Received Date:
10 June 2020 Available Online:
15 September 2020
Fund Project:
Project supported by the Major Scientific and Technological Projects of China National Petroleum Corporation (ZD2019-184-001) and Fundamental Research
Funds for the Central Universities (18CX02042A and 18CX05011A)
Abstract:
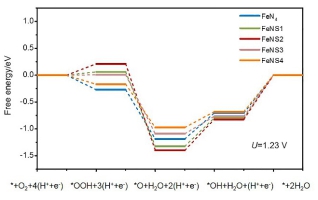
Heteratom-doped Fe-NC catalyst is promising for highly efficiently oxygen reduction reaction (ORR). In this work, density functional theory with the Vienna Ab initio Simulation Package (VASP) has been employed to systematically study the electronic structure regulation mechanism and oxygen reduction promoting mechanism on sulfur atom doped Fe-NC catalyst. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional within a generalized gradient approximation (GGA) was used in this work. The computataional hydrogen electron model was used to calculate the changes in Gibbs free energy. To consider the influence of S doping proportion, we build FeNSx models with 1~4 S atoms. The thermodynamic stability of catalysts was firstly considered based on formation energy, following by electronic structure analysis through Bader charge analysis and densities of states. Then, the oxygen adsorption ability was considered based on oxygen adsorption configurations and energies analyses. At last, reaction overpotentials were calculated based on computational hydrogen electrode model to study activity of catalytic sites. The results show that the catalyst doped with few sulfur atoms around the active sites of FeN4 could remain stable. Three possible ORR promoting mechanisms of S atoms doping were investigated. Firstly, the doping of sulfur atoms would reduce the band gap of the catalyst, thus improving the conductivity of the catalyst, which is beneficial to electrocatalytic oxygen reduction reactions. Secondly, the doping of a small amount of S atoms can improve the affinity between oxygen and the catalysts, which is also important for oxygen reduction reaction. At last, the introduction of four S atoms in the system would reduce the overpotential of ORR, thus improving the activity of the active sites to catalyze the oxygen reduction reaction. Our results predict that few S atoms doping would improve ORR performance of the Fe-NC catalyst through reducing band gap, improving ability to adsorb oxygen, and improving catalytic activity of FeN4 site. This work may give a new insight into regulation rules of heteratom doping on single atom catalysts based on carbon materials.
Figure 1.
The optimized structures of FeN4 and FeNSx Blue spheres represent nitrogen atoms, yellow spheres represent sulfur atoms, and gray spheres represent iron atoms, shown here. The number is the length of the Fe-n1 bond
Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T. A.; Hennig, R. G. J. Chem. Phys.2014, 140, 084106. doi: 10.1063/1.4865107
[21]
Guo, C.; Wei, S.; Zhou, S.; Zhang, T.; Wang, Z.; Ng, S. P.; Lu, X.; Wu, C. M. L.; Guo, W. J. ACS Appl. Mater. Interfaces2017, 9, 26107. doi: 10.1021/acsami.7b07945
[22]
Chen, Z.; Zhao, J.; Cabrera, C. R.; Chen, Z. F. Small Methods2019, 3, 1800368. doi: 10.1002/smtd.201800368
图 1
FeN4及硫原子掺杂结构示意图
Figure 1
The optimized structures of FeN4 and FeNSx Blue spheres represent nitrogen atoms, yellow spheres represent sulfur atoms, and gray spheres represent iron atoms, shown here. The number is the length of the Fe-n1 bond


 下载:
下载:




 下载:
下载:






 下载:
下载:
