li Weihua. Progress of Boron Nitride Nanosheets Used for Heavy-duty Anti-Corrosive Coatings[J]. Acta Chimica Sinica, 2020, 78(11): 1139-1149. doi: 10.6023/A20060267
氮化硼纳米片在重防腐涂层中的应用进展
摘要:
氮化硼纳米片也被称为“白色石墨烯”,是一种重要的纳米填料,具有优异的机械性、导热性、耐磨性、阻隔性、疏水性,同时也是一种新兴的性能优良的绝缘材料.被广泛应用于重防腐涂层、润滑剂、传感器等领域.基于氮化硼纳米片在金属腐蚀防护领域巨大的应用前景,本综述将从氮化硼纳米片的制备及表面官能化、氮化硼薄膜防护涂层、氮化硼纳米片/有机防护涂层、氮化硼纳米片-无机复合材料/有机防护涂层这四部分进行系统总结,重点围绕氮化硼纳米片在有机涂层中均匀分散能力以及金属腐蚀防护能力等方面等进行详细分析和介绍,同时对氮化硼纳米片基防腐涂料未来发展进行了展望.
English
Progress of Boron Nitride Nanosheets Used for Heavy-duty Anti-Corrosive Coatings
Abstract:
Boron nitride nanosheets (BNNSs), also known as "white graphene", is an important nanofiller with excellent mechanical properties, thermal conductivity, abrasion resistance, barrier properties, and hydrophobicity. It is also a new type of excellent performance insulation materials. It is widely used in heavy-duty anti-corrosion coatings, lubricants, sensors and other fields. Based on the huge application prospects of BNNSs in the field of metal corrosion protection, this article systematically summarizes the preparation and surface functionalization of BNNSs, boron nitride thin film protective coatings, BNNSs/organic protective coatings, BNNSs-inorganic materials/organic protective coatings, and focuses on the detailed analysis and existing problems of BNNSs uniformly dispersed in organic coatings and used for metal corrosion protection. The future development of BNNSs-based anticorrosive coatings is prospected.
-
Key words:
- boron nitride nanosheets
- / surface functionalization
- / metal
- / coating
- / anticorrosion
-
1. 引言
腐蚀是一个非常缓慢的化学反应, 它是指金属与腐蚀性介质(水、氧气、氯离子等)作用从而失去原来金属性质(高硬度、高强度、金属光泽)的现象, 造成了巨大的经济损失以及重大的社会影响.金属防护最有效的措施是涂层保护, 为此大量科研人员在涂层领域进行了许多研究, 开发了金属氧化物涂料[1]、有机涂料[2]、无机/有机复合涂料[3], 其中有机复合涂层因其加工简单、防护性能较强等特点成为最受欢迎的一种涂层.近年来, 在有机涂层中加入纳米填料(石墨烯、黏土、SiO2、BNNSs等)被认为是提高其长期防腐性能的有效途径.目前, 石墨烯[4]已经被广泛应用于金属防腐蚀领域.然而, 有文献报道, 在质量分数为3.5%的NaCl溶液中长时间浸泡后, 石墨烯会与金属发生“电偶腐蚀”, 促进了金属溶解.
为了探究金属的长效防腐蚀方法, 将BNNSs作为填料添加到聚合物中(如环氧树脂、聚氨酯、聚乙烯醇等)以增强复合涂层的导热率[5]、电阻率[6]、耐蚀性[7]、疏水性[8]、机械强度[9]已经成为一个非常热门的研究方向, 很多研究者在这方面投入了大量的工作.商业上买来的六方氮化硼(h-BN)几乎都未经剥离处理, 并且由于其高比表面积及层间很强的范德华相互作用, h-BN很容易在聚合物材料中发生团聚, 因此通常需要在表面进行化学修饰(包括共价[10]或非共价官能化[11])以此来提高氮化硼纳米片与聚合物基体之间的分散性、相容性[12].基于氮化硼纳米片在金属腐蚀防护领域巨大的应用潜力, 本文将从氮化硼纳米片的制备及表面官能化, 以及氮化硼薄膜防护涂层、氮化硼纳米片/有机防护涂层、氮化硼纳米片-无机复合材料/有机防护涂层在金属腐蚀防护领域的应用等方面的研究进展进行全面综述.
2. 氮化硼纳米片的制备及表面官能化
2.1 机械剥离
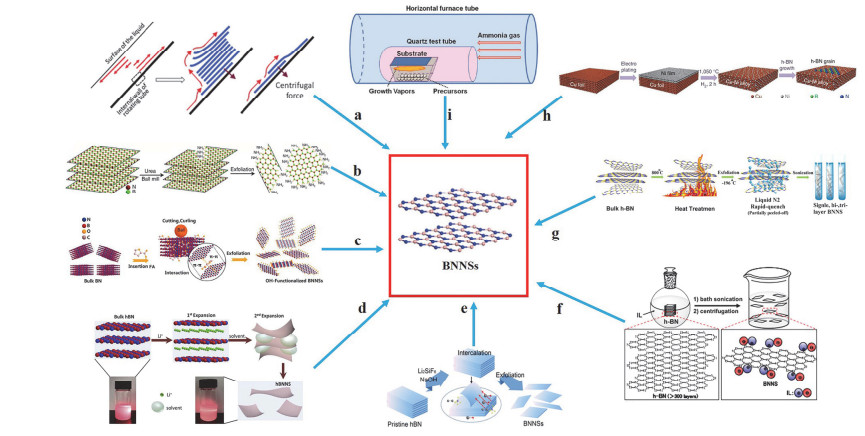
剥离层状材料最初是使用透明胶带对材料进行撕拉从而完成剥离过程, Novoselov等[13]首次通过这种方法成功剥离了单层石墨烯之后, 又通过“胶带剥离法”成功地实现了对h-BN[14]的有效剥离.他们将氮化硼粘在胶带上对折撕开, 然后撕开的一面用胶带粘住再撕开, 重复上述过程最终获得了单层、薄而透明的氮化硼纳米片.胶带剥离后的氮化硼纳米片维持了原有的横向尺寸, 并且没有破坏它内部的晶格结构, 因此成为优良导热材料[15]、电子元件[16]的绝佳候选者.由于胶带法剥离氮化硼存在很大的局限性, 不能大规模进行生产, 有文献报道通过高速剪切可以连续批量获得氮化硼纳米片. Chen等[17]报道了一种非常简单的剪切剥离工艺(图 1a), 氮化硼分散在装有N-甲基-2-吡咯烷酮(NMP)的试管中, 倾斜45°后在8000 r/min的剪切力下剥离30 min, 此时剥离的氮化硼具有明显的多层纳米片结构.
图 1
Lei等[18]在尿素的辅助下通过球磨成功剥离了氮化硼(图 1b), 这种方法的产率高达85%, 质量也非常好(绝大多数纳米片横向尺寸为100 nm, 厚度为2~2.5 nm), 并且通过球磨剥离的过程在纳米片表面引入了亲水性基团(-NH2), 因此氮化硼纳米片胶体在水中的溶度高达30 mg/mL.之后Ding等[19]改进了球磨剥离法(图 1c), 以2-呋喃甲酸(FA)为改性剂, 球磨剥离后得到氮化硼纳米片的产率高达98%.该纳米片热导率高达25.2 W•m-1• K-1, 可以用于制备三维超轻气凝胶和超薄二维导热膜.但是球磨过程也存在一个很大的问题, 强大的剪切力毫无疑问会对BNNSs的结构产生一定的破坏.
2.2 液相辅助剥离
液相辅助剥离主要分为以下两种:超声辅助剥离法、离子插层法[20].超声辅助剥离法:即直接在液体介质中对h-BN进行超声波处理来制备二维纳米片, 这种超声波产生的剪切力和气泡可以破坏层间结构并产生单层或少量的层状纳米片.大量研究表明, 许多强极性溶剂, 如二甲基甲酰胺(DMF)、N, N-二甲基乙酰胺(DMAc)、1, 2-二氯苯和乙二醇, 都能有效地超声辅助剥离h-BN, 从而获得非常稳定的分散体. Zhi等[21]将氮化硼分散在DMF中对其进行超声处理, 得到的氮化硼纳米片可以保持高度的结晶状态.作者认为超声剥离的过程中, 氮化硼表面与DMF之间存在很强的相互作用力. Cao等[22]开发了一种简便的混合溶剂策略(V氨水溶液:V异丙醇=3:2)用于大规模制备h-BN纳米片.混合溶液中经过超声、离心等过程, 所制得的氮化硼纳米片可以均匀分散在异丙醇(IPA)中, 长时间静置后也无团聚现象产生.由于硼是一种不导电的原子, 因此他们认为在Lewis酸碱作用下形成了一种配合物, 从而促使h-BN的剥离. Wang等[23]以甲磺酸(MSA)为溶剂, 采用液相剥离法制备了氮化硼纳米片(厚度小于3 nm), 相比于常用的有机溶剂, 这种质子磺酸的辅助剥离能力更强. Zhou等[24]比较了乙醇和水两种溶剂对h-BN、WS2、MoS2等二维材料的影响, 发现当乙醇和水都加入时, 二维材料的分散度和剥离效率都有很大的提高.因此, 在液相中剥离氮化硼, 极性溶剂往往在其中起着一个非常重要的作用.
由于Cao等单纯用IPA长时间超声剥离氮化硼的产率很低, Wang等[25]报道了一种改良的液相辅助剥离法(图 1d).将层状氮化硼粉末晶体分散在IPA溶液中, 然后加入了LiCl, Li+的加入使氮化硼的剥离产率提高到了55%, 在水中的分散度达到4.13 mg/mL.并且溶剂的极性和Li+对氮化硼的剥离效果影响很大.相比于其他极性溶剂(如乙醇、NMP、DMF), 氮化硼在IPA中剥离得到的纳米片最薄(厚度只有1~3 nm), 横向尺寸最大(可达几十微米).由于Li+成功插入到了氮化硼的层与层之间, 层间距急剧膨胀, 层间相互作用大大减弱, 因此对氮化硼的剥离起到了非常重要的作用.为了提高Li+插层剥离的效率, Zhao等[26]将h-BN粉末分散在Li2SiF6/NaOH的混合溶液中(图 1e), 并进行温和搅拌. SiF62-与h-BN强非共价键相互作用产生的静电斥力导致h-BN的层间膨胀[27], 同时OH-和Li+在协同作用下插入相邻h-BN晶格的层间空隙, 导致h-BN层的结构膨胀.随后, 强烈的搅拌促进了相邻h-BN层之间的相对运动, 导致纳米片在侧向力的作用下变得越来越薄.结果表明他们制得氮化硼纳米片的产率高达78.5%, 分散在水中的溶度为12.78 mg/mL, 作者认为NaOH的添加在氮化硼纳米片表面修饰了-OH, 因此可以保持良好的稳定性和分散性, 并且该方法对于其他二维材料的剥离具有良好的通用性. Güler等[28]将石墨烯与氮化硼分散在强酸溶液中(浓H2SO4和浓HNO3)得到一种杂化插层复合物, 然后在DMF中进行超声剥离.这种方法制得的氮化硼/石墨烯杂化纳米片可以广泛用于光电器件和导热材料.
离子液体(ILs)是一种熔点低于100 ℃的盐溶液, 由于其具有离子导电率高、热稳定性好、蒸汽压低等特点, 引起了人们的广泛关注[29].受到ILs在碳纳米管分散中的应用启发[30], Morishita等[31]比较了不同离子液体对h-BN剥离效果的影响, 其中在[bmim][PF6]这种离子液体协助下制备出具有微米级边缘的氮化硼纳米片. ILs吸附在BNNSs表面, 显著地增强了剥离效果(图 1f), 使BNNSs高浓度分散(约1.9 mg/mL), 产率达到约50%.单纯在液体中进行h-BN的剥离非常耗时, 因此Ding等[32]报道了一种液-气相协同剥离的方法(图 1g). h-BN粉末先经过800 ℃的高温预处理后削弱了层间的相互作用; 随后通入液氮, N2迅速撑开了氮化硼层间距离; 最后在水中超声处理2 h即可得到厚度小于5层的纳米片.该工作操作简单、时间成本较低、无缺陷纳米片的平均收率高达33%, 因此非常适合大规模生产.剥离法制氮化硼纳米片的方法已总结于表 1.
表 1
 表 1 剥离法制氮化硼纳米片Table 1. Boron nitride nanosheets prepared by the exfoliation method
表 1 剥离法制氮化硼纳米片Table 1. Boron nitride nanosheets prepared by the exfoliation method 下载:
导出CSV
下载:
导出CSV
Solvent/insertion molecules Exfoliation condition Concentration/(mg•mL-1) Dimension Ref. MSA Ultrasonication: 8 h;
Centrifuge: 4000 r/min, 90 min0.2~0.3 Thickness:<3 nm [23] C8H15N2F6P/Water Bath ultrasonication: 8 h;
Centrifuge: 3000 r/min (1220 G), 20 min1.86 Thickness: 6.3 layers
Lateral size: 1.34 μm[30] Urea/Nitrogen Ball mill;
Centrifuge: 700 r/min, 20 h30 Thickness: 2~2.5 nm Lateral size: 100 nm [18] 2-Furoic acid Ball mill 35 Thickness: 2 nm Lateral size: 1.8 μm [19] IPA/Ammonia water Ultrasonication: 40 kHz, 35 h;
Centrifuge: 3000 G4 Not supplied [22] IPA/LiCl Hydrothermal reaction for 12 hours;
Centrifuge: 500 r/min, 5 min4.13 Thickness:<10 layers Lateral size: 400 nm [25] LiSiF6/NaOH Stirring: 500 r/min, 60 h;
Centrifuge: 2000 r/min, 30 min8.35 Thickness:<5 layers Lateral size: 1~3 μm [26] Liquid N2/Water Ultrasonication: 2 h;
Centrifuge: 1000 r/min, 15 minNot supplied Thickness:<5 layers Lateral size: 1~5 μm [32] 2.3 化学气相沉积(CVD)法
CVD法, 它是利用气态物质在气相或者气固相界面上发生反应生成固态沉积物的过程.最早在20世纪60年代, 科学家通过使用二硼烷(B2H6)和氨气(NH3)作为反应的前驱体, 然后高温(600~1080 ℃)的条件下可以在Si、Ta、Mo、Ge和熔融石英等基底表面沉积BNNSs. CVD工艺制备BNNSs的工作原理非常简单:一块衬底被固定在石英管加热室中, 在引入前驱体(如氨硼烷)之前, 将该室加热至一定温度.调节气泵流速, 从而可以在相对较低的压力或大气压下实现BNNSs的快速生长.
Lee等[33]以氨硼烷(又称硼砂烷)为前驱体, 采用热化学气相沉积法(图 1h)在镍箔表面上制备了层状六方氮化硼(h-BN)薄片.原子力显微镜(AFM)显示, Ni表面的CVD-h-BN薄膜具有层状结构. h-BN薄片在Ni(100)晶面上的生长速率较大, 但在Ni(111)晶面上的生长速率不明显, 表明h-BN在Ni上的CVD生长应考虑动力学而非热力学控制. Lu等[34]也以氨硼烷为前驱体通过热CVD法在Cu-Ni合金箔上成功地合成了大尺寸h-BN薄片, 通过优化Ni在基体中的比例, 可以使每平方毫米氮化硼的成核密度显著降低到60, 并且该方法也大大提高了h-BN晶粒的生长速率, 比以前的报道大两个数量级.
为了能在Cu上制备具有高质量且层数控制的h-BN薄片, 科学家进行了大量的研究. Song等[35]首次以氨硼烷为前驱物通过CVD法在Cu表面生长了2~5层较大尺寸的h-BN薄片.他们还通过AFM证实了整个薄片92%是由单层组成. Tay等[36]报道了在常压下通过CVD法在高电抛光的Cu衬底上生长了面积非常大的(约35 μm2) h-BN薄膜的工作.铜的表面形貌和含氧溶度对h-BN薄膜的成核密度以及区域尺寸影响很大, 通过抑制成核速度, 提高侧向横向生长速度可以在一定程度上获得具有大尺寸的六边形氮化硼, 这样有效地减少了晶粒边界的缺陷. Pakdel等[37]报道了一种在传统水平管式炉中, 以硼、氧化镁、氧化铁为前驱体, 氨气为流动介质, 采用CVD法在硅/二氧化硅(Si/SiO2)衬底上直接制备BNNSs的简单有效策略(图 1i).氮化硼纳米片的厚度大多小于5 nm.该项工作的亮点之处在于非常方便地制备超疏水(水接触角超过150°)纯BNNSs涂层, 并且在不同温度梯度下获得了不同结构的纳米片. 900 ℃时合成了光滑的氮化硼涂层, 在1000 ℃以及更高的温度可以制得由部分垂直排列的纳米片组成的致密BNNSs涂层.
2.4 氮化硼纳米片表面的功能化改性
氮化硼纳米片, 是近年来最有发展前景的无机纳米材料.它独特的性能, 包括高机械强度、宽带隙、优异的导热性和热稳定性, 在各种工程领域中具有很大的潜在应用.特别是, h-BN纳米材料已被广泛用作功能性填料, 以制备高性能聚合物纳米复合材料.但是在将其用于纳米复合材料之前, 通常需要对氮化硼纳米片进行表面功能化改性, 以阻止其快速团聚的趋势, 并改善其在聚合物中的分散性和界面相容性. h-BN与氧化石墨烯(GO)最大的区别在于, GO表面存在很多含氧官能团(羟基、羧基、环氧基等), 而氮化硼表面几乎没有任何官能团, 因此对氮化硼表面进行官能化可以大大提高它的分散性.
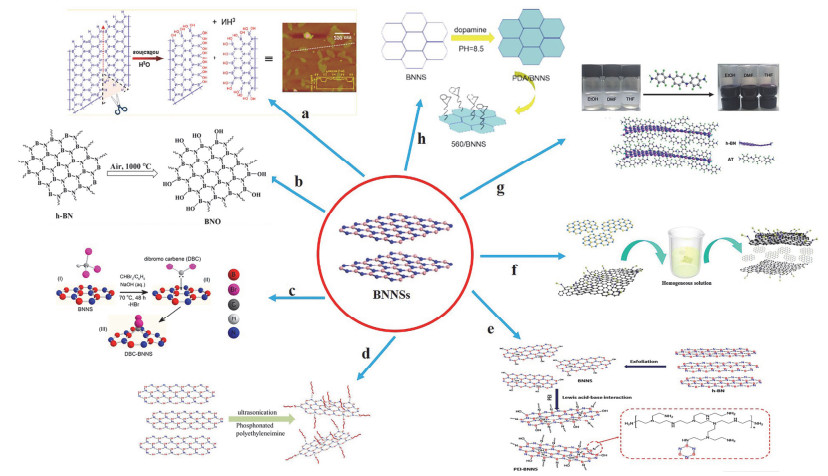
直接对氮化硼表面进行功能化的报道非常少, 大多数的工作都是通过将官能团共价吸附在氮化硼边缘的羟基上. Lin等[38]报道了一种非常简便的提高氮化硼纳米片在水中的分散方法, 他们提出将h-BN粉末直接放在水中用超声法制备羟基化氮化硼纳米片(图 2a), 超声辅助水解可以促进层状氮化硼的剥离及羟基化, 并且所获得的分散体中也证实了单层和多层氮化硼纳米片的存在. Yu等[39]提出在大气环境下将氮化硼粉末加热到1000 ℃, 即可在其表面引入羟基(图 2b), 极大地方便了后续的进一步剥离分散及表面官能化.
图 2

Sainsbury等[40]提出了一种共价官能化的方法, 使用过氧化二叔丁基在BNNSs表面首次引入烷氧基(图 2c), 再经过H2SO4/H2O2处理后, 结合的叔-丁氧基被水解成羟基.除此之外, 他们将羟基化氮化硼纳米片(OH-BNNSs)分散在聚乙烯醇(PVA)中制得纳米复合材料, 羟基化氮化硼纳米片的加入极大地提高了复合材料的力学性能, 这在很大程度上归于材料基底与羟基化氮化硼纳米片之间的均匀分布, 以及二者之间良好的相容性.
近几年, Lewis酸碱相互作用提供了同时剥离和修饰h-BN纳米片的新策略. Cai等[41]合成了磷酸化聚乙烯亚胺(P-PEI), 通过Lewis酸碱相互作用使P-PEI很容易地与h-BN纳米片表面的缺电子硼原子形成配合物, 从而更好地实现了氮化硼的共价剥离(图 2d).他们将共价改性后的纳米片添加到热塑性聚氨酯中, 制得的复合材料阻燃效果有了很大的提高.在Wu等[42]的工作当中, 他们首先将氮化硼在IPA中超声处理获得了羟基化的纳米片, 然后将水溶性支链聚乙烯亚胺通过Lewis酸碱相互作用对羟基化氮化硼纳米片进行共价改性(图 2e), 改性之后的纳米片在溶液中的分散性和稳定性都有了很大的提高.因此通过共价改性剥离得到的纳米片与其他基体(包括溶剂、树脂等)的相容性显著提高, 这大大拓宽了BNNSs在材料领域的应用范围.
非共价官能化剥离法顾名思义:就是共轭聚合物和h-BN表面之间通过π-π相互作用从而接枝到氮化硼纳米片表面.关于BNNSs与含有平面或芳香族结构的功能分子之间的非共价相互作用, 已有一些理论报道. Ding等[19]为了制备高质量、高收率的氮化硼纳米晶体, 采用2-呋喃甲酸(FA)为改性剂, 在剥离过程中, h-BN层与层之间的键通过FA芳香族中的强π-π相互作用打开.同时, FA分子吸附在h-BN表面, 通过压力作用实现原位共价修饰.此外FA也可以当做润滑剂, 减少球磨对BNNSs结构的损伤, 从拉曼光谱图中可看出剥离的纳米片显示出非常薄的结构.由于h-BN具有疏水性难以分散在水性环氧基体中, Wu等[43]选用亲水性GO作为h-BN的插层剂(图 2f), 直接增强其在水性环氧树脂中的分散性. GO与h-BN之间的π-π相互作用使h-BN均匀地堆积在GO表面, 这样一方面可以极大提高氮化硼的剥离效率, 另一方面也可以起到稳定易于团聚的氮化硼纳米片. AFM结果显示, GO/h-BN(mGO:mh-BN=1:1)的高度分布达到3.32~3.76 nm(约10到11个原子层厚), 这表明h-BN被GO成功地剥离分散了.另外GO/h-BN复合材料的π-π*吸收峰变为225 nm, 这进一步表明GO和h-BN之间存在π-π相互作用. Cui等[11]通过胺封端的苯胺三聚体(AT)成功剥离修饰了h-BN纳米片(图 2g), 使得h-BN纳米片稳定分散在有机溶剂中, 紫外可见吸收光谱和透射电镜分别证明了AT与h-BN存在着π-π相互作用以及h-BN在AT的协助下剥离成了很薄的纳米片.
有研究表明硅烷偶联剂可以实现对氧化石墨烯的表面改性, 从而大大提高了它在树脂基体中的分散度[44-46].氮化硼与石墨烯具有相似的结构, Fan等[47]成功地在氮化硼表面接枝了硅烷偶联剂(KH-560).首先他们通过π-π相互作用在氮化硼纳米片表面引入多巴胺分子, 然后多巴胺分子中邻苯二酚羟基与KH-560发生化学反应成功地合成了KH-560修饰的氮化硼纳米片(图 2h), 羟基和KH-560的修饰大大提高了氮化硼纳米片在水性丙烯酸树脂中的稳定性. Liu等[48]也证明了用(3-氨基丙基)三乙氧基硅烷(APTES)改性的氮化硼纳米片明显提高了其在环氧树脂中的分散性和界面相容性, 均匀分散并且具有一定取向排列的氮化硼纳米片促使涂层的导热系数提高了26倍, 在大功率电子元器件封装领域具有非常大的应用前景.
3. 氮化硼薄膜防护涂层
石墨烯是一种层状二维材料, 具有很高的阻隔性, 已经被用作为一种超薄且质量较轻的防腐蚀阻隔层, 但是由于石墨烯优良的导电性[49], 使之具有腐蚀促进活性从而加速金属基底的腐蚀[50].氮化硼纳米片被称为“白色石墨烯”, 是一种二维绝缘体, 比石墨烯更具热稳定性和化学稳定性, 因而可以从本质上解决金属间的电偶腐蚀这个严重的问题.最近的研究也表明, h-BN纳米片由于其高比表面积和与基底的高附着力, 可以为金属提供有效的耐腐蚀性能.
3.1 镍基复合镀层防护
Gyawali等[51]以镍为阳极铁为阴极, 采用脉冲电沉积技术将5~20 g/L的h-BN纳米片分散在氨基磺酸盐电解槽中进行电沉积, 成功地制备了镍基复合镀层.当h-BN纳米片的浓度为20 g/L时, Ni/h-BN (20 g/L)复合镀层的腐蚀速率为0.0094 mm/year, 比纯Ni涂层大约低一个数量级.他们认为复合镀层腐蚀速率降低的原因主要有以下三种: (1) Ni和h-BN纳米片本身具有很强的耐蚀性; (2) h-BN纳米片的层积改变了Ni晶体的结构, 从而提高了Ni的耐腐蚀性能; (3)可能形成了以h-BN纳米片为阴极、镍金属为阳极的腐蚀微电池, 这种腐蚀微电池有利于阳极极化, 从而降低了镍金属的腐蚀速率.
3.2 抗高温氧化
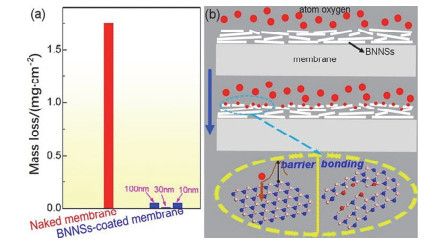
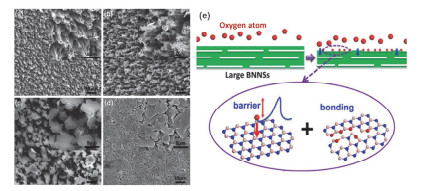
原子氧具有很强的氧化活性, 能腐蚀大多数金属和聚合物组分, 氮化硼薄膜是一种非常稳定的二维材料, 不会与大多数化学物质发生反应, 由于它出色的热稳定性和化学稳定性[52], 有望成为一种抗高温氧化的理想涂层. Liu等[53]通过CVD法在镍箔表面沉积了厚度分别为2 nm、5 nm的氮化硼薄膜.镍金属在1100 ℃的空气氛围下持续加热30 min后, 表面非常光滑, 看不到任何被氧化腐蚀的迹象, 而且在一定温度范围内随着氮化硼薄膜厚度的不断增加, 涂层的高温抗氧化性能也越来越好. Yi等[54]采用有效的流体力学方法在77 MPa的恒定压力下制备了厚度为0.5~4.2 nm, 大小为几百纳米到几微米的BNNSs, 并在尼龙表面沉积了厚度可控的BNNSs涂层.暴露在原子氧中后, 尼龙纤维被严重腐蚀, 质量损失最大(图 3a).在BNNSs涂层膜中, 由于真空过滤过程, BNNSs在平面上紧密排列, 从而形成覆盖整个膜表面的完美涂层(图 3b).氧原子腐蚀后, 薄膜仍被BNNSs覆盖, 但BNNSs涂层变得更加粗糙和疏松, 大量BNNSs开裂并从表面突出.因此, 从这些表面形貌的变化可以直观地得出BNNSs涂层可以保护聚合物膜免受氧原子的腐蚀.我们知道大部分的腐蚀都是从缺陷的位置开始的, 通过CVD法制备的氮化硼薄膜不可避免地也会对氮化硼的结构产生一定的缺陷空位, 非常有趣的是在Liu等[55]报道的工作当中, 他们利用密度泛函第一性原理计算出原子氧可以通过化学吸附修复填补氮化硼薄膜中的缺陷, 对氧分子的阻隔能力提升了4倍以上.
图 3

3.3 金属表面钝化
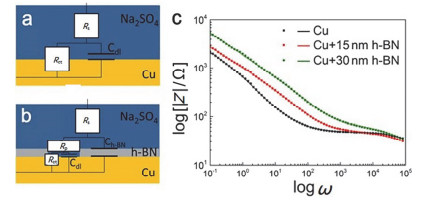
氮化硼纳米片薄膜除了具有对腐蚀性介质(如氧气、水等)具有比较好的物理阻隔性之外, 还具有钝化金属表面的作用. Zhang等[56]采用化学气相沉积法在镍箔上制备了大尺寸且厚度为15~30 nm的h-BN薄膜, 并将其作为腐蚀钝化膜转移到铜表面上.当h-BN涂层存在时, 腐蚀速率降低了4倍.电化学阻抗谱(EIS)拟合结果表明(图 4), 由于h-BN涂层的电荷转移电阻Rct显著增加, 从而大大提高了腐蚀钝化效应.作者认为, h-BN涂层上的缺陷是腐蚀性介质(Na2SO4、O2、H2O)进入铜表面的主要途径, 通过对这些缺陷进行适当的钝化处理, 可以极大地提高涂层的缓蚀性能.同样Mahvash等[57]也通过CVD法在铜箔表面沉积单层氮化硼薄膜, 证实了在薄膜的防护下铜的腐蚀速率较裸铜降低了一个数量级, 充分证明了该超薄(约0.45 nm)涂层对铜的腐蚀具有很好的缓蚀作用.
图 4

3.4 抗海洋微生物污染
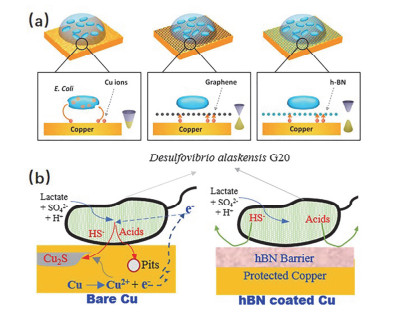
金属在海洋大气中的腐蚀很大一部分原因是由于海水中的微生物附着在金属表面所产生的, 解决这个问题的传统方法是在金属表面涂覆一层防污涂料, 然而防污涂层在使用的过程当中不可避免地会对环境造成很大的破坏[58]. Parra等[59]研究了石墨烯、氮化硼薄膜对铜表面生物的抗菌能力, 他们发现石墨烯、氮化硼薄膜在很大程度上抑制了细菌与底层铜基底之间的相互作用, 并且它们的二维层状结构也是防止发生物理接触的有效屏障(图 5a), 因此大大降低了铜表面生物腐蚀的概率. Chilkoor等[60]证明了用CVD法制备的单层氮化硼薄膜能够有效抑制硫酸盐还原菌G20对铜产生的微生物腐蚀.沉积氮化硼薄膜后, 能有效地抑制浮游细胞的腐蚀效应, 阳极电流比裸铜降低了36倍, 对细菌产生的代谢物具有91%的缓蚀效果.但是与石墨烯薄膜不同的是, 氮化硼薄膜能够有效抑制阴极还原和电偶腐蚀效应(图 5b).
图 5

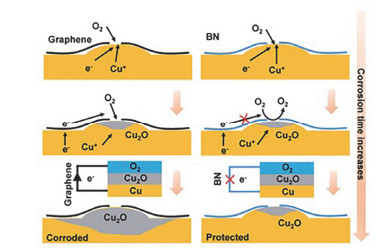
3.5 长效防腐
Shen等[61]为研究石墨烯(G)及氮化硼(h-BN)涂层对金属基底的长期耐腐蚀性能, 通过CVD法在铜表面分别沉积一层石墨烯和氮化硼, 将试样在相对湿度为60%的大气环境中放置160 d, 发现在长期的腐蚀测试实验中h-BN/Cu试样的耐蚀性能比G/Cu要好很多.为了进一步研究石墨烯及氮化硼涂层对铜金属的防护性能, 他们认为高导电的石墨烯可以将电子传输给氧原子, 充当电化学反应的阴极, 在很大程度上加速了金属腐蚀的速率.然而由于六方氮化硼是绝缘体, 切断了电子传输的路径, 铜的腐蚀速率大大减小(图 6).
图 6

Li等[62]利用CVD法在铜箔表面生长了一层7~8 nm(约为20层)的氮化硼薄膜, 在250 ℃的空气氛围下对试样热处理100 h, 光学显微镜下看到裸铜箔表面完全变为黑色, 而氮化硼涂层试样表面只有一层淡淡的浅褐色氧化膜, 充分表明氮化硼涂层在铜箔表面起着非常优异的抗氧化作用.并且相比于裸铜金属氮化硼涂层试样具有较低的腐蚀电流密度, 直接证明了氮化硼薄膜显著降低了铜的腐蚀速率.
4. 氮化硼纳米片/有机防护涂层
有机涂层是一种目前使用最为广泛的金属防护手段, 为了不断提高涂层的耐蚀性能, 研究者将纳米材料(石墨烯、SiO2、TiO2、Al2O3、ZnO)添加到聚合物中大大提高了涂层阻隔腐蚀性介质的性能[63-66].近年来将h-BN纳米片与聚合物树脂进行复合使用已经成为重防腐涂层领域的一大研究热点, 这种涂层不仅能够保留氮化硼纳米片优异的抗渗性能、机械性能、导热性能, 同时还兼具了聚合物树脂强附着力、高力学性能等特点, 因此在金属防护方面具有很大的应用前景.但是氮化硼纳米片层间存在很强的π-π相互作用力, 导致它很容易自动发生团聚, 难以均匀分散在树脂基底中, 极大地影响了复合涂层的耐蚀性能[11].
Husain等[67]以剥离的h-BN和聚乙烯醇(PVA)为原料制备了复合涂层, 并探究了复合涂层在模拟海水的环境下对316不锈钢耐蚀性能的影响.通过对比纯PVA涂层, 他们发现h-BN的加入大大提高了316不锈钢的耐蚀性能, 在质量分数为3.5%的NaCl溶液中浸泡30 d后腐蚀电流密度仅为5.14×10-8 A•cm-2.但是由于该工作没有解决氮化硼易于团聚的问题, 涂层在保护过程当中不可避免地会发生严重的点蚀现象, 导致该涂层不能实现对316不锈钢的长效保护.受Husain等[67]工作的启发, Yi等[68]对比了纯PVA薄膜和BNNSs-PVA复合涂层抗氧化腐蚀的能力.暴露在氧原子的氛围中, 纯PVA薄膜出现严重腐蚀并且表面也变得非常粗糙, 但是BNNSs(1.0%)-PVA复合涂层表面较为光滑、致密(图 7a~7d).因此BNNSs的加入可以大大提高PVA的抗氧化腐蚀性能.他们认为BNNSs抗氧原子腐蚀机理如下: (1) BNNSs的片状结构能对氧原子的穿透起到一定的物理阻隔作用(图 7e), 保护下面的聚合物不受腐蚀; (2)氧原子诱导的单层h-BN氧化是通过h-BN晶格中N逐渐被O所取代而发生的[69], 并且因为h-BN是一种具有N空位的结构, 所以它本身就存在一种有效的氧治愈机制[70].
图 7

由于石墨烯的分子不渗透性, 无论是作为超薄保护涂层还是作为有机涂层的纳米填料, 石墨烯都被认为是一种理想的金属保护材料[71].然而, 最近的研究表明, 石墨烯作为一种二维纳米填料, 并不能为金属基底提供长期保护, 原因在于石墨烯本身具有一定的腐蚀促进活性[72, 73]. Sun等[74]研究比较了纯PVB涂层、rGO/PVB复合涂层、BNNSs/PVB复合涂层的耐腐蚀性能, 在质量分数为3.5%的NaCl溶液中浸泡四个月后含1.0% BNNSs纳米填料的BNNSs/PVB复合涂层的防护性能是纯PVB涂层的67000倍, 而且BNNSs作为保护涂层的纳米填料与石墨烯不同, 在涂层完整性受损时, BNNSs不会加速金属/涂层界面处的金属腐蚀, 因此在重防腐涂层领域BNNSs有着比石墨烯更宽广的前景.
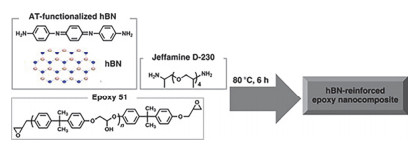
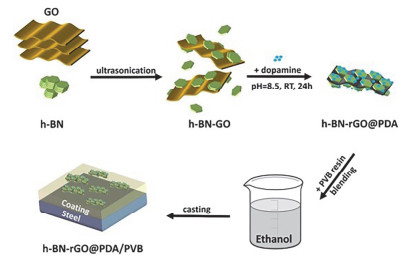
Chen等[75]以氨基苯胺三聚体(AT)作为氮化硼纳米片的分散剂, 通过π-π相互作用促进了h-BN纳米片在环氧树脂基底中的分散, 然后制备了含有不同h-BN质量分数的复合涂层(图 8).随着h-BN含量的不断提高, 复合涂层的接触角也显著增加, 最高可达112.89°±1.39°, 说明EP/h-BN涂层具有非常好的疏水性能.与纯环氧树脂涂层相比, 当h-BN的质量分数为1%时, 复合涂层的拉伸强度可提高6.6% (52.74 MPa), 模量可提高5.5% (1416 MPa).
图 8
相比于溶剂型环氧涂层, 水性环氧涂层具有非常鲜明的优点, 不会产生有毒的易挥发溶剂, 对环境非常友好, 是未来重防腐涂层发展的一个重要研究方向.但是由于传统的水性环氧涂层体系中包含亲水基团和表面活性剂, 不能有效地抑制氧、水气、其他腐蚀性离子的渗透[76], 因此不能为金属基底提供长效的保护.将氮化硼纳米片作为填料添加到水性环氧涂层体系中可以大幅度提高水性环氧复合涂层的阻隔性和耐蚀性能. Gu等[77]报道了一种石墨烯在水性环氧树脂中均匀分散的方法, 即通过羧化苯胺三聚体(CAT)与石墨烯之间的π-π相互作用来实现对石墨烯的共价改性.受此启发, Cui等[7]借助CAT在水中剥离得到了h-BN纳米片, 制备了不同含量的h-BN纳米片水性环氧涂层.通过吸水率测试及开路电位测试, 1% h-BN纳米片的加入显著降低了复合涂层的吸水率及腐蚀倾向, 这是因为h-BN的均匀分散不仅增加了环氧树脂的密度, 而且减少了复合涂层表面的孔洞和缝隙, 进而抑制了水和腐蚀性离子在涂层中的扩散.
在我国南海海域由于长期处于一种高温、高湿、高盐、强紫外辐射的环境中, 海洋工程装备面临着严重的腐蚀考验. Zhao等[78]制备了甲基丙烯酸糠酯(FAM)非共价键功能化的氮化硼纳米片, 将FAM/h-BN纳米片作为环氧大豆油甲基丙烯酸树脂(ESOM)紫外光固化涂料的活性稀释剂, 他们发现含0.75% h-BN纳米片涂层的阻抗是空白涂层的200倍, 因此该工作可以为制备一种耐蚀性能优异的紫外光固化涂料提供一个很好的借鉴方法, 以供潜在的海上应用.
为了探究二维纳米片材料在水性环氧树脂涂层中的防腐机理, Yu等[79]选取了三种导电率不同的二维纳米片材料, 即石墨烯(G)、氧化石墨烯(GO)、六方氮化硼纳米片(BNNSs), 并通过3-氨基丙基三乙氧基硅烷(APTES)对以上材料进行官能化, 分别研究了三种材料的耐蚀性能及防腐机理.通过塔菲尔极化曲线测试, 改性后的氮化硼纳米片有机复合涂层体系呈现出较高的腐蚀电位(E=-0.608 V), 较低的腐蚀电流密度(icorr=4.960×10-8 A•cm-2), 保护效率达到了99.58%.官能化氧化石墨烯(FG)在涂层基底中的均匀分布增大了腐蚀介质进入金属表面的路径, 但是由于FG的导电率非常优异, 当涂层底层的FG与金属接触时, 阳极(Fe)反应失去的电子很容易通过FG与腐蚀介质(H2O和O2)接触完成一个电化学腐蚀(也被称为“电偶腐蚀”)的过程, 从而加速铁的腐蚀(图 9a); 官能化氮化硼(F-h-BN)的均匀分散不仅有效阻止了腐蚀性介质的渗透, 并且作为一种良好的绝缘体还有效地防止了“电偶腐蚀”的发生(图 9b).
图 9

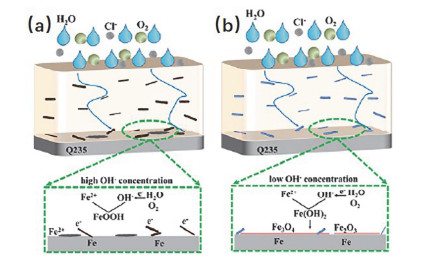
为了实现氮化硼有机复合涂层长效的耐蚀性能, Cui等[80]用聚2-丁基苯胺(PBA)作为非共价分散剂来对h-BN进行化学改性.他们的工作创新之处在于PBA不仅有助于h-BN的剥离, 还可以作为腐蚀抑制剂, 对金属基底的防护起到了双重保险.一方面氮化硼纳米片的随机分布会产生“迷宫效应”, 从而增大了腐蚀介质进入涂层的空间位阻; 另一方面PBA的添加, 不仅促进了氮化硼纳米片在环氧树脂中的无序分散, 还作为活性物质促进了铁金属表面钝化膜的形成, 从而实现了对铁金属的长效保护. Zou等[81]用Ar+NH3(体积比为4:1)这种等离子体气体对在氮化硼纳米片表面引入了-OH、-NH2, 然后与丙烯酸树脂混合制得复合涂层.改性之后的氮化硼纳米片/丙烯酸树脂涂层的腐蚀电流密度比未改性的大约低13倍, 作者认为耐蚀性能的提高归功于: -OH、-NH2促进了h-BN的剥离, 也改善了h-BN在丙烯酸涂料中的分散性; 此外等离子体处理后可以增大层间距, 有利于在h-BN层间储存更多的Fe2+, 从而防止Fe2+的迁移和Fe2O3的形成(图 10).
图 10

5. 氮化硼纳米片-无机复合材料/有机防护涂层
氮化硼纳米片与其他无机材料复合之后对金属的腐蚀防护呈现了很好的协同效应, 大大增强了涂层的耐蚀性能. Huang等[82]受贻贝强粘附力的启发, 利用聚多巴胺(PDA)将氧化石墨烯和氮化硼纳米片(h-BN)结合在一起, 制备出了一种新型的纳米复合材料h-BN-rGO@ PDA(图 11).比较h-BN-rGO@PDA/PVB、GO/PVB、h-BN/PVB在质量分数为3.5%的NaCl溶液中对Q235碳钢的抗腐蚀能力, 他们发现由于h-BN-rGO@PDA的加入, 涂层的耐蚀性能比纯PVB提高了2个数量级, 并且也大于GO/PVB、h-BN/PVB, 这归功于h-BN-rGO@ PDA在PVB涂层中具有比h-BN或GO更好的物理屏障能力.同样, Wu等[43]报道了用GO改性的氮化硼纳米片来提高水性环氧树脂涂层抗腐蚀能力的工作. GO/h-BN(mGO:mh-BN=1:1)/水性环氧树脂(WBE)复合涂层与纯WBE涂层相比具有显著的耐蚀性能, GO/h-BN(mGO:mh-BN=1:1)纳米复合材料的均匀分散以及GO和h-BN对腐蚀性介质(H2O、O2和Cl-)的协同阻隔效应可以用来解释这一现象.
图 11

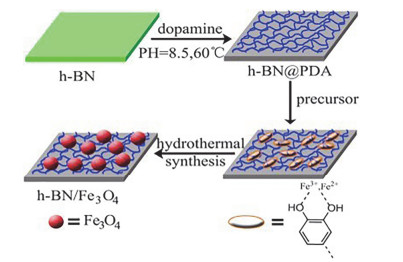
Zhang等[83]报道了在多巴胺的协助下通过水热合成方法成功地在氮化硼纳米片表面负载了Fe3O4颗粒(图 12), 层状结构的h-BN纳米片和Fe3O4纳米颗粒的结合对环氧复合涂层的防腐蚀性能具有显著的协同作用, 因为Fe3O4纳米颗粒本身具有一定的缓蚀性, 并且它的加入也提高了涂层的机械性能和稳定性能.
图 12
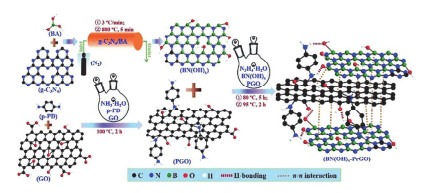
Li等[84]以尿素为原料, 采用热缩聚和煅烧两步法在氮化硼表面引入了羟基, 然后再与对苯二胺(Pr)改性的GO通过亲核取代反应成功制得BN(OH)-PrGO复合材料(图 13). BN(OH)与PrGO的良好协同作用大大改善了BN(OH)-PrGO/PU涂层的阻隔性能, 使得BN(OH)-PrGO成为热塑性聚氨酯(PU)优异的防腐蚀填料. BN(OH)-PrGO的质量分数为3%时, 聚氨酯涂层的拉伸强度提高了62%, 储存模量提高了95%, 防腐蚀效率达到了99.96%.
图 13
6. 总结与展望
通过近十年的发展, 氮化硼纳米片已经慢慢成为各行各业运用较为广泛的一种纳米材料, 与传统的氮化硼材料相比, 氮化硼纳米片具有更大的比表面积、更低的密度、更高的热稳定性、化学稳定性和耐腐蚀性, 从而吸引着全世界的科研工作者都在对它进行研究.本综述主要介绍了氮化硼纳米片的制备及表面官能化、以及氮化硼薄膜防护涂层、氮化硼纳米片/有机防护涂层、氮化硼纳米片-无机复合材料/有机防护涂层在金属腐蚀防护领域的应用.
氮化硼纳米片作为一种性能优异的二维材料, 不但可以对腐蚀性介质起到非常好的阻隔作用, 还能为金属提供长效的腐蚀防护, 因为它本身是一种带隙较大的绝缘体, 不会发生“电偶腐蚀”从而导致涂层失效.然而, 氮化硼纳米片在金属腐蚀防护领域也存在着一些难以解决的问题, 仍然面临着巨大的挑战:如何对氮化硼纳米片进行表面功能化以提高它在树脂涂层当中的分散度; 如何实现氮化硼纳米片在树脂中的定向平行排列; 如何制备缺陷少、质量高的氮化硼纳米片.为了解决氮化硼纳米片存在的这些问题, 还需要进行大量的研究.
-
-
[1]
Barati, N.; Meletis, E. I. Mater. Today Commun. 2019, 19, 1. doi: 10.1016/j.mtcomm.2018.12.001
-
[2]
Richards, C. A. J.; McMurray, H. N.; Williams, G. Corros. Sci. 2019, 154, 101. doi: 10.1016/j.corsci.2019.04.005
-
[3]
Samiee, R.; Ramezanzadeh, B.; Mahdavian, M.; Alibakhshi, E. J. Clean Prod. 2019, 220, 340. doi: 10.1016/j.jclepro.2019.02.149
-
[4]
丁锐, 陈思, 吕静, 桂泰江, 王晓, 赵晓栋, 刘杰, 李秉钧, 宋立英, 李伟华, 化学学报, 2019, 77, 1140. doi: 10.6023/A19050174Ding, R.; Chen, S.; Lv, J.; Gui, T.-J.; Wang, X, ; Zhao, X.-D.; Liu, J.; Li, B.-J.; Song, L.-Y.; Li, W.-H. Acta Chim. Sinica 2019, 77, 1140(in Chinese). doi: 10.6023/A19050174
-
[5]
王海旭, 杨光, 程天舒, 王宁, 孙蓉, 汪正平, 化学学报, 2019, 77, 316. doi: 10.6023/A18110456Wang, H.-X.; Yang, G.; Cheng, T.-S.; Wang, N.; Sun, R.; Wang, Z.-P. Acta Chim. Sinica 2019, 77, 316(in Chinese). doi: 10.6023/A18110456
-
[6]
Sugino, T.; Kawasaki, A. S.; Tanioka, K.; Shirafuji, J. Appl. Phys. Lett. 1997, 71, 2704. doi: 10.1063/1.120183
-
[7]
Cui, M. J.; Ren, S. M.; Chen, J.; Liu, S.; Zhang, G. G.; Zhao, H. C.; Wang, L. P.; Xue, Q. J. Appl. Surf. Sci. 2017, 397, 77. doi: 10.1016/j.apsusc.2016.11.141
-
[8]
Zhao, H. R.; Ding, J. H.; Yu, H. B. New. J. Chem. 2018, 42, 14433. doi: 10.1039/C8NJ03113D
-
[9]
Zhang, D. D.; Zhao, D. L.; Yao, R. R.; Xie, W. G. RSC Adv. 2015, 5, 28098. doi: 10.1039/C5RA00312A
-
[10]
Weng, Q. H.; Wang, X. B.; Wang, X.; Bando, Y.; Golberg, D. Chem. Soc. Rev. 2016, 45, 3989. doi: 10.1039/C5CS00869G
-
[11]
Cui, M. J.; Ren, S. M.; Qin, S.; Xue, Q. J.; Zhao, H. R.; Wang, L. P. RSC Adv. 2017, 7, 44043. doi: 10.1039/C7RA06835B
-
[12]
Zhi, C. Y.; Bando, Y.; Tang, C. C.; Golberg, D. Mater. Sci. Eng. R-Rep. 2010, 70, 92. doi: 10.1016/j.mser.2010.06.004
-
[13]
Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666. doi: 10.1126/science.1102896
-
[14]
Rao, C. N. R.; Nag, A. J. Inorg. Chem. 2010, 27, 4244.
-
[15]
Yu, C. P.; Zhang, J.; Tian, W.; Fan, X. D.; Yao, Y. G. RSC Adv. 2018, 8, 21948. doi: 10.1039/C8RA02685H
-
[16]
Wang, J. G.; Ma, F. C.; Liang, W. J.; Sun, M. T. Mater. Today Phys. 2017, 2, 6. doi: 10.1016/j.mtphys.2017.07.001
-
[17]
Chen, X. J.; Dobson, J. F.; Raston, C. L. Chem. Commun. 2012, 48, 3703. doi: 10.1039/c2cc17611d
-
[18]
Lei, W. W.; Mochalin, V. N.; Liu, D.; Qin, S.; Gogotsi, Y.; Chen, Y. Nat. Commun. 2015, 6, 8849. doi: 10.1038/ncomms9849
-
[19]
Ding, J. H.; Zhao, H. R.; Yu, H. B. 2D Mater. 2018, 5, 045015. doi: 10.1088/2053-1583/aad51a
-
[20]
Nicolosi, V.; Chhowalla, M.; Kanatzidis, M. G.; Strano, M. S.; Coleman, J. N. Science 2013, 304, 1420.
-
[21]
Zhi, C.; Bando, Y.; Tang, C.; Kuwahara, H.; Golberg, D. Adv. Mater. 2009, 21, 2889. doi: 10.1002/adma.200900323
-
[22]
Cao, L.; Emami, S.; Lafdi, K. Mater. Express 2014, 4, 165. doi: 10.1166/mex.2014.1155
-
[23]
Wang, Y.; Shi, Z. X.; Yin, J. J. Mater. Chem. 2011, 21, 11371. doi: 10.1039/c1jm10342c
-
[24]
Zhou, K. G.; Mao, N. N.; Wang, H. X.; Peng, Y.; Zhang, H. L. Angew. Chem. Int. Ed. 2011, 50, 10839. doi: 10.1002/anie.201105364
-
[25]
Wang, N.; Yang, G.; Wang, H. X.; Yan, C. Z.; Sun, R.; Wong, C. P. Mater. Today 2019, 27, 33. doi: 10.1016/j.mattod.2018.10.039
-
[26]
Zhao, H. R.; Ding, J. H.; Shao, Z. Z.; Xu, B. Y.; Zhou, Q. B.; Yu, H. B. ACS Appl. Mater. Interfaces 2019, 11, 37247. doi: 10.1021/acsami.9b11180
-
[27]
Yan, H. L.; Yu, P.; Han, G. C.; Zhang, Q. H.; Gu, L.; Yi, Y. P.; Liu, H. B.; Li, Y. L.; Mao, L. Q. Angew. Chem. Int. Ed. 2019, 58, 746. doi: 10.1002/anie.201809730
-
[28]
Guler, O.; Guler, S, H. Optik 2016, 127, 4630. doi: 10.1016/j.ijleo.2016.02.033
-
[29]
Zhou, X. S.; Wu, T. B.; Ding, K. L; Hu, B. J.; Hou, M. Q.; Han, B. X. Chem. Commun. 2010, 46, 386. doi: 10.1039/B914763B
-
[30]
Gunasekaran, S. G.; Dharmendirakumar, M. High Perform. Polym. 2014, 26, 274. doi: 10.1177/0954008313511349
-
[31]
Morishita, T.; Okamoto, H.; Katagiri, Y.; Matsushita, M.; Fukumori, K. Chem. Commun. 2015, 51, 12068. doi: 10.1039/C5CC04077A
-
[32]
Ding, J. H.; Zhao, H. C.; Wang, Q. L.; Peng, W. J.; Yu, H. B. Nanotechnology 2017, 28, 475602. doi: 10.1088/1361-6528/aa8e3d
-
[33]
Lee, Y. H.; Liu, K. K.; Lu, A. Y.; Wu, C. Y.; Lin, C. T.; Zhang, W. J.; Su, C. Y.; Hsu, C. L.; Lin, T. H. RSC Adv. 2012, 2, 111. doi: 10.1039/C1RA00703C
-
[34]
Lu, G. Y.; Wu, T. R.; Yuan, Q. H.; Wang, H. S.; Wang, H. M.; Ding, F. F.; Xie, X. M.; Jiang, M. H. Nat. Commun. 2015, 6, 6160. doi: 10.1038/ncomms7160
-
[35]
Song, L.; Ci, L. J.; Lu, H.; Sorokin, P. B.; Jin, C. H.; Ni, J.; Kvashnin, A. G.; Kvashnin, D. G.; Lou, J.; Yakobson, B. I.; Ajayan, P. M. Nano Lett. 2010, 10, 3209. doi: 10.1021/nl1022139
-
[36]
Tay, R. Y.; Griep, M. H.; Mallick, G.; Tsang, S. H.; Singh, R. S.; Tumlin, T.; Teo, E. H. T.; Karna, S. P. Nano Lett. 2014, 14, 839. doi: 10.1021/nl404207f
-
[37]
Pakdel, A.; Zhi, C. Y.; Bando, Y.; Nakayama, T.; Golberg, D. ACS Nano 2011, 5, 6507. doi: 10.1021/nn201838w
-
[38]
Lin, Y.; Williams, T. V.; Xu, T. B.; Cao, W.; Elsayed-Ali, H. E.; Connell, J. W. J. Phys. Chem. C 2011, 115, 2679.
-
[39]
Yu, B.; Xing, W. Y.; Guo, W. W.; Qiu, S. L.; Wang, X.; Lo, S. M.; Hu, Y. J. Mater. Chem. A 2016, 4, 7330. doi: 10.1039/C6TA01565D
-
[40]
Sainsbury, T.; Satti, A.; May, P.; Wang, Z. M.; McGovern, I.; Gunko, Y. K.; Coleman, J. J. Am. Chem. Soc. 2012, 134, 18758. doi: 10.1021/ja3080665
-
[41]
Cai, W.; Hong, N. N.; Feng, X. M.; Zeng, W. R.; Shi, Y. Q.; Zhang, Y.; Wang, B. B.; Hu, Y. Chem. Eng. J. 2017, 330, 309. doi: 10.1016/j.cej.2017.07.162
-
[42]
Wu, Y. Q.; He, Y.; Zhou, T. G.; Chen, C. L.; Zhong, F.; Xia, Y. Q.; Xie, P.; Zhang, C. Prog. Org. Coat. 2020, 142, 105541. doi: 10.1016/j.porgcoat.2020.105541
-
[43]
Wu, Y. Q.; He, Y.; Chen, C. L.; Zhong, F.; Li, H. J.; Chen, J. Y.; Zhou, T. G. Colloid Surf. A-Physicochem. Eng. Asp. 2020, 587, 124337. doi: 10.1016/j.colsurfa.2019.124337
-
[44]
Li, J.; Cui, J. C.; Yang, J. Y.; Ma, Y.; Qiu, H. X.; Yang, J. H. Prog. Org. Coat. 2016, 99, 443. doi: 10.1016/j.porgcoat.2016.07.008
-
[45]
Pourhashem, S.; Vaezi, M. R.; Rashidi, A.; Bagherzadeh, M. R. Prog. Org. Coat. 2017, 111, 47. doi: 10.1016/j.porgcoat.2017.05.008
-
[46]
Raza, M. A.; Rehman, Z. U.; Ghauri, F. A. Thin Solid Films 2018, 663, 93. doi: 10.1016/j.tsf.2018.07.046
-
[47]
Fan, Y. Z.; Yang, H. Z.; Fan, H. S.; Liu, Q.; Lv, C.; Zhao, X.; Yang, M. X.; Wu, J. J.; Cao, X. M. Materials 2020, 13, 2340. doi: 10.3390/ma13102340
-
[48]
Liu, Z.; Li, J. H.; Liu, X. H. ACS Appl. Mater. Interfaces 2020, 12, 6503. doi: 10.1021/acsami.9b21467
-
[49]
顾林, 丁纪恒, 余海斌, 化学进展, 2016, 28, 737.Gu, L.; Ding, J.-H.; Yu, H.-B. Prog. Chem. 2016, 28, 737(in Chinese).
-
[50]
Cui, G.; Bi, Z. X.; Zhang, R. Y.; Liu, J. G.; Yu, X.; Li, Z. L. Chem. Eng. J. 2019, 373, 104. doi: 10.1016/j.cej.2019.05.034
-
[51]
Gyawali, G.; Adhikari, R.; Kim, H. S.; Cho, H. B.; Lee, S. W. ECS Electrochem. Lett. 2013, 2, C7.
-
[52]
Britun, V. F.; Kurdyumov, A. V.; Petrusha, I. A. Mater. Lett. 1999, 41, 83. doi: 10.1016/S0167-577X(99)00108-1
-
[53]
Liu, Z.; Gong, Y. J.; Zhou, W.; Ma, L. L.; Yu, J. J.; Idrobo, J. C.; Jung, J.; MacDonald, A. H.; Vajtai, R.; Lou, J.; Ajayan, P. M. Nat. Commun. 2013, 4, 1.
-
[54]
Yi, M.; Shen, Z. G.; Zhao, X. H.; Liang, S. S.; Liu, L. Appl. Phys. Lett. 2014, 104, 143101. doi: 10.1063/1.4870530
-
[55]
Liu, K.; Zhang, G. G.; Pu, J. B.; Ma, F.; Wu, G. Z.; Lu, Z. H. Ceram. Int. 2018, 44, 13888. doi: 10.1016/j.ceramint.2018.04.236
-
[56]
Zhang, J.; Yang, Y. C.; Lou, J. Nanotechnology 2016, 27, 364004. doi: 10.1088/0957-4484/27/36/364004
-
[57]
Mahvash, F.; Eissa, S.; Bordjiba, T.; Tavares, A. C.; Szkopek, T.; Siaj, M. Sci Rep 2017, 7, 42139. doi: 10.1038/srep42139
-
[58]
Miller, R. J.; Adeleye, A. S.; Page, H. M.; Kui, L.; Lenihan, H. S.; Keller, A. A. J. Nanopart. Res. 2020, 22, 129. doi: 10.1007/s11051-020-04875-x
-
[59]
Parra, C.; Montero-Silva, F.; Henríquez, R.; Flores, M.; Garín, C.; Ramírez, C.; Moreno, M.; Correa, J.; Seeger, M.; Haberle, P. ACS Appl. Mater. Interfaces 2015, 7, 6430. doi: 10.1021/acsami.5b01248
-
[60]
Chilkoor, G.; Karanam, S. P.; Star, S.; Shrestha, N.; Sani, R. K.; Upadhyayula, V. K. K.; Ghoshal, D.; Koratkar, N. A.; Meyyappan, M.; Gadhamshetty, V. ACS Nano 2018, 12, 2242. doi: 10.1021/acsnano.7b06211
-
[61]
Shen, L. T.; Zhao, Y. D.; Wang, Y.; Song, R. B.; Yao, Q.; Chen, S. S.; Chai, Y. J. Mater. Chem. A 2016, 4, 5044. doi: 10.1039/C6TA01604A
-
[62]
Li, L. H.; Xing, T.; Chen, Y.; Jones, R. Adv. Mater. Interfaces 2014, 1, 1300132. doi: 10.1002/admi.201300132
-
[63]
Percival, S. J.; Melia, M. A.; Alexander, C. L.; Nelson, D. W.; Schindelholz, E. J.; Spoerke, E. D. Surf. Coat. Int. 2020, 383, 125228. doi: 10.1016/j.surfcoat.2019.125228
-
[64]
Zhang, X. F.; Chen, Y. Q.; Hu, J. M. Corros. Sci. 2020, 166, 108452. doi: 10.1016/j.corsci.2020.108452
-
[65]
Sharifalhoseini, Z.; Entezari, M. H.; Davoodi, A.; Shahidi, M. J. Ind. Eng. Chem. 2020, 83, 333. doi: 10.1016/j.jiec.2019.12.006
-
[66]
Ghomi, E. R.; Khorasani, S. N.; Kichi, M. K.; Dinari, M.; Ataei, S.; Enayati, M. H.; Koochaki, M. S.; Neisiany, R. E. Colloid. Polym. Sci. 2020, 298, 213. doi: 10.1007/s00396-019-04597-0
-
[67]
Husain, E.; Narayanan, T. N.; Taha-Tijerina, J. J.; Vinod, S.; Vajtai, R.; Ajayan, P. M. ACS Appl. Mater. Interfaces 2013, 5, 4129. doi: 10.1021/am400016y
-
[68]
Yi, M.; Shen, Z. G.; Liu, L.; Liang, S. S. RSC Adv. 2015, 5, 2983. doi: 10.1039/C4RA09156F
-
[69]
Simonov, K.; Vinogradov, N. A.; Ng, M. L.; Vinogradov, A.; Mårtensson, N.; Preobrajenski, A. B. Surf. Sci. 2012, 606, 564. doi: 10.1016/j.susc.2011.11.031
-
[70]
Petravic, M.; Peter, R.; Kavre, I.; Li, L. H.; Chen, Y.; Fan, L. J.; Yang, Y. W. Phys. Chem. Chem. Phys. 2010, 12, 15349. doi: 10.1039/c0cp00984a
-
[71]
Prasai, D.; Tuberquia, J. C.; Harl, R. R.; Jennings, G. K.; Bolotin, K. I. ACS Nano 2012, 6, 1102. doi: 10.1021/nn203507y
-
[72]
Sun, W.; Wang, L. D.; Wu, T. T.; Pan, Y. Q.; Liu, G. C. Carbon 2014, 79, 605. doi: 10.1016/j.carbon.2014.08.021
-
[73]
Camilli, L.; Yu, F.; Cassidy, A.; Hornekaer, L.; Boggild, P. 2D Mater. 2019, 6, 022002. doi: 10.1088/2053-1583/ab04d4
-
[74]
Sun, W.; Wang, L. D.; Wu, T. T.; Pan, Y. Q.; Liu, G. C. J. Electrochem. Soc. 2016, 163, C16. doi: 10.1149/2.0301602jes
-
[75]
Chen, J.; Chen, B.; Li, J. Y.; Tong, X.; Zhao, H. C.; Wang, L. P. Polym. Int. 2017, 66, 659. doi: 10.1002/pi.5296
-
[76]
Pathan, S.; Ahmad, S. J. Mater. Chem. A 2013, 1, 14227. doi: 10.1039/c3ta13126b
-
[77]
Gu, L.; Liu, S.; Zhao, H. C.; Yu, H. B. ACS Appl. Mater. Interfaces 2015, 7, 17641. doi: 10.1021/acsami.5b05531
-
[78]
Zhao, H. C.; Ding, J. H.; Yu, H. B. ChemistrySelect 2018, 3, 11277. doi: 10.1002/slct.201802079
-
[79]
Yu, J. J.; Zhao, W. J.; Liu, G.; Wu, Y. M.; Wang, D. L. Surf. Topogr.-Metrol. Prop. 2018, 6, 034019. doi: 10.1088/2051-672X/aad5ab
-
[80]
Cui, M. J.; Ren, S. M.; Qin, S. L.; Xue, Q. J.; Zhao, H. C.; Wang, L. P. Corros. Sci. 2018, 131, 187. doi: 10.1016/j.corsci.2017.11.022
-
[81]
Zou, B. J.; Chang, X. J.; Yang, J. X.; Wang, S. C.; Xu, J. L.; Wang, S. R.; Samukawa, S.; Wang, L. Prog. Org. Coat. 2019, 133, 139. doi: 10.1016/j.porgcoat.2019.04.040
-
[82]
Huang, H. W.; Huang, X. F.; Xie, Y. H.; Tian, Y. Q.; Jiang, X.; Zhang, X. Y.; Prog. Org. Coat. 2019, 130, 124. doi: 10.1016/j.porgcoat.2019.01.059
-
[83]
Zhang, C. L.; He, Y.; Li, F.; Di, H. H.; Zhang, L.; Zhan, Y. Q. J. Alloy. Compd. 2016, 685, 743. doi: 10.1016/j.jallcom.2016.06.220
-
[84]
Li, X. Y.; Bandyopadhyay, P.; Kshetri, T.; Kim, N. H.; Lee, J. H. J. Mater. Chem. A 2018, 6, 21501. doi: 10.1039/C8TA08351G
-
[1]
-

表 1 剥离法制氮化硼纳米片
Table 1. Boron nitride nanosheets prepared by the exfoliation method
Solvent/insertion molecules Exfoliation condition Concentration/(mg•mL-1) Dimension Ref. MSA Ultrasonication: 8 h;
Centrifuge: 4000 r/min, 90 min0.2~0.3 Thickness:<3 nm [23] C8H15N2F6P/Water Bath ultrasonication: 8 h;
Centrifuge: 3000 r/min (1220 G), 20 min1.86 Thickness: 6.3 layers
Lateral size: 1.34 μm[30] Urea/Nitrogen Ball mill;
Centrifuge: 700 r/min, 20 h30 Thickness: 2~2.5 nm Lateral size: 100 nm [18] 2-Furoic acid Ball mill 35 Thickness: 2 nm Lateral size: 1.8 μm [19] IPA/Ammonia water Ultrasonication: 40 kHz, 35 h;
Centrifuge: 3000 G4 Not supplied [22] IPA/LiCl Hydrothermal reaction for 12 hours;
Centrifuge: 500 r/min, 5 min4.13 Thickness:<10 layers Lateral size: 400 nm [25] LiSiF6/NaOH Stirring: 500 r/min, 60 h;
Centrifuge: 2000 r/min, 30 min8.35 Thickness:<5 layers Lateral size: 1~3 μm [26] Liquid N2/Water Ultrasonication: 2 h;
Centrifuge: 1000 r/min, 15 minNot supplied Thickness:<5 layers Lateral size: 1~5 μm [32]  下载: 导出CSV
下载: 导出CSV
-















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 92
- 文章访问数: 4461
- HTML全文浏览量: 1251
 下载:
下载:
