引用本文:
黄清明. 亚晶格能量团簇构建及晶场调控对四方LiYF4:RE上转换发光机制的影响研究[J]. 化学学报,
2020, 78(9): 968-979.
doi:
10.6023/A20050154 Citation:
Huang Qingming. Study on the Upconversion Luminescence Mechanism of Tegtragonal LiYF4: RE with Sublattice Energy Cluster Construction and Crystal Field Manipulation[J]. Acta Chimica Sinica,
2020, 78(9): 968-979.
doi:
10.6023/A20050154
Received Date:
09 May 2020 Available Online:
15 September 2020
Fund Project:
Project supported by the Natural Science Foundation of Fujian Province (No. 2017J01688)
Abstract:
Lanthanide ions doped tetragonal LiYF4 has became an investigative focus of upconversion luminescence (UCL) materials for its well properties of multi-photon UCL and as a comparable matrix material with hexagonal NaYF4. While the cause for its well performance on short bands emission is still unrevealed. After the exploration of crystal structure characteristic of tetragonal LiYF4, a hexagonal circle sublattice structure of Y3+ with 0.3710 nm interval between adjacent Y3+ ions and larger than 0.5 nm interval between meta-position and para-position Y3+ ions were revealed. The energy transfer of rare earth ions are easy take place around the hexagonal circles or among the cluster of five adjacent trivalent ions. Base on the sublattice structure characteristic of tetragonal LiYF4, we have an idea to study UCL mechanism systematically of tetragonal LiYF4:RE by the construction of sublattice energy cluster 1M-xYb (M=Er, Ho, Tm) and the manipulation of crystal field symmetry by introducing different amount Yb3+ ions and Sc3+ or Hf4+ ions, respectively. Hydrothermal method was employed to prepare LiY0.98-xYbxEr0.02F4, LiY0.98-xYbxHo0.02F4, LiY0.995-xYbxTm0.005F4, LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples. A typical preparation process demonstrate as follows, at first, (1-x) mmol Y(NO3)3 (0.2 mol/L), x mmol (x=0.2, 0.5, 0.7 and 0.9) Yb(NO3)3 (0.20 mol/L) and Er(NO3)3 (0.02 mmol) solution was dropwise added into 20 mL deionized (DI) water with 1 mmol EDTA to form a solution under vigorous stirring for 30 min. Secondly, 3.0 mL LiOH (1.0 mol/L) and 4.0 mL NH4HF2(1.0 mol/L) aqueous solution were dropwise added to the solution under thorough stirring for 30 min until the solution completely became a white emulsion, the pH value of the emulsion is 3~4. Finally, the white emulsion was slowly transferred into a 50 mL Teflon-lined autoclave, sealed and heated at 190℃ for 18 h. The final products were collected by centrifugation, and then washed with DI water several times. The collected samples were dried at 60℃ over night. X-ray powder diffraction (XRD) and Rietveld refinement method were employed to reveal the variation of crystal structure, field emission scanning electron microscopy (FESEM) and field emission transmission electron microscopy (FETEM) were employed to the analysis of crystal morphology and crystal structure. UCL performance was analyzed by Edinburgh fluorescence spectrophotometer FSP920. After investigation, we found excited energy levels distribution of different RE ions is diverse, and the level matching with Yb3+ are different too, it result in different luminescence quenching of energy cross relaxation, so the different sublattice energy clusters 1Er-2Yb, 1Ho-2Yb and 1Tm-4Yb of different active rare earth ions can be constructed for the best UCL performance. The cystal field symmetry of tetragonal LiYF4:Yb/Er were manipulated successfully by 6 mol% Sc3+ or 4 mol% Hf4+ doping, and UCL intensity were enhanced about 50% with 6 mol% Sc3+, while the UCL intensity were weaken after Hf4+ doping. After Sc3+ or Hf4+ doping, there are only three Yb3+ ions in the five trivalence ions cluster that can't realize two-photon cooperation upconversion synchronous electron population of 4F5/2 excited state level of Er3+ ions and 2G7/2 or 4Fo5/2 excited state level of Sc3+ or Hf4+ respevticely, and then Sc3+ and Hf4+ ions become a quenching center in the asymmetric crystal field that is conversed with them doped hexagonal NaYF4:Yb/Er that Sc3+ and Hf4+ ions were taken as energy storage ions and dramatically enhanced UCL performance. In this work, the UCL mechanism of sublattice energy cluster construction and crystal field manipulation were revealed that may be an inspiration for high efficient UC luminescence materials design and preparation.
Figure 2.
(a, b) XRD patterns of LiY0.98-xYbxEr0.02F4 and LiY0.98-xYbxHo0.02F4 series samples, (c, d) lattice constants changing trend of LiY0.98-xYbxEr0.02F4 series sample.
Figure 4.
(a, c, e) Upconversion luminescence spectra of LiY0.98-xYbxEr0.02F, LiY0.98-xYbxHo0.02F4 and LiY0.995-xYbxTm0.005F4 series samples, respectively. (b, d, f) emissions intensity changing trends of LiY0.98-xYbxEr0.02F4, LiY0.98-xYbxHo0.02F4 and LiY0.995-xYbxTm0.005F4 series samples with the rising of Yb3+ doping concentration, respectively.
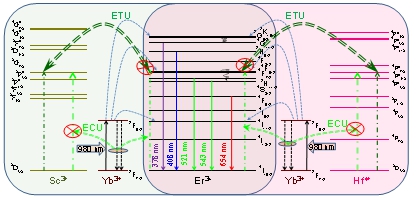
Figure 6.
The schematic of upconversion luminescence of Er3+, Ho3+ and Tm3+ by the construction of 1M-xYb (M=Er3+, Ho3+ and Tm3+) sublattice energy clusters.
Figure 7.
(a~d) cell parameters and bond distance changing trends of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 with the rising of Sc3+ and Hf4+ doping concentration, respectively.
Figure 8.
(a, c) Upconversion luminescence spectra of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively. (b, d) emissions intensity changing trends of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 with the rising of Sc3+ and Hf4+ doping concentration, respectively.
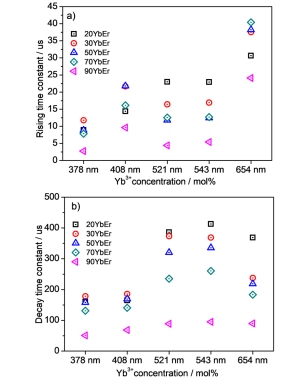
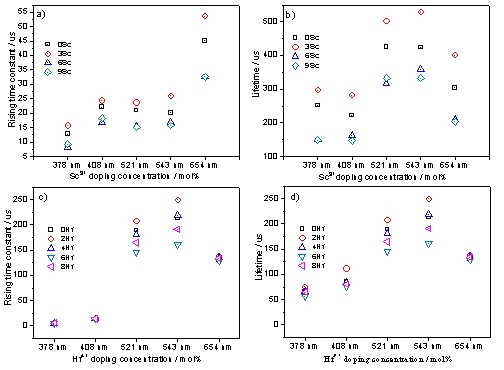
Figure 9.
(a, c) Emissions rising time constant changing trend of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively. (b, d) emissions lifetime changing trend of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively.
Ullah, S.; Hazra, C.; Ferreira-Neto, E. P.; Silva, T. C.; Rodrigues-Filho, U. P.; Ribeiro, S. J. L. Crystengcomm2017, 19, 3465. doi: 10.1039/C7CE00809K
Borges, M. E.; Sierra, M.; Mendez-Ramos, J.; Acosta-Mora, P.; Ruiz-Morales, J. C.; Esparza, P. Sol. Energy Mater. Sol. Cells2016, 155, 194. doi: 10.1016/j.solmat.2016.06.010
[22]
Kakavelakis, G.; Petridis, K.; Kymakis, E. J. Mater. Chem. A2017, 5, 21604. doi: 10.1039/C7TA05428A
Zhao, J.; Jin, D.; Schartner, E. P.; Lu, Y.; Liu, Y.; Zvyagin, A. V.; Zhang, L.; Dawes, J. M.; Xi, P.; Piper, J. A.; Goldys, E. M.; Monro, T. M. Nat. Nanotechnol.2013, 8, 729. doi: 10.1038/nnano.2013.171
[43]
刘明丽, 吴琪, 史慧芳, 安众福, 黄维, 化学学报, 2018, 76, 246. doi: 10.6023/A17110504Liu, M.; Wu, Q.; Shi, H.; An, Z.; Huang, W. Acta Chim. Sinica2018, 76, 246(in Chinese). doi: 10.6023/A17110504
Figure 2
(a, b) XRD patterns of LiY0.98-xYbxEr0.02F4 and LiY0.98-xYbxHo0.02F4 series samples, (c, d) lattice constants changing trend of LiY0.98-xYbxEr0.02F4 series sample.
Figure 4
(a, c, e) Upconversion luminescence spectra of LiY0.98-xYbxEr0.02F, LiY0.98-xYbxHo0.02F4 and LiY0.995-xYbxTm0.005F4 series samples, respectively. (b, d, f) emissions intensity changing trends of LiY0.98-xYbxEr0.02F4, LiY0.98-xYbxHo0.02F4 and LiY0.995-xYbxTm0.005F4 series samples with the rising of Yb3+ doping concentration, respectively.
Figure 6
The schematic of upconversion luminescence of Er3+, Ho3+ and Tm3+ by the construction of 1M-xYb (M=Er3+, Ho3+ and Tm3+) sublattice energy clusters.
Figure 7
(a~d) cell parameters and bond distance changing trends of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 with the rising of Sc3+ and Hf4+ doping concentration, respectively.
Figure 8
(a, c) Upconversion luminescence spectra of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively. (b, d) emissions intensity changing trends of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 with the rising of Sc3+ and Hf4+ doping concentration, respectively.
Figure 9
(a, c) Emissions rising time constant changing trend of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively. (b, d) emissions lifetime changing trend of LiY0.68-xYb0.3Er0.02ScxF4 and LiY0.68-xYb0.3Er0.02HfxF4 series samples, respectively.


 下载:
下载:







 下载:
下载:











 下载:
下载:
