
图 1.
Phainanoids A~F的结构
Figure 1.
Structures of Phainanoids A~F

天然产物Phainanoids 是岳建民教授课题组从海南细枝叶下珠中分离的具有高度取代的三萜类化合物, 它们包含独特的3-氢-螺[苯并呋喃-2, 1′-环丁烷]-3-酮和1, 6-二氧杂螺[4.4]壬烷-2-酮两个结构片段(图 1). 这类天然产物具有非常显著的免疫抑制活性, 其中最为突出的是Phainanoid F, 其T淋巴细胞增殖抑制活性的IC50值为(2.04±0.01) nmol/L, B淋巴细胞增殖抑制活性的IC50值小于(1.60±0.01) nmol/L[1].
天然产物Phainanoids的结构包含了10个环系和不少于13个手性中心. 初步的构效关系分析表明, 4, 5-螺环醚和/或5, 5-螺环内脂片段对于免疫抑制活性具有至关重要的影响[1]. 然而由于天然产物Phainanoids在自然资源中极其稀缺, 每公斤干植物中仅含有1~4 mg, 结合其新颖独特的结构骨架和显著的生物活性, Phainanoids吸引了越来越多的研究小组对其进行合成以及进一步生物学评价的研究.
2017年, 董广斌教授课题组发展了钯催化分子内的烯基化偶联反应, 非对映选择性地构建了Phainanoids的4, 5-螺环片段, 其中羰基氧原子与钯催化剂的相互稳定作用是实现非对映选择性的关键(图 2a)[2]. 南发俊教授课题组报道了通过呋喃氧化环化策略构建Phainanoids的1, 6-二氧杂螺[4.4]壬烷-2-酮结构片段(图 2b)[3].

为了探索合成天然产物Phainanoids的关键反应, 我们设计了简化的Phainanoids左翼4, 5-螺环片段化合物19的合成路线作为模型研究. 对于模型化合物19的合成需要解决两个问题: (1)构建具有环张力的多取代环丁烷结构; (2)非对映选择性地构建4, 5-螺环结构. 一直以来, 过渡金属催化的串联环化反应以其高效性、原子经济性以及立体选择性成为有机化学的热点, 尤其在构建环型分子领域发展了多种多样的合成方法学[4]. 2007年, Murakami教授课题组报道了铑(I)催化的4-炔-1-酮类结构与苯硼酸发生4-exo-trig类型芳基化环化反应, 构建环丁醇结构的方法学[5]. 在此方法学的基础上, 我们对模型化合物19进行了逆合成分析(见图 3).
模型化合物19中的五元醚环可以通过格氏试剂9与环丁酮8进行加成反应, 随后发生分子内的芳香亲核取代反应而构建. 我们设想此加成反应的非对映选择性可以通过格氏试剂9中的镁离子与化合物8中羰基氧原子以及邻位烷氧基螯合, 按照Cram’s chelation模型发生立体选择性加成而实现[6]. 环丁酮8可以由环丁烯6中的双键通过臭氧解反应而获得. 炔酮化合物5在铑(I)催化条件下, 与苯硼酸发生芳基化环化反应, 构建环丁烯化合物6. 最后, 炔酮化合物5可以由选择性羰基保护的Wieland-Miescher酮1经过Birch还原和烷基化反应制备.
我们的合成路线以已知化合物1为起始原料[7], 首先在锂/液氨条件下发生Birch还原, α, β不饱和酮结构被还原成烯醇负离子, 所得到的烯醇负离子中间体与1-溴-2-丁炔发生烷基化反应, 由于炔烃侧链与角甲基存在1, 3-直立效应, 非对映选择性的制备了化合物3[8]. 下面尝试在三取代羰基邻位引入甲基构建季碳中心, 以获得炔酮底物5. 由于二取代羰基邻位碳原子相对于三取代羰基邻位碳原子位阻小, 更容易发生甲基化反应, 因此需要先引入双键以避免该位点进行甲基化反应. 首先尝试了Saegusa氧化的方法, 然而未能在二取代羰基邻位引入双键[9]. 接下来, 在双(三甲基硅烷基)氨基钾(KHMDS)/苯基溴化硒条件下, 对化合物3中二取代羰基邻位进行苯硒基化, 随后在双氧水/吡啶条件下, 对苯硒基化合物进行氧化消除得到双键化合物4[10]. 最后在KHMDS/碘甲烷条件下进行甲基化反应, 在锂/液氨条件下还原掉不饱和双键, 得到炔酮化合物5.

Reagents and Conditions: a) Li (3.5n1), NH3(l), -40 ℃, 12 h; H2O (1.5n1), -40 ℃, 30 min, 2 (2.0n1), THF, -40 ℃, 12 h; b) KHMDS (1.5n3), THF, 0 ℃, 2 h, then HMPA (1 mL/mmol), PhSeBr (2.0n3), -78 ℃, 1 h; c) H2O2 (3.0n3), pyridine (10.0n3), THF, 0 ℃, 5 h; d) KHMDS (1.5n4), THF, 0 ℃ to 50 ℃, 2 h, then MeI (3.0n4), 0 ℃ to 25 ℃, 12 h; e) Li (3.n4), NH3(l), -40 ℃, 12 h. KHMDS=Potassium bis(trimethylsilyl)amide, HMPA=Hexamethylphosphoramide.
获得化合物5之后, 我们对炔酮5和苯硼酸在铑(I)催化剂存在的条件下, 通过芳基化环化反应构建具有环张力的多取代环丁烯6的反应条件进行了筛选. 氩气保护条件下, 当炔酮5和苯硼酸在[Rh(OH)(cod)]2催化剂载量(n催化剂/n5)为1.25%, 以1, 4-二氧六环/水(V1, 4-二氧六环∶V水=100∶1)为溶剂, 20 ℃搅拌18 h, 产物环丁烯6的柱层析分离收率为21%(表 1, Entry 1). 当提高铑(I)催化剂载量至2.5%时, 收率提高至36%(表 1, Entry 2). 同时提高铑(I)催化剂载量至2.5%, 苯硼酸至5n5时, 收率达到42%(表 1, Entry 3). 对反应温度进行考察, 当反应温度提高至35 ℃时, 收率提高到56%(表 1, Entry 4), 然而在该条件下, 炔酮底物5仍有少量剩余. 最后我们发现, 当将载量2.5%的[Rh(OH)(cod)]2催化剂和5n5苯硼酸分两批加入至炔酮底物5中, 在35 ℃条件下分别反应12 h, 以73%的柱层析收率获得产物环丁烯6(表 1, Entry 5).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | n([Rh(OH)(cod)]2)/n5 | n[PhB(OH)2]/n5 | Temp./℃ | Yieldb/% |
| 1 | 1.25% | 2 | 20 | 21 |
| 2 | 2.5% | 2 | 20 | 36 |
| 3 | 2.5% | 5 | 20 | 42 |
| 4 | 2.5% | 5 | 35 | 56 |
| 5c | 2.5% | 5 | 35 | 73 |
| a Reactions were conducted on a 0.3 mmol scale in 1.5 mL dioxane/H2O(V∶V, 100∶1) in a 10 mL vial under Ar atmosphere for 18 h unless otherwise noted; b Yield of isolated product 6; c Reactions were conducted on a 0.3 mmol scale in 1.5 mL dioxane/H2O(V∶V, 100∶1) in a 10 mL vial under Ar atmosphere. 1.25%n5 [Rh(OH)(cod)]2 and 2.5n5 PhB(OH)2 were added first, the reaction was stirred for 12 h, then another 1.25%n5 [Rh(OH)(cod)]2 and 2.5n5 PhB(OH)2 were added, the reaction was stirred for another 12 h. | ||||
在二异丙基乙基胺条件下, 对环丁烯6中的羟基进行乙氧基甲基(EOM)保护得到产物7, 对双键进行臭氧解得到环丁酮8. 邻烷氧基取代的环丁酮8与新鲜制备的格氏试剂9发生加成反应, 得到了Felkin-Anh模型预测的立体选择性加成产物环丁醇10[11], 而非Cram’s chelation-control模型中镁离子与羰基氧原子和邻位烷氧基螯合所预测的立体选择性加成产物, 其中环丁醇10的相对立体构型经过单晶衍射实验的验证(图 5).
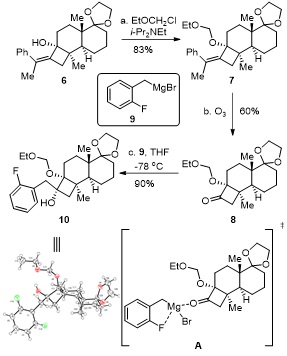
Reagents and Conditions: a) EtOCH2Cl (2.0n6), i-Pr2NEt (3.0n6), THF, 25 ℃, 12 h; b) O3, DCM, -78 ℃, then Me2S (3.5n7), -78 ℃, 1 h; c) 9 (6.0n8), THF, -78 ℃, 3 h.
文献报道的格氏试剂对邻烷氧基环丁酮结构加成的反应有两例(图 6). 1993年, Holger Butenschön报道了甲基碘化镁与邻甲氧基环丁酮11按照Cram’s chelation模型立体选择性加成, 得到环丁醇12(图 6a)[12]. 2000年, Louis S. Hegedus报道了乙烯基溴化镁与邻甲氧基环丁酮13按照Cram’s chelation模型立体选择性加成, 得到环丁醇14(图 6b)[13].

结合上面两例报道, 我们推测格氏试剂9与邻烷氧基环丁酮8未按照Cram’s chelation-control模型进行立体选择性加成是由于格氏试剂9中镁离子与氟原子进行了分子内螯合[14], 从而抑制了加成反应过程中镁离子与羰基氧原子进行螯合后, 继续与羰基邻位的烷氧基螯合(图 5中间体A). 为验证此推测, 我们将底物8分别与苄基溴化镁和苯基溴化镁发生加成反应, 得到了按照Felkin-Anh模型进行立体选择性加成的产物环丁醇15和16, 从图 7中反应过渡态B可以看出, 由于底物8中环系的刚性, 格氏试剂中的镁离子未能与羰基邻位的氧原子发生螯合, 因此R基团从位阻相对较小的一侧发生加成反应, 分别得到化合物15和16.

Reagents and Conditions: a) BnMgBr (6.0n8), THF, -78 ℃, 3 h; b) PhMgBr (6.0n8), THF, -78 ℃, 3 h.
结合上述文献报道和实验结果, 我们认为格氏试剂9与邻烷氧基环丁酮8未按照Cram’s chelation-control模型进行立体选择性加成的原因可以从两个方面进行阐述: (1)主要由于环丁酮8的结构相对刚性, 增加了镁离子与羰基氧原子和邻位烷氧基螯合过渡态的能垒; (2)其次格氏试剂9中镁离子与氟原子进行了分子内螯合, 从而抑制了镁离子与羰基邻位烷氧基的螯合.

Reagents and Conditions: a) t-BuOK (3.0n10), THF, 60 ℃, 10 h; b) CSA (0.1n17), THF, 60 ℃, 10 h. CSA=Camphorsulfonic acid.
最后, 环丁醇10在叔丁醇钾的条件下进行分子内的芳香亲和取代反应, 得到4, 5-螺环化合物17[15], 其结构经过单晶衍射实验验证. 在樟脑磺酸的催化下, 脱除EOM和缩酮保护基得到模型化合物的非对映异构体18.
我们应用铑(I)催化芳基化环化反应和格氏试剂加成反应以及分子内芳香亲核取代反应为关键反应, 对天然产物Phainanoids的4, 5-螺环骨架合成进行了探索. 以已知化合物1为起始原料, 合成了模型化合物的非对映异构体18. 其中格氏试剂9与邻烷氧基环丁酮8未按照Cram’s chelation-control模型进行立体选择性加成, 结合文献报道和相关实验, 我们认为原因可以从两个方面进行阐述: (1)环丁酮8相对刚性的结构, 增加了镁离子与羰基氧原子和邻位烷氧基螯合过渡态的能垒; (2)格氏试剂9中镁离子与氟原子进行了分子内螯合, 从而抑制了镁离子与环丁酮8中羰基邻位烷氧基的螯合作用.
Fan, Y.-Y.; Zhang, H.; Zhou, Y.; Liu, H.-B.; Tang, W.; Zhou, B.;Zuo, J.-P.; Yue, J.-M. J. Am. Chem. Soc. 2015, 137, 138.
Xie, J.; Wang, J.; Dong, G. Org. Lett. 2017, 19, 3017.
Zhang, C. L.; Nan, F. J. Tetrahedron Lett. 2017, 58, 4357.
Reviews:(a) Mascareñas, J. L.; Varela, I.; López, F. Acc. Chem. Res. 2019, 52, 465.(b) Kwiatkowski, M. R.; Alexanian, E. J. Acc. Chem. Res. 2019, 52, 1134.(c) Montgomery, J. Angew. Chem., Int. Ed. 2004, 43, 3890.(d) Bates, R. W.; Satcharoenb, V. Chem. Soc. Rev. 2002, 31, 12.(e) Inglesby, P. A.; Evans, P. A. Chem. Soc. Rev. 2010, 39, 2791.(f) Marinetti, A.; Jullien, H.; Voituriez, A. Chem. Soc. Rev. 2012, 41, 4884.(g) Aubert, C.; Fensterbank, L.; Garcia, P.; Malacria, M.; Simonneau, A. Chem. Rev. 2011, 111, 1954.(h) Ojima, I.; Tzamarioudaki, M.; Li, Z.; Donovan, R. J. Chem. Rev. 1996, 96, 635.(i) Aubert, C.; Buisine, O.; Malacria, M. Chem. Rev. 2002, 102, 813.(j) Grigg, R.; Sridharan,V. J. Organomet. Chem. 1999, 576, 65.(k) Xiao M. Y.; Zhu S.; Shen Y. B.; Wang L.; Xiao J. N. Chin. J. Org. Chem. 2018, 38, 328.(l) Li J. X.; Lin S.; Huang R. K.; Li C.; Yang S. R. Chin. J. Org. Chem. 2019, 39, 1417.(m) Geng D. G.; Chin. J. Org. Chem. 2019, 39, 301.(n) Qian X. Y.; Xiong P.; Xu H. C. Acta Chim. Sinica 2019, 77, 879.(o) Xu J.; Zhang S. F.; Luo Y.; Zhang, L.; Zhang, F.; Huang, T. J.; Song, Q. L. Acta Chim. Sinica 2019, 77, 932.(p) Liao F. M.; Du Y.; Zhou F.; Zhou J. Acta Chim. Sinica 2018, 76, 862.(q) Wang, W. G.; Huang, S.; Yan, S. K.; Sun, X. J.; Tung, C. H.; Xu, Z. H. Chin. J. Chem. 2020, 38, 445.(r) Zhou, L. J.; Xu, B.; Ji, D. T.; Zhang, Z. M.; Zhang, J. L. Chin. J. Chem. 2020, 38, 577.
Miura, T.; Shimada, M.; Murakami, M. Tetrahedron 2007, 63, 6131.
(a) Cram, D. J.; Kopecky, K. R. J. Am. Chem. Soc. 1959, 81, 2748.(b) Mengel, A.; Reiser, O. Chem. Rev. 1999, 99, 1191.(c) Chen, X.; Hortelano, E. R.; Eliel, E. L. J. Am. Chem. Soc. 1990, 112, 6130.(d) Fan, X. Y.; Walsh, P. J. Acc. Chem. Res. 2017, 50, 2389.(e) Bartolo, N. D.; Read, J. A.; Valentín, E. M.; Woerpel, K. A. Chem. Rev. 2020, 120, 1513.(f) Stanton, G. R.; Koz, G.; Walsh, P. J. J. Am. Chem. Soc. 2011, 133, 7969.(g) Ye, J. L.; Huang, P. Q.; Lu, X. J. Org. Chem. 2007, 72, 35.(h) Bailey, W. F.; Reed, D. P.; Clark, D. R.; Kapur, G. N. Org. Lett. 2001, 3, 1865.
Ciceri, P.; Demnitz, F. W. J. Tetrahedron Lett. 1997, 38, 389.
Ali, A.; Guile, S. D.; Saxton, J. E.; Thornton-Pett, M. Tetrahedron 1991, 41, 6407.
(a) Ito, Y.; Hirao, T.; Saegusa, T. J. Org. Chem. 1978, 43, 1011.(b) Toyooka, N.; Okumura, M.; Nemoto, H. J. Org. Chem. 2002, 67, 6078.
Moustafa, G. A. I.; Saku, Y.; Aoyama, H.; Yoshimitsu, T. Chem. Commun. 2014, 50, 15706.
Chérest, M.; Felkin, H.; Prudent, N. Tetrahedron Lett. 1968, 9, 2199.
Butenschn, H. Chem. Ber. 1994, 127, 137.
Hegedus, L. S.; Ranslow, P. B. Synthesis 2000, 7, 953.
(a) Yamazaki, T.; Ando, M.; Kitazume, T.; Kubota, T.; Omura, M. Org. Lett. 1999, 1, 905.(b) Yamazaki, T.; Kawashita, S.; Kitazume, T; Kubota, T. Chem. Eur. J. 2009, 15, 11461.(c) Tenza, K.; Northen, J. S.; O'Hagan, D.; Slawin, A. M. Z. J. Fluorine Chem. 2004, 125, 1779.(d) Sazonov, P. K.; Oprunenko, Y. F.; Khrustalev, V. N.; Beletskaya, I. P. J. Fluorine Chem. 2011, 132, 587.(e) Kulawiec, R. J.; Holt, E. M.; Lavin, M.; Crabtree, R. H. Inorg. Chem. 1987, 26, 2559.(f) Ooi, T.; Kagoshima, N.; Maruoka, K. J. Am. Chem. Soc. 1997, 119, 5754.(g) Kawachi, A.; Tani, A.; Machida, K.; Yamamoto, Y. Organometallics 2007, 26, 4697.(h) Bizet, V.; Cahard, D. Chimia. 2014, 68, 378.(i) Carrell, H. L.; Glusker, J. P.; Piercy, E. A.; Stallings, W. C.; Zacharias, D. E.; Davis, R. L.; Astbury, C.; Kennard, C. H. L. J. Am. Chem. Soc. 1987, 109, 8067.
Nakamura, K.; Ohmori, K.; Suzuki, K. Angew. Chem., Int. Ed.2017, 56, 182.

图 2 董教授和南教授对于Phainanoids的模型合成研究
Figure 2 Professor Dong’s and Nan’s model study towards Phainanoids

图 4 底物5的制备
Figure 4 Preparation of the substrate 5
Reagents and Conditions: a) Li (3.5n1), NH3(l), -40 ℃, 12 h; H2O (1.5n1), -40 ℃, 30 min, 2 (2.0n1), THF, -40 ℃, 12 h; b) KHMDS (1.5n3), THF, 0 ℃, 2 h, then HMPA (1 mL/mmol), PhSeBr (2.0n3), -78 ℃, 1 h; c) H2O2 (3.0n3), pyridine (10.0n3), THF, 0 ℃, 5 h; d) KHMDS (1.5n4), THF, 0 ℃ to 50 ℃, 2 h, then MeI (3.0n4), 0 ℃ to 25 ℃, 12 h; e) Li (3.n4), NH3(l), -40 ℃, 12 h. KHMDS=Potassium bis(trimethylsilyl)amide, HMPA=Hexamethylphosphoramide.

图 5 以化合物6合成化合物10的路线
Figure 5 Synthesis of compound 10 from compound 6
Reagents and Conditions: a) EtOCH2Cl (2.0n6), i-Pr2NEt (3.0n6), THF, 25 ℃, 12 h; b) O3, DCM, -78 ℃, then Me2S (3.5n7), -78 ℃, 1 h; c) 9 (6.0n8), THF, -78 ℃, 3 h.

图 6 已报道的格式试剂对邻甲氧基环丁酮的加成
Figure 6 Reported α-methoxyl cyclobutone added by Grignard reagent

图 7 格式试剂对邻烷氧基环丁酮8的加成
Figure 7 α-Alkoxyl cyclobutone 8 added by Grignard reagent
Reagents and Conditions: a) BnMgBr (6.0n8), THF, -78 ℃, 3 h; b) PhMgBr (6.0n8), THF, -78 ℃, 3 h.

图 8 化合物18的合成
Figure 8 Synthesis of compound 18
Reagents and Conditions: a) t-BuOK (3.0n10), THF, 60 ℃, 10 h; b) CSA (0.1n17), THF, 60 ℃, 10 h. CSA=Camphorsulfonic acid.
表 1 炔酮 5 的铑催化芳基化环化条件筛选 a
Table 1. Optimization of the rhodium(I)-catalyzed arylative cyclization of alkynone 5a
|
|
||||
| Entry | n([Rh(OH)(cod)]2)/n5 | n[PhB(OH)2]/n5 | Temp./℃ | Yieldb/% |
| 1 | 1.25% | 2 | 20 | 21 |
| 2 | 2.5% | 2 | 20 | 36 |
| 3 | 2.5% | 5 | 20 | 42 |
| 4 | 2.5% | 5 | 35 | 56 |
| 5c | 2.5% | 5 | 35 | 73 |
| a Reactions were conducted on a 0.3 mmol scale in 1.5 mL dioxane/H2O(V∶V, 100∶1) in a 10 mL vial under Ar atmosphere for 18 h unless otherwise noted; b Yield of isolated product 6; c Reactions were conducted on a 0.3 mmol scale in 1.5 mL dioxane/H2O(V∶V, 100∶1) in a 10 mL vial under Ar atmosphere. 1.25%n5 [Rh(OH)(cod)]2 and 2.5n5 PhB(OH)2 were added first, the reaction was stirred for 12 h, then another 1.25%n5 [Rh(OH)(cod)]2 and 2.5n5 PhB(OH)2 were added, the reaction was stirred for another 12 h. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: