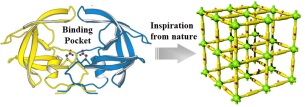
图 1.
借鉴仿生理念构建多孔有机聚合物基催化材料
Figure 1.
The exploration POPs as a platform for the construction of biomimetic catalysts

在化学品生产过程中, 约有85%的转化涉及催化反应, 因此催化科学与技术被认为是现代化学工业的基石.利用催化过程提高反应选择性, 是实现资源优化利用的重要途径, 也是目前催化学科所面临的关键性挑战之一[1].众所周知, 生物体系中发生的化学反应通常具有极高的催化活性和选择性, 这激发了研究者寻找能够模拟生物分子组成和结构进而提升当前催化体系性能的兴趣[2].酶是一类生物催化剂, 具有多个分隔空腔, 能够使反应物在受限的纳米空间中选择性地快速进行反应.这些性能与酶活性部位的结构特点密不可分, 酶的结构特点主要包含以下几部分[3]: (1)活性部位周围的氨基酸残基能与底物通过非共价键作用键合; (2)活性部位构象具有柔性; (3)活性部位的裂隙具有高度疏水性.这些因素同时影响酶的催化作用, 主要是隔离的环境提供了底物的预组织和邻近、高的局部浓度、氨基酸残基之间的协同作用和稳定过渡态的可能性.尽管构建复杂的酶主体分子非常具有诱惑力, 但要了解酶的高效性, 需要使用组成和结构明确的体系来系统分析上述因素, 评估哪些因素对其高性能起关键作用, 进而设计出高性能的催化材料.
传统的催化材料骨架一般是刚性的, 并且不同催化功能中心通常以无序、隔离的状态存在, 因而, 难以在空间上耦合并建立结构和性能的联系.为了设计类酶催化材料, 实现多功能位点在特定微环境中的协同, 这对材料的可设计性提出了极高的要求.多孔有机聚合物的快速发展为这一目标的实现提供了机遇.这类材料是由有机结构单元通过共价键链接而成的高分子多孔材料, 根据材料的结晶性可分为无定形多孔有机聚合物(POPs)和结晶共价有机框架材料(COFs)[4].由于其有机单元的可设计性、网格结构的多样性及可调节性, 能够同时对初次和高次结构进行精密合成控制, 因此, 多孔有机聚合物已逐渐发展为有机材料家族的新成员, 为复杂的结构设计和量身定制的功能开发提供了可能性, 这无疑为实现仿生催化材料制备提供了强大的制备平台(图 1).本文将着重介绍我们课题组基于活性中心次级环境调控、柔性骨架设计以及疏水反应腔构建这些仿生理念, 在多孔有机聚合物基催化材料设计方面的研究工作.深入阐述结构与功能的相关性, 讨论有待解决的挑战性关键问题, 并展望多孔有机聚合物在催化领域的未来发展方向.
一般来说, 酶的优异活性和选择性来源于其多个功能位点间的协同作用, 酶的活性部位主要分为结合部位和催化部位.在反应过程中, 酶与底物在非共价键的作用下结合来活化反应物、稳定过渡态以及降低反应能垒, 进而促进催化反应.非共价相互作用导向的催化被认为是一种具有发展潜力催化剂设计策略, 并已在分子催化体系得到了一定的发展.目前催化材料的设计研究仍然局限在将直接起催化作用的位点引入到各种载体中.然而, 如何模拟酶将多个功能位点引入到催化材料中用以提升材料性能, 是大家关注的热点问题之一.为了实现各个功能中心的有效协同, 功能位点在材料中的相对空间位置就显得尤为重要.为了阐明位点空间位置的重要性, 我们利用POPs分子层面的可设计性, 通过骨架基元设计引入辅助基团, 构建出系列含有羟基和季鏻盐双催化中心的多孔有机聚合物(图 2)[5], 并研究它们在CO2环加成反应中的催化性能[6].结果表明:羟基的引入以及羟基与季鏻盐相对位置对催化效率有重要影响.通过理论计算, 我们发现, 在反应过程中羟基和季鏻盐上的溴离子同时作用在一个分子上, 通过羟基与环氧化合物的氢键作用, 实现对其活化; 并利用溴离子完成开环以及CO2插入, 两个功能中心太远或太近都不利于彼此协同.当羟基处在季鏻盐的间位时大幅降低了反应的活化能, 相比于不含羟基的材料, 其活化能降低了约1.4倍(96.75 kJ•mol-1 vs. 134.64 kJ•mol-1), 为此, 催化性能随之提升了2.5倍.
除了引入辅助基团的策略之外, 溶剂筛选是我们最常用的调控反应次级环境的手段.许多反应在N-甲基吡咯烷酮(NMP)等高沸点的非质子化溶剂中表现出优异的性能, 但是, 由于其难挥发的不足, 这些溶剂的使用对产物分离带来极大的困难, 不仅耗时、耗能甚至还会在分离过程中导致副反应的发生.如果能在固体催化材料中创造合适的溶剂化环境, 采用分离友好型溶剂作为反应介质, 就能有效地解决上述分离困难.但是, 要实现这个目标, 需要对溶剂分子与催化中心的距离以及比例进行精准调控.为此, 我们利用POPs组成可调性以及聚合方式的多样性特点, 通过将乙烯基功能化的溶剂分子, 如二甲基亚砜(DMSO)、NMP以及离子液与4-乙烯基苯磺酸按一定比例共聚, 实现对磺酸基团周围溶剂化环境的调控[7].以果糖到5-羟甲基呋喃(HMF)作为模型反应为例, 催化剂的性能随着溶剂分子单元与磺酸基团数的比例增加而提高.粉末X-射线衍射(PXRD)测试结果显示, 只有当聚合物中溶剂分子的量达到一定程度时才能使果糖分子被完全溶剂化.这种溶剂化作用主要源自于果糖与聚合物中的溶剂基团强的氢键作用, 可以破坏果糖分子内的氢键使其晶体结构消失.采用四氢呋喃(THF)为溶剂, 以离子液与4-乙烯基苯磺酸30:1(物质的量比)比例共聚得到的材料为催化剂(PSS-30IL-SO3H)时, HMF的产率高达98.8%.而传统的磺酸树脂Amberlyst-15和分子催化剂(对甲苯磺酸)在相同的反应条件下产率均低于10%.值得一提的是, 反应结束后, 该反应体系与一般多相催化剂类似可以通过简单的过滤实现重复利用, 而产物HMF可以从低沸点的THF溶剂中分离得到, 从而大幅简化了分离过程(通常该反应需要在DMSO、NMP等高沸点溶剂或离子液中进行), 解决了这一重要生物质转化反应所面临的困境, 并为研发其他高性能催化材料提供新思路
上述例子虽然能很好地将溶剂化环境引入到催化材料中, 但是这些含溶剂基团单体的制备过程繁冗, 极大限制了其实际应用.为了解决这个问题, 我们提出了另一种设计策略:即将由溶剂分子组成的线性聚合物和含有催化中心的共价有机骨架材料(COFs)通过主客体组装手段, 成功将溶剂化效应引入到多相催化材料中.例如, 由聚乙烯基吡咯烷酮(PVP)和含磺酸基团的COF([SO3H]x-COF)所组成的复合材料(PVP@[SO3H]x- COF), 在果糖到HMF的转化中表现出优异的催化性能, 产率高达99%(图 3)[8].这种组合对于主体材料和客体线性聚合物都没有特定的要求, 具有更强的普适性, 且制备简单, 因此能成功解决催化剂规模化生产的问题.

总之, 本部分研究通过引入辅助基团或溶剂化环境的设计策略, 实现了对功能中心次级环境的调控, 解决了多相催化材料中多催化组分空间隔离问题, 提升了催化性能, 拓宽了催化材料的设计思路, 对设计高效多相协同催化体系具有重要的指导意义.
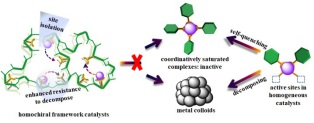
研究表明, 酶的活性部位是柔性的, 在与底物结合时, 酶分子的活性部位构象会发生改变, 且在催化过程中是一个动态的过程[9].而传统的多孔材料如活性碳、硅基材料是刚性的, 负载往往会导致催化中心运动受限、活性位点空间隔离, 难以实现分子内协同, 极大限制了其性能发挥(图 4).因此, 提高固载化体系中功能中心结构域的柔性和自由度是提升固载化体系性能的关键.这里, 我们利用多孔有机聚合物骨架刚柔可设计性, 针对金属有机催化、通过柔性多孔聚膦配体设计合成, 成功构建出系列高效催化体系.

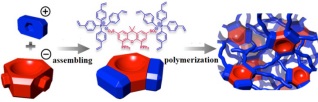
众所周知, 配体的种类和用量在调节反应活性和选择性方面起到决定性的作用, 如氢甲酰化反应(最大规模的均相催化反应, 年产约2000万吨)[10].在这类反应中, 由于反应活性物种的形成需多配体协同, 使得反应过程中需要大大过量的配体(配体与金属的比例约为50:1, 物质的量比).然而, 传统的催化材料固载化方法通常会导致催化中心空间隔离, 无法有效协同, 极大地限制了固载化配体体系的发展.如何提高材料中配体的空间连续性及链接点的柔性是配体多相化后催化性能维持的关键和基础所在.为此, 我们发展了一种普适性的方法, 即通过乙烯基功能化配体的自聚, 并借助乙烯基聚合后所得烷基链具有柔性的特点, 最大限度提高了配体在材料中的浓度、连续性以及链接点的柔性.利用该方法我们首先构建了多孔聚三苯基膦, 将乙烯基功能化的三苯基膦在溶剂热条件下通过自由基聚合得到了多孔聚三苯基膦, 其比表面达到1100 m2•g-1(图 5)[11].静态固体31P核磁证明:在溶剂存在下, 得到的这个聚三苯基膦具有优异的柔动性, 具体表现为, 加入乙醇后, 固态材料的各向异性消失, 出现一个尖锐的P核磁信号.负载Rh后, 在长链末端烯烃的氢甲酰化反应中表现出媲美于均相催化体系的性能, 远优于传统固载型体系.进一步研究表明:三苯基膦配体在材料中的空间连续性以及聚合物的柔性对其固载后催化性能的维持起了至关重要的作用[12].同时, 为了提高催化的选择性, 我们将这种合成策略拓展至双膦配体, 成功制备了系列柔性多孔聚双膦配体, 提高了氢甲酰化反应的区域选择性[13].并且, 为了进一步优化反应性能, 我们将柔性聚配体设计理念与催化中心微环境调控相结合, 构建出仿生反应器.借鉴酶的分子环境, 利用离子对组装的方法将乙烯基功能化的阳离子模板(季鏻盐)和阴离子配体(Xantphos)组装, 得到外层亲水、内层疏水的限域反应腔的超分子结构基元(图 6).这种设计可显著提高醛的区域选择性, 所得到的多孔超分子聚合物负载Rh之后, 在1-辛烯氢甲酰化反应中直链醛与支链醛的比例较未修饰的配体体系提升了16倍, 产物壬醛的正异构之比达到40[14].

这种策略不仅解决了配体固载后活性降低的问题, 还能有效提高催化中心的稳定性.但是, 在金属有机催化过程中, 配体与金属之间的键是动态的, 在反应过程中会发生歧化反应导致部分金属物种会团聚形成纳米粒子, 使催化体系失活(图 7)[15].虽然加入过量的配体能有效抑制配合物分解, 但是过多的配体会抑制反应, 因此这类体系的催化活性和稳定性很难达到平衡.我们利用柔性多孔聚配体中的邻近配体可协同, 又由于聚合物张力难以形成配位饱和结构的特点, 实现了催化活性和稳定性的同时提升.以手性的亚磷酰胺(phosphoramidite)为例, 引入Rh之后, 在2-乙酰氨基丙烯酸甲酯不对成加氢反应中, 以多孔聚亚磷酰胺为修饰配体的体系在活性和稳定性上较相应的均相体系都有显著提升[16].具体实验结果表明, 在相同条件下, 均相体系在反应物转化率为30%时基本失活, 而多相体系则能达到完全转化.
为此, 本部分研究发展了一种具有普适性的有机配体多相化的方法, 揭示了材料中配体的空间连续性以及链接点的柔性与催化性能之间的内在联系, 解决了固载化后多配体协同问题, 为设计绿色、高效的多相金属有机催化反应奠定了基础.
在已知结构的酶分子中, 除非水分子本身是底物分子, 与底物分子结合的活性部位裂隙可以有效阻隔水分子的进入.因此酶的活性部位通常被称之为“疏水口袋”, 活性部位的疏水性增进了酶与底物的结合, 是酶具有高效性的原因之一.近年来, 有关超疏水材料的研究引起了人们极大的关注[18].自然界中一些生物体的优异结构和特性, 给人类在不断开发新型材料过程中带来灵感和启发.例如荷叶表面具有极强的疏水性, 使得水流带走其表面的尘土达到自清洁的效果.生物体的某些部位具有超疏水性的秘密, 主要源自于其特殊的微结构, 即这些部位存在着非常复杂的多重纳米和微米级的多级孔结构.由此启发下, 研究者们总结得出, 材料中多级孔结构的引入可以增强聚合物界面对气体的吸附能力增强其浸润性, 进而提升材料的抗水解能力[19].我们通过控制溶剂热聚合的条件, 将多级孔引入到聚合物中, 提升了材料的疏水性, 构建出超疏水的聚二乙烯基苯[20].
众所周知, 许多高效的催化剂都面临着水解导致失活的困扰, 这严重限制了它们的实际应用.在工业生产中很难做到绝对的无水环境, 并且很多反应过程中水是反应的副产物, 因此, 如何实现提高催化剂的水解稳定性同时不改变催化性能, 将具有深远的实际意义.为此, 我们首次将上述原理应用到催化体系的稳定性设计方面, 通过构筑多级孔结构, 增强材料的疏水性, 一步解决易水解均相催化体系稳定性和分离问题[21].通过调控乙烯基功能化的亚磷酸酯配体的聚合条件, 成功制备具有多级孔的超疏水聚亚磷酸酯, 展示出长久的催化稳定性, 实现了内烯烃的高效转化.在连续反应过程中均相催化剂逐渐失活, 而超疏水的多孔聚亚磷酸酯相则一直保持了稳定的活性.该项工作为我们展示了如何利用特殊微结构来提高催化剂水解稳定性, 这为设计具有其它新颖性质的催化剂提供了新方法(图 8).值得注意的是, 超疏水骨架的构筑除了能阻止水进入孔道、稳定骨架, 还能有效富集疏水反应物、转移亲水产物, 进而打破反应平衡、提高催化效率.例如, 烯烃在超疏水聚亚磷酸酯孔道的富集使其表现出比均相催化体系更快的反应速率; 在Pd催化的丙烯醇脱水反应中可以看到, 超疏水的聚亚磷酸酯因能快速将副产物水从体系排出较均相催化体系表现出更高的活性和稳定性.超疏水催化材料这种富集疏水反应物转移亲水产物的现象具有普适性.我们将Noyori等发展的手性二胺(1, 2-二苯基乙二胺, TsDPEN)固载在超疏水PDVB (polymerized divinyl benzene)中, 负载Ru之后, 在水相的苯已酮氢转移表现出比均相催化剂更好的活性, 这主要归因于这种超疏水催化材料反应物和产物的选择性吸附(图 9)[22].


另外, 许多酸催化反应都伴随着水的产生, 而水极易附着在酸中心上, 大幅降低酸强度甚至还会导致副产物的产生.针对这些问题, 我们课题组通过在多级孔的超疏水聚合物骨架PDVB上引入磺酸基或酸性离子液, 成功构建了系列疏水的酸催化剂并在几类经典的酸催化反应中表现出优异的性能[23].控制实验和表征结果显示, 反应物在催化剂孔道富集以及副产物水及时从疏水孔道排出, 是使这些催化材料具有高活性的原因.值得一提的是, 在果糖到HMF的转化中发现, 由于产生的水能迅速从催化剂中转移, 避免了产物HMF进一步的催化转化, 进而大幅提高了反应的选择性[24].为了进一步提高酸强度, 我们通过在超疏水的全氟多孔聚合物骨架中引入磺酸基团, 制得了疏水的具有超强酸特性的多孔聚合物.基于其超强酸性、多孔性、热稳定性, 该催化材料在连续的苯甲醇和苯的傅氏烷基化反应中表现出接近100%的转化率和烷基化产物选择性, 而商品化催化剂Amberlyst-15和Nafion NR50则在相同反应条件下很快失活[25].同时, 我们还构建了系列超疏水的多孔有机聚合物基碱催化剂都表现了比相应均相体系更优异的催化性能[26].
本部分系列研究表明, 调控催化剂表面亲疏水性, 可以有效调节催化剂表面物质的局部浓度, 从而促进或者抑制反应的进行, 这种简单易行的调控手段为催化剂的设计与性能提升提供了一种十分简便的方案.
我们总结了借鉴仿生理念, 从活性中心次级环境调控、功能中心结构域柔性调控以及疏水孔道环境构建这几方面出发成功构建了系列高效的催化体系.这些设计概念的引入大幅提高了活性中心的活性、稳定性甚至产物的选择性, 为构建高性能的催化材料提供了新思路.尽管我们在活性中心微环境调控上取得了一定的进展, 但是向自然界学习设计更为精巧的催化材料还有很长一段路要走.开展模拟细胞结构及其催化研究一直是科学家研究的目标之一.细胞作为生命体中基本单元, 其内部包含多个空间上彼此分离的细胞器, 分别进行不同的酶催化反应, 这些反应在时间和空间上巧妙地耦合起来, 最终高选择性地合成大分子, 这是目前人工催化体系所不能实现的.细胞优异的催化性能就在于它包含着多级微纳界面和限域空间, 在调控反应物的传输、反应速率、反应平衡状态及选择性方面发挥了关键性和决定性的作用.发展多催化活性中心及其微反应环境的调控方法是催化领域中一个重要而富有挑战的课题.多孔有机聚合物因其分子层面的可设计性为组装拥有不同微化学环境的催化活性中心提供可能性, 同时, 链接方式以及拓扑结构的多样性能使这些活性中心既保持合适的空间距离又不相互干扰, 确保多步催化反应在时间和空间上能有效耦合, 进而有可能为类细胞催化反应器的构建提供了强大的设计平台.
Rothenberg, G. Catalysis:Concepts and Green Applications, Wiley-VCH, Weinheim, Germany, 2011.
(a) Breslow, R. Acc. Chem. Res. 1995, 28, 146;(b) Sun, Q.; Aguila, B.; Ma, S. Chem 2018, 4, 2736.
Benkovic, S. J.; Hammes-Schiffer, S. Science 2003, 301, 1196.
(a) Das, S.; Heasman, P.; Ben, T.; Qiu, S. Chem. Rev. 2017, 117, 1515;(b) Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Chem. Soc. Rev. 2015, 44, 6018;(c) Sun, Q.; Aguila, B.; Song, Y.; Ma, S. Acc. Chem. Res. 2020, 53, 812;(d) Sun, Q.; Dai, Z.; Meng, X.; Wang, L.; Xiao, F.-S. ACS Catal. 2015, 5, 4556;(e) Song, Y.; Sun, Q.; Aguila, B.; Ma, S. Adv. Sci. 2019, 6, 1801410;(f) Wang, S.; Sun, Q.; Chen, W.; Tang, Y.; Aguila, B.; Pan, Y.; Zheng, A.; Yang, Z.; Wojtas, L.; Ma, S.; Xiao, F.-S. Matter 2020, 2, 416;(g) Xiang, Z.; Dai, Q.; Chen, J.-F.; Dai, L. Adv. Mater. 2016, 28, 6253;(h) Wang, K.; Yang, L.-M.; Wang, X.; Guo, L.; Cheng, G.; Zhang, C.; Jin, S.; Tian, B.; Cooper, A. Angew. Chem. Int. Ed. 2017, 56, 14149;(i) Peng, Z. K.; Ding, H. M.; Chen, R. F.; Gao, C.; Wang, C. Acta Chim. Sinica 2019, 77, 681(in Chinese). (彭正康, 丁慧敏, 陈如凡, 高超, 汪成, 化学学报, 2019, 77, 681);(j) Liu, J. G.; Zhang, M. Y.; Wang, N.; Wang, C. G.; Ma, L. L. Acta Chim. Sinica 2020, 78, 311(in Chinese). (刘建国, 张明月, 王楠, 王晨光, 马隆龙, 化学学报, 2020, 78, 311.
Hu, K.; Tang, Y.; Cui, J.; Gong, Q.; Hu, C.; Wang, S.; Dong, K.; Meng, X.; Sun, Q.; Xiao, F.-S. Chem. Commun. 2019, 55, 9180.
(a) Sun, Q.; Ma, S.; Dai, Z.; Meng, X.; Xiao, F.-S. J. Mater. Chem. A 2015, 3, 23871;(b) Sun, Q.; Jin, Y.; Aguila, B.; Meng, X.; Ma, S.; Xiao, F.-S. ChemSusChem 2017, 10, 1160.
Sun, Q.; Wang, S.; Aguila, B.; Meng, X.; Ma, S.; Xiao, F.-S. Nat. Commun. 2018, 9, 3236.
Sun, Q.; Tang, Y.; Aguila, B.; Wang, S.; Xiao, F.-S.; Thallapally, P. K.; Al-Enizi, A. M.; Nafady, A.; Ma, S. Angew. Chem. Int. Ed. 2019, 58, 8670.
Nestl, B. M.; Hauer, B. ACS Catal. 2014, 4, 3201.
Franke, R.; Selent, D.; Börner, A. Chem. Rev. 2012, 112, 5675.
Sun, Q.; Jiang, M.; Shen, Z.; Jin, Y.; Pan, S.; Wang, L.; Meng, X.; Chen, W.; Ding, Y.; Li, J.; Xiao, F.-S. Chem. Commun. 2014, 50, 11844.
Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Catal. Today 2017, 298, 40.
Sun, Q.; Dai, Z.; Liu, X.; Sheng, N.; Deng, F.; Meng, X.; Xiao, F.-S. J. Am. Chem. Soc. 2015, 137, 5204.
Dong, K.; Sun, Q.; Tang, Y.; Shan, C.; Aguila, B.; Wang, S.; Meng, X.; Ma, S.; Xiao, F.-S. Nat. Commun. 2019, 10, 3059.
Chen, F.; Wang, S.; Sun, Q.; Xiao, F.-S. ChemCatChem 2020, 12, 3285.
Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Chem. Mater. 2017, 29, 5720.
Dai, Z.; Sun, Q.; Chen, F.; Pan, S.; Wang, L.; Meng, X.; Li, J.; Xiao, F.-S. ChemCatChem 2016, 8, 812.
(a) Sun, Q.; He, H.; Gao, W.-Y.; Aguila, B.; Wojtas, L.; Dai, Z.; Li, J.; Chen, Y.-S.; Xiao, F.-S.; Ma, S. Nat. Commun. 2016, 7, 13300;(b) Sun, Q.; Aguila, B.; Perman, J.; Butts, T.; Xiao, F.-S.; Ma, S. Chem 2018, 4, 1726.
Su, B.; Tian, Y.; Jiang, L. J. Am. Chem. Soc. 2016, 138, 1727.
Zhang, Y.; Wei, S.; Liu, F.; Du, Y.; Liu, S.; Ji, Y.; Yokoi, T.; Tatsumi, T.; Xiao, F.-S. Nano Today 2009, 4, 135.
Sun, Q.; Aguila, B.; Verma, G.; Liu, X.; Dai, Z.; Deng, F.; Meng, X.; Xiao, F.-S.; Ma, S. Chem 2016, 1, 628.
Sun, Q.; Jin, Y.; Zhu, L.; Wang, L.; Meng, X.; Xiao, F.-S. Nano Today 2013, 8, 342.
(a) Liu, F.; Meng, X.; Zhang, Y.; Ren, L.; Nawaz, F.; Xiao, F.-S. J. Catal. 2010, 271, 52;(b) Liu, F.; Kong, W.; Qi, C.; Zhu, L.; Xiao, F.-S. ACS Catal. 2012, 2, 565;(c) Liu, F.; Wang, L.; Sun, Q.; Zhu, L.; Meng, X.; Xiao, F.-S. J. Am. Chem. Soc. 2012, 134, 16948;(d) Liu, F.; Huang, K.; Zheng, A.; Xiao, F.-S.; Dai, S. ACS Catal. 2018, 8, 372.
Wang, L.; Wang, H.; Liu, F.; Zheng, A.; Zhang, J.; Sun, Q.; Lewis, J. P.; Zhu, L. F.; Meng, X.; Xiao, F.-S. ChemSusChem 2014, 7, 402.
Sun, Q.; Hu, K.; Leng, K.; Yi, X.; Aguila, B.; Sun, Y.; Zheng, A.; Meng, X.; Ma, S.; Xiao, F.-S. J. Mater. Chem. A 2018, 6, 18712.
(a) Liu, F.; Li, W.; Sun, Q.; Zhu, L.; Meng, X.; Guo, Y.-H.; Xiao, F.-S. ChemSusChem 2011, 4, 1059;(b) Zhang, Y.-L.; Liu, S.; Liu, S.; Liu, F.; Zhang, H.; He, Y.; Xiao, F.-S. Cat. Commun. 2011, 12, 1212.

图 1 借鉴仿生理念构建多孔有机聚合物基催化材料
Figure 1 The exploration POPs as a platform for the construction of biomimetic catalysts


图 4 通过柔性多孔聚配体构筑来增强配体协同的设计
Figure 4 Schematic illustration of flexible frameworks construction to promote the cooperation between ligands

图 5 柔性多孔聚三苯基膦合成示意图
Figure 5 Schematic illustration of the construction of flexible porous polymeric triphenylphosphine

图 6 多孔聚离子对超分子聚合物合成示意图
Figure 6 Schematic illustration of the formation of ion pair-directed supramolecular assemblies and their subsequent construction of porous framework


图 8 通过多级孔结构构筑增强材料疏水性提升均相催化体系水解稳定性示意图以及催化长久稳定性[21]
Figure 8 Resistance against hydrolytic degradation of water-sensitive homogeneous catalysts can be dramatically boosted by the formation of superhydrophobic porous frameworks. Compared with their homogenous counterparts, the resultant catalysts exhibit superior performance in terms of catalytic stability[21]

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


 下载:
下载: