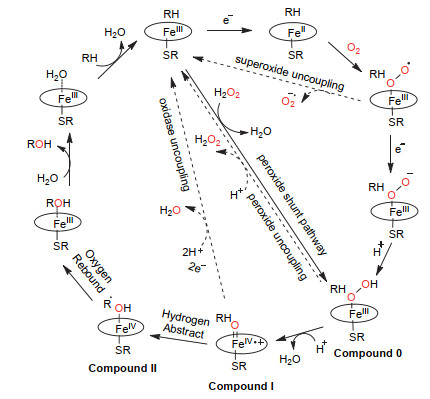
图 1.
细胞色素P450单加氧酶催化的烷烃羟化反应机理
Figure 1.
The mechanism of alkanes hydroxylation catalyzed by Cytochrome P450 monooxygenases

烷烃是石油和天然气的主要成分, 尤其是甲烷在天然气中的含量达到70%~95%.醇类化合物是重要的液态燃料、功能化学品比如作为溶剂等、以及一些高附加值化合物的合成前体等.烷烃转化制备醇类化合物, 是油气资源化利用的有效途径之一.由烷烃到醇的转化通常采用间接路线, 这是因为烷烃C—H键结合能很高(398~440 kJ/mol)[1], 往往需要强氧化剂的参与; 而对应的醇产物比烷烃更容易被氧化, 导致过度氧化反应生成醛或羧酸.例如工业上制备甲醇是将甲烷先转化为合成气, 然后加氢还原, 尽管这一反应在经济上是可行的, 但是存在能耗高、反应条件苛刻等不足.在温和条件下实现烷烃的选择性直接羟化能够有效克服上述问题, 一直是该领域的研究热点[2, 3].另外对于中长链烷烃直接氧化羟化反应的区域选择性(碳链长度≥C3)和立体选择性(碳链长度≥C4)控制, 更是合成化学领域的一个极大挑战.
自然界中的一些氧化金属酶, 比如甲烷单加氧酶(Methane Monooxygenases)[4]、氨单加氧酶(Ammonia Monooxygenase)[5]、烷烃羟化酶AlkB[6]、以及细胞色素P450单加氧酶(Cytochromes P450 Monooxygenase, P450s或CYP)[7]等, 对不活泼C—H键具有良好的催化活性和化学选择性, 并且一些酶对特定天然底物有着优异的区域/立体选择性, 显示出其作为C—H键选择氧化催化剂方面的巨大潜力, 受到合成化学家的广泛关注.其中, 细胞色素P450单加氧酶具有来源广泛、异源表达重组技术成熟易获取、蛋白分子量较小易于改造等优点.天然P450酶能催化杂原子氧化、环氧化、惰性C—H键羟化反应等多种氧化反应, 是公认的多功能生物氧化催化剂[8].
近年来工程化P450酶在合成应用方面的潜力被进一步发掘, 在天然产物如青蒿素前体青蒿酸等的生物合成[9~11]、环丙化反应[12]、C—N键[13]和C—B键[14]生成反应等方面取得重要进展.通过基因工程、酶工程、以及化学调控等技术手段对细胞色素P450单加氧酶进行分子改造, 提高其催化活性并扩展其底物适用范围, 对开发具有潜在应用价值的烷烃选择性羟化生物催化剂具有重要意义[15~18].
细胞色素P450单加氧酶来源广泛, 广泛存在于动物、植物、细菌以及真菌等生物体中, 已被鉴定的基因序列超过41000种, 它是一类以半胱氨酸为轴向配体的亚铁血红素(heme: iron protoporphyrin IX)蛋白超级家族[19].在底物羟化反应过程中, 细胞色素P450酶需要还原辅酶NAD(P)H提供两个电子, 经过两步连续的一电子转移氧化, 以氧气为氧化剂将P450的血红素活性中心氧化为高价铁氧物种Compound I, 后者作为实际的氧化剂羟化烷烃.最后结果如总反应式所示, 实现将氧气的一个氧原子插入到底物中, 而另一个氧原子还原为水: RCH3+NAD(P)H+O2+H+→RCH2OH+ NAD(P)++H2O.
底物同P450酶的结合对于引发反应催化循环非常重要, Poulos等通过研究来自Pseudomonas putida的CYP101A1(P450cam)的晶体结构[20a]发现:在无底物结合时P450cam的底物口袋呈现开放状态(open state), 当底物进入后口袋呈关闭状态(close state), 这种P450氧化结构域(heme domain)的构象变化, 通过蛋白-蛋白间的相互作用传递到它的还原结构域(reduced domain), 从而触发从还原辅酶NAD(P)H到血红素催化中心的电子转移引发催化反应循环[20b].
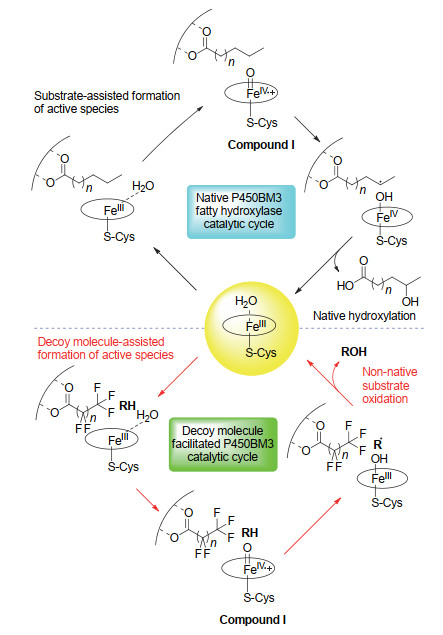
从氧化还原反应角度来说:在休止状态(resting state)即开放状态构象下P450酶的血红素反应中心三价铁有一个轴向配位的H2O分子[21], 使其处于稳定的六配位低自旋态.底物结合破坏H2O分子的配位形成五配位高自旋态, 氧化还原电位发生正迁移, 三价铁离子接受来自NAD(P)H的一个电子, 被还原为二价铁离子[22].二价铁与氧分子结合形成三价铁超氧自由基, 随后接受来自NAD(P)H的另一个电子形成三价铁过氧化物负离子态, 再经过质子化, 生成FeⅢ-OOH中间体(compound 0). Compound 0接受第二个质子经过O—O键异裂生成反应活性物种(Por·+)FeⅣ=O (Compound I). Compound I摄取烷烃C—H键的一个氢原子形成FeⅣ—OH中间体(Compound II)和碳自由基, 这一过程称为氢原子“拔取”(Hydrogen abstract).最后羟基自由基与底物再结合得到羟化产物, 即氧原子“回弹”(Oxygen rebound)(图 1)[23, 24].由于羟化产物与底物的化学结构存在差异, 蛋白空腔与结合底物之间的特异性使得产物醇一般不能再次进入P450的催化空腔, 从而阻止了过度氧化反应的发生, 实现选择羟化.
如图 1所示, P450酶的催化循环过程中也存在着可能的副反应途径, 导致未形成烷烃羟化产物的无效NAD(P)H消耗, 被称为非耦合反应(uncoupling reaction).比如三价铁超氧自由基如果没有及时接收到第二个电子, 就无法阻止超氧阴离子自由基的离开, 令血红素活性中心回到初始的五配位三价铁状态(superoxide uncoupling); 而如果Compound 0的质子化发生在与铁相连的氧原子上, 则会导致过氧化氢(H2O2)生成, 同样使NADPH提供的电子未能有效耦合Compound I的形成(peroxide uncoupling); 即便高价铁氧物种Compound I顺利生成, 也可能由于烷烃处在不利于氢原子被“拔取”的位置导致Compound I的“氧”被还原为水分子(oxidase uncoupling)[25].在需要耦合NAD(P)H来实现其催化功能的P450酶的氧化反应中, 一般将氧化产物生成量与NAD(P)H消耗量的百分比定义为NAD(P)H的耦合效率(coupling efficiency of NADPH)[26, 27].鉴于其价格较高, NAD(P)H的耦合效率高意味着反应的经济性.相反则不但造成NAD(P)H的无谓消耗, 而且可能在非耦合途径中产生超氧自由基和H2O2等活泼氧物种, 对反应体系也是潜在危害.因而NAD(P)H耦合效率是一个用来评估P450酶、尤其是P450工程酶的重要参数.
另外当H2O2在体系中达到一定浓度时, 从氧化还原反应机理上来说, 它也可以直接将三价铁氧化成Compound I.由于H2O2的氧是-1价还原态, 所以其在起到替代氧气作为末端氧化剂的同时, 也不再需要NAD(P)H提供电子(图 1).这被称为P450催化循环中的过氧化物旁路(peroxide shunt pathway)[25].对于高度依赖辅酶NAD(P)H和还原伴侣电子传递链来实现其催化功能的P450酶来说, 这条途径无疑大大简化了它的催化循环, 从有机合成应用的角度来说具有一定潜力.但实际上, 绝大多数NAD(P)H依赖型P450酶在H2O2存在下的活性极低甚至没有活性.直到最近我们开发了H2O2依赖型P450过加氧酶催化小分子烷烃羟化反应体系之前[28], 还没有以H2O2作为末端氧化剂的P450天然酶或者工程酶能够催化烷烃羟化反应的报道.
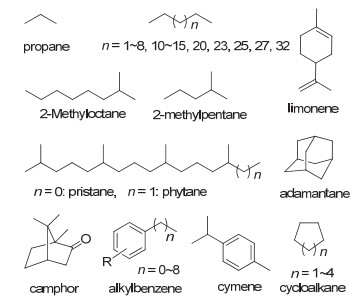
已报道的以烷烃作为天然底物的细胞色素P450单加氧酶, 主要来自于CYP153家族和CYP52家族以及其他P450家族中的部分酶.这些P450酶的底物范围很广, 几乎涵盖除气态烷烃以外的各种正烷烃、一些支链烷烃、部分环烷烃和多元环烃等(图 2).
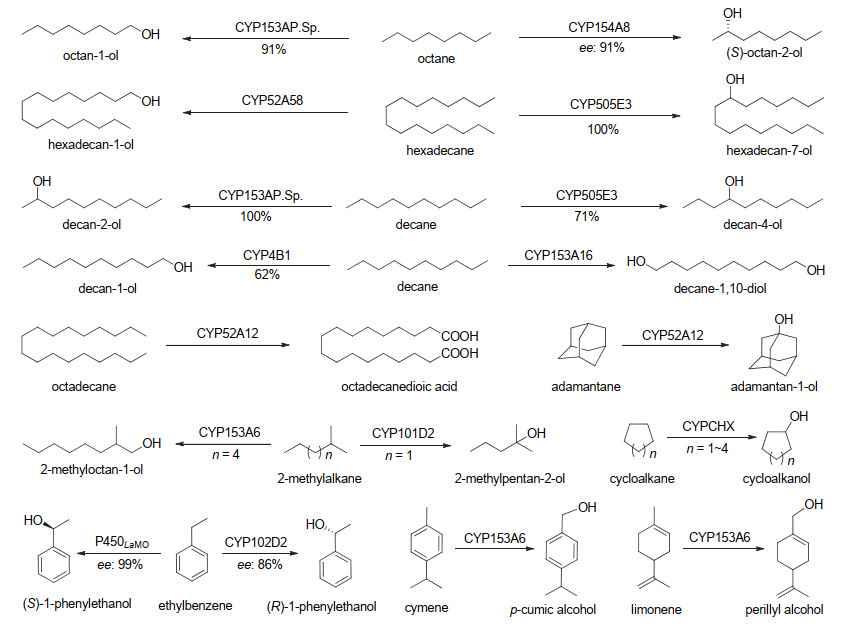
CYP153家族的P450酶大多来源于革兰氏阳性烷烃降解细菌, 以正烷烃(C5~C16)为底物, 主要生成末端(ω-位)和亚末端(ω-1位)羟化产物, 且都具有高度区域选择性, 但在大肠杆菌中重组表达的CYP153同工酶的催化转化率不高(表 1, 图 3).比如CYP153AP.Sp.催化C5~C8正烷烃选择性羟化生成伯醇[29a], 反应几乎是区域专一性的(100%), 即便较低的正辛烷的选择性也有90%;正壬烷(C9: 59%)和正癸烷(C10: 100%)羟化生成2-醇的区域选择性也很高.但催化反应的总转化数(TTN)仅为20~110. CYP153A16有着相似的底物偏好性, 对C5~C8的正烷烃也主要生成伯醇.有趣的是这个酶催化正壬烷和正癸烷都得到双末端羟化产物二醇, 显示其同样可以接受极性的烷基醇作为底物, 反应最好的TTN也仅为80. CYP153A6氧化C6~C11正烷烃得到区域专一性的伯醇[29b]. CYP153A6也能接受支链烷烃如2-甲基辛烷、以及一些较复杂结构的烃类作为底物, 比如柠檬烯和对伞花烃, 同样得到高度区域选择性羟化产物, 表明其具有更好的底物多样性. CYP153A13催化辛烷氧化生成1-辛醇的TTN也仅有233[29c].
 下载:
导出CSV
下载:
导出CSV
| Enzyme | Alkanes | Final Product | [Final Product]a | PFRb | TTNc |
| CYP153AP.Sp.[29a] | Pentane (C5) | 1-Pentanol (100%d) | 30 | — | 20 |
| Hexane (C6) | 1-Hexanol (100%d) | 62 | — | 41 | |
| Heptane (C7) | 1-Heptanol (100%d) | 103 | — | 69 | |
| Octane (C8) | 1-Octanol (90%d) | 165 | — | 110 | |
| Nonane (C9) | 2-Nonanol (59%d) | 114 | — | 76 | |
| Decane (C10) | 2-Decanol (100%d) | 99 | — | 66 | |
| CYP153A16[29a] | Pentane (C5) | 1-Pentanol | 34 | — | 22 |
| Hexane (C6) | 1-Hexanol | 24 | — | 16 | |
| Heptane (C7) | 1-Heptanol | 81 | — | 54 | |
| Octane (C8) | 1-Octanol | 120 | — | 80 | |
| Nonane (C9) | Nonane-1, 9-diol | 113 | — | 75 | |
| Decane (C10) | Decane-1, 10-diol | 65 | — | 43 | |
| CYP153A6[29b] | Hexane (C6) | 1-Pentanol | — | 0.74 | — |
| Heptane (C7) | 1-Hexanol | — | 21.2 | — | |
| Octane (C8) | 1-Heptanol | — | 60.8 | — | |
| Nonane (C9) | 1-Octanol | — | 49.7 | — | |
| Decane (C10) | 1-Nonanol | — | 13.8 | — | |
| Undecane (C11) | 1-Undecanol | — | 0.4 | — | |
| 2-Methyloctane (C9) | 2-Methyl-1-octanol | — | 35 | — | |
| Limonene (C10) | Perillyl alcohol | — | 31.2 | — | |
| p-Cymene (C10) | p-Cumic alcohol | — | 38.9 | — | |
| CYP153A13[29c] | Octane (C8) | 1-Octanol | 116 | — | 233 |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. d Regioselectivity. | |||||
为解决CYP153纯酶体系催化活力低的问题, Lee等通过构建共表达CYP153AM.aq基因的全细胞工程菌株, 成功实现月桂烷(C12)到1-月桂醇(1.5 g/L)、肉豆蔻烷(C14)到1-肉豆蔻醇(2.0 g/L)、以及月桂烷的双羟化到1, 12-月桂二醇(3.76 g/L)的克级制备转化[30a]. Smit等则通过添加细胞破碎粗提物稳定P450蛋白、优化P450酶与还原伴侣蛋白比率、辅以合适的脱氢酶催化辅因子再生等策略, 将CYP153A6催化辛烷生成1-辛醇的TTN提高到制备量级的45828[30b].
鉴于CYP153酶的底物集中在中、短链烷烃范围, 有研究指出P450很可能以一种协同作用的模式与烷烃羟化酶AlkW1一起在烷烃降解菌中分别完成对不同链长烷烃的氧化降解[31].这也意味着有可能以CYP153基因作为亲本开发气态烷烃小分子选择性羟化酶, 从而为烷烃选择性羟化中最大难题之一的甲烷选择羟化提供可选方案.
在真菌中发现的CYP52家族P450酶, 特别是来源于Candida maltosa、Candida tropicalis和Candida apicola的CYP52酶, 主要羟化中长直链烷烃(C12~C18)的末端甲基(表 2, 图 3).其中典型的例子包括来自Candida maltosa的CYP52A3, CYP52A4, CYP52A5, CYP52A9[32], 以及来源于酿酒酵母的CYP52A58[33], 这些酶将正十二烷(C12)和正十六烷(C16)氧化为主要产品1-十二醇和1-十六醇, 但也产生少量的十二/十六烷醛、十二/十六烷酸、α, ω-十二/十六烷二醇、ω-羟基十二/十六烷酸和α, ω-十六烷二酸等过度氧化副产物.这说明一些CYP52家族酶的初级氧化产物如烷基醇、烷基醛和烷基酸也可以作为它们的底物[32].从Candida tropicalis ATCC 20336中克隆的5个CYP52基因(CYP52A12, CYP52A13, CYP52A14, CYP52A17, CYP52A18)即在正十八烷烃的两个末端碳上连续发生氧化, 得到α, ω-十八烷二元羧酸作为主要产物[34].有的CYP52家族酶能以脂肪酸作为天然底物, 比如来自于Candida bombicola ATCC 22214的CYP52E3, CYP52M1和CYP52N1将脂肪酸(C16~C18)氧化为二元羧酸, 这可能是生物合成槐糖脂(一种多用途生物表面活性剂)的关键步骤[35].尽管很多研究都证实多种来源的CYP52酶能氧化中长链烷烃的ω-位, 但对其酶学性质的研究并没有像CYP153那样深入, 很少报道相应的酶催化反应动力学参数[36].值得注意的是, Ciuffetti等报道来自丝状真菌Graphium sp. ATCC 58400的CYP52L1可以氧化丙烷[37], 这在P450超级家族中极为少见, 可能是迄今为止唯一使用气态烷烃作为天然底物的P450酶的例子.基于CYP52酶在烷烃和脂肪酸ω-位的选择氧化优势开发全细胞催化剂, 生产高价值的羟基脂肪酸或二元羧酸, 在生物合成技术领域已开始受到更多关注[38].
下载:
导出CSV
| Enzyme | Alkanes | Final Product | PFRb | TTN c |
| CYP52A3, A4, A5, A9[32] | n-Dodecane (C12) | 1-Dodecanol | 3~44 | 6~88 |
| n-Hexadecane (C16) | 1-Hexadecanol | 12~49 | 24~98 | |
| CYP52A58[33] | n-Hexadecane (C16) | 1-Hexadecanol | 0.6 | — |
| CYP52A12, A13, A14, A17, A18[34] | n-Octadecane (C18) | α, ω-Dicarboxylic acid | — | — |
| CYP52L1[37] | Propane | 1-Propanol | — | — |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. | ||||
除了CYP153和CYP52家族之外, 还有来自不同家族的P450酶个体也能催化烷烃选择性羟化, 且底物范围更宽, 反应位点更多样化(表 3, 图 3).
下载:
导出CSV
| Enzyme | Alkanes | Final Product | [Final Product]a | PFRb | TTNc |
| CYP154A8[39a] | Heptane(C7) | (S)-2-Heptanol (84%d) | 1900 | 1.13 | 2800 |
| Octane(C8) | (S)-2-Octanol (91%d) | 2200 | 4.60 | 3200 | |
| Nonane(C9) | (S)-2-Nonanol (84%d) | 3000 | 3.73 | 4400 | |
| Decane(C10) | (S)-2-Decanol (63%d) | 1200 | 0.37 | 1700 | |
| CYP63A2[40] | n-Alkane(C9~C12, C15~C19) | ω-Alkanol (9-30%e) | — | — | — |
| CYP96A15[41a] | n-Alkane (C27, C29, C31) | Secondary alcohol and ketone | — | — | — |
| CYP116B5[42] | C14, C16, C24, C36 | Primary alcohol | — | — | — |
| CYP4B1[43] | Heptane (C7) | 1-Heptanol (96%f) | — | 33 | — |
| Octane (C8) | 1-Octanol (86%f) | — | 26 | — | |
| Nonane (C9) | 1-Nonanol (71%f) | — | 31 | — | |
| Decane (C10) | 1-Decanol (62%f) | — | 11 | — | |
| CYP505E3[44a] | Decane (C10) | 7-Decanol (71%f) | — | — | 8013 |
| Dodecane (C12) | 7-Dodecanol (71%f) | — | — | 1969 | |
| Tetradecane (C14) | 7-Tetradecanol (74%f) | — | — | 138 | |
| Hexadecane (C16) | 7-Hexadecanol (100%f) | — | — | 138 | |
| CYP124[45a] | Phytane & Pristane | ω-Oxidation | — | — | — |
| CYPCHX[45b] | Cyclopentane | Cyclopentanol | 10239 | — | — |
| Cyclohexane | Cyclohexanol | 9834 | — | — | |
| Cycloheptane | Cycloheptanol | 1933 | — | — | |
| Cyclooctane | Cyclooctanol | 3069 | — | — | |
| CYP101D2[47] | Camphor | 5-exo-Hydroxycamphor (99%f) | — | 2008i (99%g) | — |
| Adamantane | 1-Adamantanol (98%f) | — | 84.1i (62%g) | — | |
| Cyclooctane | Cyclooctanol (99%h) | — | 167i (75%g) | — | |
| Hexane | 2-OH/3-OH (56%:44%f) | — | 3.0i (9%g) | — | |
| 2-Methylpentane | 2-methyl-2-pentanol (55%f) | — | 14.7i (28%g) | — | |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. d ee: enantiomeric excess percent. e Alkanes oxidation ratio. f Regioselectivity. g NADPH coupling efficiency. h Chemoselectivity, cyclooctenone (1%). i PFR in nmol·(nmol P450)-1· min-1. | |||||
线性烷烃仍然是这些P450酶的主要天然底物.来源于Nocardia farcinica IFM 10152的CYP154A8能够催化C7~C12正烷烃亚末端(ω-1位)羟化, 生成高度区域选择性(>90%)和立体选择性(up to 91% ee)的(S)-2-醇[39a]. Urlacher小组将CYP154A8与合适的还原伴侣(redox partner)分别整合进重组的大肠杆菌(Escherichia coli)和恶臭假单胞菌(Pseudomonas putida)全细胞系统中, 发现均产生(S)-2-辛醇.溶剂耐受性更好的后者产醇浓度达到16 mmol·L-1, ee值为87%.向反应体系中添加醇脱氢酶后, (S)-2-辛醇的ee值提升至99%[39b].来自促木质素分解白腐真菌Phanerochaete chrysosporium的CYP63A2酶, 分别氧化烷基酚的正烷基侧链(C3~C9)和中、长链烷烃(C9~C12和C15~C19)生成ω-羟化产物, 氧化转化率为9%~30%, 并且也能催化多环芳烃氧化[40]. CYP96A15是合成植物表皮细胞角质层腊的关键酶, 研究揭示其催化中链烷烃羟化生成2-醇和2-酮[41a].水稻中WSL5基因编码的CYP96B5酶在稻叶角质层腊的合成过程中氧化长链烷烃(C29)生成伯醇[41b].来自Acinetobacter radioresistens S13的CYP116B5酶能够氧化链长为C14、C16、C24及C36等烷烃生成伯醇[42].哺乳动物来源的CYP4B1酶则能氧化中等链长的烷烃(C7~C10)得到ω-和ω-1位羟化产物[43].
与绝大多数的P450选择性羟化碳链末端或亚末端不同, 来自土曲霉Aspergillus terreus的CYP505E3被证明能够催化中链烷烃(C10, C12, C14, C16)的ω-7位羟化(表 3, 图 3).反应区域选择性大约70%, 对正十六烷高达100%, 并且最高的反应TTN达到8000 (C10)[44a]. CYP505E3是第一个高选择性氧化烷烃碳链中间位置的P450酶.据报道只有极少数的P450脂肪酸羟化酶能够实现类似位置的氧化, 来自Tepidiphilus thermophiles的CYP116B46氧化正癸酸和月桂酸的ω-5位羟化[44b]; 来自白腐真菌Phanerochaete chrysosporium的CYP505D6氧化月桂醇和月桂酸以ω-2和ω-3位羟化产物为主, 同时得到ω-1, ω-4到ω-7位的羟化副产物[44c].一般来说, 这种碳链中间位置的区域选择性羟化很难通过化学方法学来实现, 是生物催化剂P450酶的独特优势, 表明其在合成应用方面有巨大潜力.
来源于Mycobacterium tuberculosis的CYP124氧化植烷和降植烷的ω-位支链甲基, 选择性产生伯醇[45a].来自Acidovorax sp. CHX100的CYPCHX酶能氧化环烷烃(C5~C8)生成对应的醇[45b].来自Labrenzia aggregata IAM 12614的CYP116B4(P450LaMO)的底物谱极为广泛, 不但催化烷基苯侧链的不对称羟化[1-苯基乙醇化学选择性82%, (S)-构型对映选择性99%], 而且催化杂芳香醚的脱甲基反应和(杂)芳香硫醚的亚磺化反应[46a].来自Deinococcus apachensis的CYP102D2显示出与P450LaMO互补的高度对映选择性的苄基型羟化活性[(R)-1-苯基乙醇的ee值86%] [46b].来自Novosphingo-bium aromaticivorans的CYP101D2与P450cam一样均对莰酮(camphor)具有高度底物专一性, 5-exo-羟基莰酮的化学选择性高达99%, 产物生成速率(product formation rate: PFR)为2008 nmol·(nmol P450)-1·min-1, 与还原辅酶NADPH的耦合效率达到99%[47]. CYP101D2也能氧化金刚烷、环辛烷、己烷和2-甲基戊烷生成相应的醇, 不过PFR和NADPH耦合效率明显较低.
随着P450酶的基因发掘、功能结构鉴定的不断发展, 天然P450酶底物谱和氧化位点的范围不断扩大, 在烷烃选择性羟化反应方面显示出巨大应用潜力.同时对于工业量级的应用来说, 大多数天然P450仍然存在着反应速率慢和转化效率低等瓶颈问题.并且天然P450的底物谱也依然有一定限制, 基本不能氧化气态烷烃小分子(C1~C4).因此对天然P450烷烃羟化酶的工程化改造是十分必要的.
利用典型的大肠杆菌基因表达系统, 较易获得可溶性的细菌来源P450酶.因其本身即具有很高的催化活性, 细菌P450酶被广泛用于蛋白质分子工程研究以开发生物氧化催化剂[48].针对烷烃羟化进行工程化改造的P450酶也主要来源于细菌, 两个著名的例子分别是基于P450cam的理性设计和由来自Bacillus megaterium的CYP102A1(P450BM3)出发的定向进化策略.
理想中酶的理性设计是指基于蛋白质分子的三维结构信息和催化机理, 综合运用理论计算和分子动力学模拟等方法来指导酶分子的改造, 获得具有预期反应活性或选择性的突变酶.尽管酶分子在催化过程中有着相当复杂的动态特性, 但通过在计算辅助下确定关键的催化位点, 进而通过模拟考察氨基酸突变的影响, 可以为后续的实验研究提供理论支持.在此过程中结合对酶特定反应机制的理解, 往往收到事半功倍的效果.
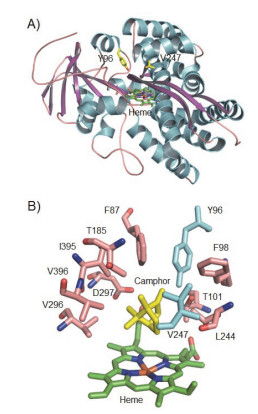
Wong小组经过一系列的烷烃底物适应性改造, 从P450cam出发, 成功构建能够羟化乙烷的P450cam突变酶, 这一研究过程堪称理性设计小分子烷烃羟化P450酶的经典案例(表 4). P450cam是较早开始即被广泛研究的P450模式酶之一[49], 对莰酮的底物特异性极强, 也能氧化大量相关化合物, 但对简单烷烃活性较差. Wong等分析了P450cam的高分辨率晶体结构(图 4)[20a], 认为将位于P450cam活性中心附近、带有亲水性侧链的酪氨酸Y96突变为疏水性的丙氨酸(Y96A)和苯丙氨酸(Y96F), 将能提高底物口袋的疏水性, 改善酶结合疏水性烷烃分子的能力, 进而提高氧化活性.结果完全验证了这一设想:突变体Y96A和Y96F氧化线性烷烃(正戊烷、正己烷和正庚烷)的PFR比野生型P450cam提高100倍以上, 但对支链烷烃(2-甲基戊烷、3-甲基戊烷和2-甲基己烷)提高较小(3~10倍)[50].
下载:
导出CSV
| P450cam enzyme | PFRb of the yielded alcoholsc (CE%d) | ||||
| n-Hexane | 3-Methylpentane | n-Butane | Propane | Ethane | |
| Wild-Type | 0.4 (2.0) | 27.7 (21.3) | 0.4 (0.5) | 0.5 (0.5) | — |
| Y96A | 89.3 (23.0) | 99.1 (52.0) | — | — | — |
| Y96F | 109.8 (47.0) | 122.6 (54.7) | 42 (42) | 2.2 (12) | — |
| Y96A/V274A | 122.2 (30.2) | 199.0 (63.6) | — | — | — |
| Y96F/V274L | 176.1 (66.8) | 42.3 (50.6) | 186 (75) | 43 (39) | — |
| F87W/Y96F/T101L/V247L | — | — | 750 (95) | 110 (32) | — |
| F87W/Y96F/T101L/V247L/L244M (EB) | — | — | 520 (90) | 176 (66.2) | — |
| EB/L294M/T185M/L1358P/G248A | — | — | — | 505 (85.6) | 78.2 (10.5) |
| a Summarized result for P450cam-catalyzed hydroxylation of alkanes according to references 26, 50~53. b PFR: product formation rate in nmol·(nmol P450)-1· min-1. c The yielded alcohols are 2-hexanol and 3-hexanol for n-hexane, 3-methyl-2-pentanol and 3-methyl-3-pentanol for 3-methyl-pentane, 2-butanol for n-butane, 2-propanol for propane, and ethanol for ethane. d CE: Coupling efficiency of NADPH, CE%=PFR/(NADPH consuming rate)×100%. | |||||
随后Wong等采用了调控酶底物口袋大小策略, 即通过调节关键位点氨基酸残基的大小以适应体积不同的烷烃底物.底物口袋变大的突变体Y96A/V247A对较大体积的3-甲基戊烷活性增强, 而底物口袋减小的突变体Y97F/V247L更适合氧化小体积的正己烷[51].基于这一设计思路, 研究人员在Y97F/V247L突变体基础上, 向底物口袋周边连续引入较大的氨基酸残基, 不断缩小底物口袋容积以适应更小的气态烷烃分子, 先后得到适合正丁烷的F87W/Y96F/T101L/V247L四点突变体[52]、更适合丙烷的F87W/Y96F/T101L/V247L/ L244M(EB)[53]、能氧化乙烷的EB/L294M/T185M/ L1358P/G248A, 首次实现了P450酶对乙烷的选择性羟化[26].晶体学研究显示P450cam突变酶活性中心附近仅结合3个水分子, 比野生型减少一半[54].这表明缩小的底物口袋疏水性增强, 有利于疏水性烷烃小分子进入到P450酶的底物口袋, 促进了氧化反应进行.
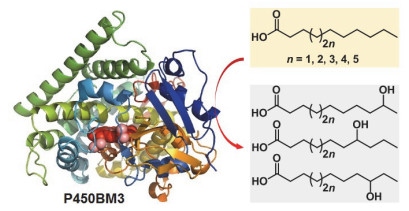
P450BM3是一个被广泛研究的P450模式酶, 也是已知催化效率最高的P450酶之一[25], 主要氧化脂肪酸(C12~C20), 同样链长的脂肪酰胺、脂肪醇和脂肪酸酯等的亚末端位置(ω-1, ω-2, ω-3)到相应的醇, 但不能氧化相应链长(C12~C20)的线性烷烃[55](图 5). Adam等报道P450BM3能氧化较短链烷烃(C6~C9)和环烷烃(C6和C8), 并且产物对映选择性高达99%[56]. Schmid等基于三维晶体结构分析, 构建了P450BM3的突变体F87V/L188Q/ A74G, 氧化辛烷得到2-、3-、4-位取代辛醇, NADPH消耗速率是野生型的700倍[57], 表明通过对活性口袋关键氨基酸突变能够改变P450BM3的催化活性和选择性.需要指出的是, 虽然NADPH的消耗速率可在一定程度上体现P450酶的催化活力, 考虑到催化过程中可能发生的各种NADPH非耦合反应(图 1), 目前在P450酶的工程化改造研究更多地采用NADPH耦合效率作为一个关键参数, 来评价P450突变酶的活性(表 4, 表 5)[26, 27, 50~53, 58~60].这显然要比单纯依靠NADPH消耗速率的评价更为准确和全面.
 下载:
导出CSV
下载:
导出CSV
| P450BM3 variants | Ethane | Propane | n-Octane | ||||||||
| Rateb | TTNc | CE%d | Rate | TTN | CE% | Rate | TTN | CE% | |||
| 139-3 | — | — | — | 12 | 500 | 12 | 480 | 1000 | 22 | ||
| J | — | — | — | 30 | 800 | 5 | 660 | 3000 | 23 | ||
| 9-10A | — | — | — | 23 | 1100 | 5 | 540 | 3000 | 21 | ||
| 1-12G | — | — | — | 160 | 6000 | 100 | 150 | 7500 | 37 | ||
| 53-5H | 0.4 | 50 | 0.06 | 370 | 5000 | — | 660 | 8000 | — | ||
| 35-E11 | 0.4 | 250 | 0.08 | 210 | 5650 | 17.4 | 420 | 8000 | — | ||
| P450PMOR1 | — | — | — | 455 | 35600 | 94.4 | — | — | — | ||
| P450PMOR2 | — | — | — | 370 | 45800 | 98.2 | — | — | — | ||
酶分子定向进化技术的概念肇始于20世纪90年代初, 主要是在实验室模拟自然进化过程, 通过随机突变和重组, 构建人为突变体文库, 然后利用高通量筛选获取特定功能的目标突变酶.虽然从理论上来说, 定向进化不需要酶结构方面信息, 为任何酶的改造提供了最大可能.但是定向进化的效率仍然受到建立合适的高通量筛选策略的制约, 而结合理性设计能够减少随机突变产生的过高筛选量, 加速实验室酶分子进化速度.
Arnold教授在提出和发展定向进化技术之初即开始以细胞色素P450作为改造的目标酶之一, 而她开发高效P450BM3丙烷单加氧酶的过程可以说是定向进化技术中不同策略的一次集中完美展示(表 5).
Arnold小组首先通过易错PCR构建P450BM3的随机突变文库, 并以硝基苯基辛基醚(8-pnpane)作为正辛烷类似物建立高通量筛选体系, 经过两轮定向进化得到辛烷羟化活性高于野生型5倍的突变酶[61].由于8-pnpane与正辛烷分子在结构和尺寸上存在差异, 导致无法通过增加突变轮次来进一步提高突变酶活性. Arnold等改用了8-pnpane筛选与NADPH消耗速率相结合的筛选方法, 经过五轮进化, 得到包含11个突变位点的139-3突变酶, 改善了C3~C8烷烃的羟化活性[58].如前面讨论中提到的NADPH消耗速率并不能完全等同于烷烃羟化的PFR, 因为不同烷烃与P450BM3的结合能力有差别.作者为此开发了针对丙烷分子、以二甲基醚为模型底物、基于Purpald显色法检测脱甲基产物甲醛的高通量筛选策略.以139-3为亲本, 通过DNA改组和易错PCR结合得到活性继续改善的突变体J和9-10A.继而通过对P450BM3与棕榈酰基甘氨酸底物结合的晶体结构分析[22], 鉴定出两个可能影响底物口袋大小的关键氨基酸残基A328和A82.然后利用定点饱和突变分别得到对区域选择性和反应活性均有显著影响的9-10A_A328V和9-10A_A82L突变体, 再通过DNA改组得到1-12G突变酶, 对丙烷和辛烷的初始反应速率分别达到160 min-1和150 min-1, 2-丙醇和2-辛醇的区域选择性接近90%[59].
从9-10A出发, 在对活性中心周围5 Å以内的11个氨基酸残基进行迭代饱和突变后, 得到含有15个突变位点的53-5H, 对丙烷和乙烷初始反应速率分别达到370 min-1和0.4 min-1, TTN分别达到5000和50.另一个含有17个突变位点的35-E11对丙烷和乙烷初始反应速率分别达到210 min-1和0.4 min-1, TTN分别达到6000和250[60].然后从35-E11出发, 采用基于区域结构的定向进化策略, 对P450BM3的氧化结构域(Heme domain)和还原结构域(FMN domain和FAD domain)分别进行改造, 既优化了与底物结合Heme domain, 也优化了负责电子传输的FMN和FAD domain.得到2个P450BM3丙烷单加氧酶P450PMOR1和P450PMOR2, 丙烷羟化的初始反应速率分别达到455 min-1和370 min-1, TTN高达35600和45800, NADPH耦合效率几乎完美(94.4%和98.2%), 各项参数媲美甚至超过P450BM3对天然底物脂肪酸的氧化[27].同源建模P450PMO的三维结构与野生型P450BM3和突变酶139-3比较, 底物口袋明显缩小, 与丙烷结合的很好.这也从另一方面说明调控底物口袋大小的理性设计方法的可行性.
Urlacher小组考察了P450BM3的单或双位点突变体催化环烷烃(C8~C12)羟化反应, 发现所有突变体对环辛烷都有很好的转化率(52%~90%, 野生型P450BM3为8%)和中等程度的催化效率[62].这说明P450BM3的固有底物口袋相当柔软, 只需要1到2个氨基酸突变就能改变其底物选择性和催化活性.但是转化率和催化效率随环增大下降明显, 可能是由于P450BM3的固有狭长底物口袋不利于接受宽大底物.
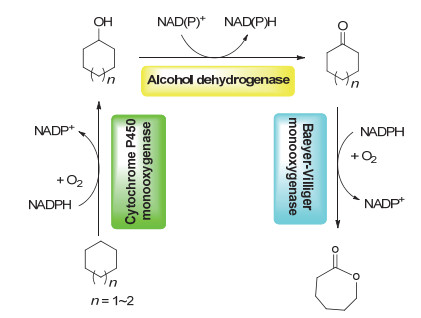
从环烷烃利用氧分子制备环烷酮是一个原子经济性反应, Groger与合作者利用P450BM3突变酶和醇脱氢酶(alcohol dehydrogenase)组合, 巧妙利用催化循环中辅酶NADPH的再生, 成功实现这一转化[63].这一策略也被用于从线性烷烃合成对应的酮, 并同时实现对映体醇产物的酶学拆分[64]. Opperman在上述反应体系中引入拜尔-维力格单加氧酶(Baeyer-Villiger monooxygenase), 实现了多酶催化环烷烃(C6~C8)到环状内酯的三步串联合成(图 6)[65].
控制C—H键羟化反应的区域选择性和立体选择性是合成有机化学的巨大挑战[66]. Reetz小组报道了野生型P450BM3及其突变体催化单取代环己烷羟化同时生成两个手性中心[67].野生型酶给出了78%的β位区域选择性, 主产物对映异构体选择性达到97%, 而突变体F87A和A328F分别给出α位(71%)和β位(71%)产物.二取代环己烷反应同时给出3个手性中心, 但仅有A328F突变体的选择性比野生型酶好.
来自Sphingomonas sp. HXN-200的P450pyr能够羟化很多疏水性底物, 氧化正辛烷高选择性生成正辛醇[68~70]. Li小组从距离P450pyr酶活性中心/底物/底物通道6 Å范围的22个关键氨基酸残基开始, 采用迭代饱和突变策略(Iterative saturation mutagenesis, ISM)[71], 历经六轮定向进化, 结合针对目标产物的高通量比色分析法(colorimetric high-throughput screening assay), 筛选到氧化正辛烷到(S)-2-辛醇的高效P450pyrSM1突变酶, 区域选择性大于99%, ee值高达98%[72].这些研究表明, 定向进化和理性设计是优化P450酶的催化活性和选择性的有效策略.
除了强大的酶工程技术, 一些科学家另辟蹊径, 发展了新的方法以拓展P450酶的催化应用空间, 比如Grengl、Arnold、Sherman等小组报道的底物工程(substrate engineering)策略[73~75]、Watanabe小组发展的诱饵分子(decoy molecule)策略[48]、以及我们小组最近提出的双功能小分子协同P450催化(dual-functional small-molecule co-catalysis)策略[76, 77].这些策略的共同特点在于利用酶与天然底物之间的“锁-钥”特别分子识别关系, 通过化学活化方式对P450酶的催化功能进行改造.
对非天然底物的局限性是酶催化应用需要改进的方面之一.一些非天然底物由于存在敏感基团或者缺少与酶具有特异性识别作用的基团而不能被催化.底物工程策略针对这些非天然底物, 采用有机合成反应中常用的导入保护基或定位基的方法, 利用酶分子对天然底物分子中特定官能团的有效识别, 在非天然底物分子结构中引入相同或近似的官能团作为定位基团(也称锚定基团)或保护基团, 建立或者增强这些非天然底物与酶分子之间的特异性相互作用, 实现对该反应底物特定位置的选择性氧化.反应结束后再脱除定位基团或锚定基团, 从而获得目标产物(图 7).底物工程策略已在P450酶催化非天然底物氧化反应中得到大量应用[73~75, 77], 然而由于烷烃分子的结构特点难以引入上述特定功能基团, 该策略并不适用于P450酶催化的烷烃羟化反应.
诱饵分子策略同样借鉴了酶对底物的特异性识别能力, 并利用了P450酶的由底物结合触发的独特催化反应机制.如图 8(上)所示, 天然底物与P450酶的特异性识别和结合是诱导P450产生反应活性物种Compound I的第一步, 其相当于P450酶的“开关”.在大多数情况下, 直接加入非天然底物无法起到“开关”作用. Watanabe等利用由与天然底物相同或者相似特征官能基团修饰的诱饵分子, 充当引发P450酶催化循环的“开关”, 诱使P450生成Compound I, 从而实现对体系中共存的非天然底物的氧化[图 8(下)].诱饵分子策略对无法引入功能性官能团的底物尤其有效, Watanabe及其合作者利用开发的不同类型诱饵分子, 比如短链脂肪酸、全氟化脂肪酸及其酰基氨基酸衍生物、脂肪酰基氨基酸衍生物等, 在诸如苯乙烯、苯及其衍生物以及小分子烷烃等非天然底物氧化方面开展了大量工作[48, 77~79].诱饵分子策略也被应用到P450之外的其他酶催化非天然底物反应中:比如以各种链长烷烃作为诱饵分子, 激活光脱羧酶CvFAP催化的非天然脂肪酸底物的脱羧反应以制取烷烃[80]; 以庚酸等短链羧酸作为诱饵分子, 活化脂肪酸水合酶催化非天然底物末端烯烃的不对称水合反应[81].这些研究显示了诱饵分子策略拓展酶底物多样性(substrate promiscuity)方面的广泛应用潜力.
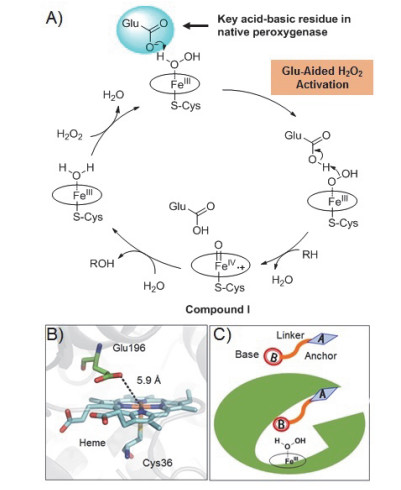
双功能小分子协同P450催化策略利用了天然P450酶催化循环中的H2O2旁路途径(图 1), 在利用酶与底物形成的特异性识别基础上引入协同催化概念, 模拟天然过氧化物酶(Peroxidase)和天然过加氧酶(Peroxygenase)的活性位点与催化机制[82], 利用一端带有与P450酶分子产生特异性识别作用的锚定基团、另一端带有可以协同活化H2O2的酸碱催化基团的双功能小分子, 将以氧分子作为末端氧化剂的NADPH依赖型P450BM3单加氧酶复杂系统, 改造为以H2O2为末端氧化剂的P450过加氧酶简单体系.作为绿色经济的氧化剂, H2O2广泛用于有机合成和化工领域的催化氧化.在P450酶催化中使用H2O2替代O2, 不需要辅酶NAD(P)H及其复杂的还原伴侣电子传递体系, 大大简化了P450酶的催化循环, 有望提高其实际催化应用潜力(图 9)[83].近来, 我们课题组证明了这种人工P450过加氧酶可用于一系列P450非天然底物的氧化反应, 比如烯烃环氧化、硫醚亚磺化、烷烃羟基化、芳烃羟基化和芳香醚脱甲基化[76, 84, 85]等, 表现出独特的催化性质.
Watanabe小组和Reetz小组分别独立报道了利用全氟化脂肪酸作为诱饵分子活化P450BM3酶, 氧化各种小分子烷烃到相应的醇[86, 87].在Watanabe等报道的丙烷和丁烷羟化反应体系中, 在十九氟正癸烷酸(PFC10)和十七氟正壬烷酸(PFC9)存在下, 2-丙醇和2-丁醇的PFR分别达到70和110 μmol·(μmol P450)-1·min-1, 2-丙醇与1-丙醇的产物物质的量比为10:1[86]. Reetz小组报道的反应体系催化活性看起来更好, 甚至能够氧化甲烷[87a].作者后来证明虽然该体系确实能羟化丙烷, 但在甲烷氧化反应中检测到的甲醇可能来自气相色谱的噪声信号[87b]. Watanabe等证明在高压反应条件下P450BM3-PFC10诱饵分子体系能氧化乙烷到乙醇, 但PFR较低[0.67 μmol·(μmol P450)-1·min-1] [88]. Ariyasu等通过开发新的高压反应装置提高了小分子烷烃氧化反应的转化效率[89]. Kawakami等证明在0 ℃的低温反应条件下, 丁烷氧化反应的NADPH耦合效率达到96%[90]. Cong等通过全氟化脂肪酸与天然氨基酸的酰基化反应进一步优化了诱饵分子结构[79].全氟化脂肪酰基氨基酸衍生物作为第二代诱饵分子与P450BM3的结合能力提高了一个数量级, 同时增强了氧化小分子烷烃的能力.在最好的诱饵分子PFC9-L-Leu(十七氟正壬烷酰胺基亮氨酸)存在下, 2-丙醇的PFR提高4倍达到256 μmol·(μmol P450)-1·min-1, 接近P450PMO的水平[27](370 min-1).结合葡萄糖脱氢酶促进的NADPH再生系统, 实现了常温常压下乙烷的羟化反应[45 μmol·(μmol P450)-1·min-1], PFR接近P450cam突变体[78 nmol· (nmol P450)-1·min-1][26](表 6).
下载:
导出CSV
| Decoy molecule | Alkanes | Final product | PFRb | TTNc | Reference |
| PFC10 | Propane | 2-Propanol | 70 | 700 | [86] |
| PFC9 | n-Butane | 2-Butanol | 110 | 1100 | [86] |
| PFC9 | Cyclohexane | Cyclohexanol | 112 | 1120 | [86] |
| PFC10d | Cyclohexane | Cyclohexanol | 484 | — | [91] |
| PFC10e | Methane | Methanol | — | 2472 | [87] |
| PFC11 | Propane | 2-Propanol | — | 1021 | [87] |
| PFC7 | n-Butane | 2-Butanol | — | 3632 | [87] |
| PFC11 | n-Hexane | 2-/3-Hexanol=77:23g | — | 525 | [87] |
| PFC9 | n-Octane | 2-/3-/4-Octanol=10:42:48g | — | 1184 | [87] |
| PFC10f | Ethane | Ethanol | 0.67 | — | [88] |
| PFC10f | Propane | 2-Propanol | 72 | — | [88] |
| PFC10f | Propane | 1-Propanol | 1.7 | — | [88] |
| PFC9-L-Leu | Ethane | Ethanol | 45 | — | [79] |
| PFC9-L-Leu | Propane | 2-Propanol | 256 | — | [79] |
| a Wild type P450BM3 enzyme is used unless otherwise stated. b PFR: Product formation rates in μmol·(μmol P450)-1·min-1. c TTN: Total turnover numbers. d A P450BM3 mutant KT2 is used. e The authors made a corrigendum about the system being unavailable for methane oxidation but available for other alkanes hydroxylation, see ref. 87b. f The reactions are carried out under high pressure conditions. g Molar ratio of alcohol products. | |||||
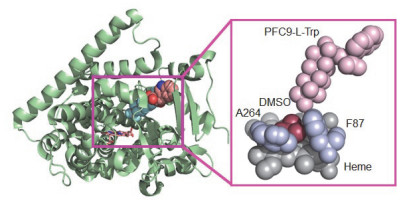
得益于第二代诱饵分子与P450BM3的强力结合, Cong等首次成功制备了P450BM3结合诱饵分子(PFC9-L-Trp:十七氟正壬烷酰胺基色氨酸)的高分辨率共结晶晶体结构(图 10左), 建立了诱饵分子活化P450机制的分子基础[79].晶体结构清楚地表明诱饵分子重构了P450的底物口袋, 而轴向配位的DMSO分子由于同丙烷分子的大小非常接近, 暗示重构后的底物口袋与丙烷分子的适配性很好(图 10右), 这与该反应体系中丙烷氧化活性最好的实验结果是一致的.另外, 对P450酶在诱饵分子存在下的紫外吸收光谱研究表明, 当诱饵分子与P450结合时, 与血红素活性中心铁离子轴向配位的水分子向远端移动, 导致中心三价铁从低自旋变成高自旋态, 能够接受1个电子被还原到二价铁, 从而引发活性物种compound I的生成[86].综合晶体学和催化反应动力学研究结果表明, 诱饵分子通过与P450酶的结合, 引发了催化循环, 同时改造了底物口袋使P450能更好结合小分子底物, 促进了小分子烷烃羟化反应发生.
Bell等将诱饵分子策略与酶工程技术结合, 发现速率增强的P450BM3变异体KT2在诱饵分子PFC9存在下, 环己醇的PFR增强7倍, 组合催化效果比野生型提高700倍[91].诱饵分子不但加快PFR, 而且影响反应的立体选择性, 但一般不影响反应的区域选择性.这些研究为P450酶催化烷烃羟化反应提供了新的策略.甲烷直接氧化制甲醇, 是烷烃氧化的终极挑战之一[18], 诱饵分子策略与酶工程技术的结合, 有可能为实现这一目标提供新的工具.
尽管H2O2在生物酶模拟体系中经常被用做催化烷烃氧化反应的末端氧化剂, 然而在天然酶催化的同样反应中却很少见.目前已知唯一能够利用H2O2氧化小分子烷烃(C3~C6)的天然酶是来自Agrocybe aegerita的过加氧酶AaeUPO [92].我们小组将双功能小分子协同P450催化策略与酶工程技术结合, 首次成功获得了对丙烷等小分子烷烃(C3~C6)具有高羟化活性和选择性的工程P450过加氧酶[28].从P450BM3出发, 通过对影响酶与H2O2以及烷烃底物结合的F87位点、对C—H键羟化具有潜在影响的T268位点、以及对底物口袋大小具有明显调节作用的A82和A184位点的循序叠加突变, 建立了一个小容量的突变体文库.然后在双功能小分子Im-C6-Phe(6-咪唑基己酰胺基苯丙氨酸)存在下, 筛选小分子烷烃(C3~C6)羟化的有益突变体.双突变体F87A/T268I与F87V/T268I的2-丙醇PFR分别达到635和499 μmol·(μmol P450)-1·min-1, 与Arnold小组报道的NADPH依赖型P450工程酶高效催化体系相当[27].催化反应TTN达到1582和1264, 接近天然过加氧酶AaeUPO的两倍[92].然而三突变体F87A/T268I/184V和F87A/T268I/184I仅对催化活性有轻微提升.同时在不添加双功能小分子的对照实验中, 所有突变体均无明显活性, 表明双功能小分子对催化起到决定性作用.另外, 突变体F87A/T268I/A82T的2-丁醇PFR达到1042 μmol·(μmol P450)-1·min-1, 远高于天然丁烷单加氧酶(BMO)[93], 优于NADPH依赖的P450cam工程酶[52]和天然过加氧酶AaeUPO[92], 最优突变体F87A/T268I/A184I的TTN达到2289.该催化系统对正戊烷和正己烷同样显示优异羟化活性(表 7).这项研究为开发小分子烷烃选择性羟化P450酶提供了新的途径和思路.
下载:
导出CSV
| P450BM3 variants | 2-Propanol | 2-Butanol | 1-/2-/3-Pentanol | 2-Hexanol | |||||||
| PFRc | TONd | PFRc | TONd | PFRc | TONd | PFRc | TONd | ||||
| F87A/T268I | 635 | 1582 | 902 | 1882 | 318 | 1056 | 297 | 775 | |||
| F87V/T268I | 499 | 1264 | 913 | 2009 | 233 | 1092 | 171 | 370 | |||
| F87A/T268I/A82T | 473 | 1353 | 1042 | 2253 | 442 | 1341 | 464 | 1003 | |||
| F87A/T268I/A184V | 619 | 1626 | 992 | 2262 | 391 | 1319 | 450 | 1203 | |||
| F87A/T268I/A184I | 630 | 1775 | 979 | 2289 | 383 | 1463 | 439 | 1286 | |||
| F87A/T268I/A82T/A184V | 481 | 1246 | — | — | 516 | 1640 | 600 | 1322 | |||
| a Reaction conditions: P450BM3 (0.5 μmol·L-1), H2O2 (60 mmol·L-1), Im-C6-Phe (500 μmol·L-1), in pH 8.0 phosphate buffer at 25 ℃. b All the control reactions did not show obvious activity of hydroxylation in the absence of Im-C6-Phe. c PFR: Product formation rates were estimated as μmol·(μmol P450)-1·min-1 over a 1-min reaction. d TON: Turnover numbers were estimated over a 30-min reaction. | |||||||||||
总之, P450酶通过遗传进化的不同亚型, 可以较好应对烷烃羟化中的几个主要难题, 即化学选择性(易发生连续过度氧化)、区域选择性和立体选择性.同时, 蛋白质分子工程等分子生物技术和化学活化策略的发展, 为发展人工P450烷烃羟化酶体系提供了更多工具.预示着细胞色素P450酶在开发新的烷烃选择性氧化体系方面的巨大潜力.当前也面临着以下一些挑战: (1)如何改造P450酶以期最终实现高难度的甲烷羟化:已知唯一例子是以PhIO作为末端氧化剂用高浓度的CYP153A6 (50~250 μmol·L-1)进行化学计量反应, 但TTN仅为0.02~0.05[94]. (2)如何提高P450酶的催化活性以适应工业生产的要求. (3)如何解决H2O2存在下P450酶的稳定性.第三个问题又和第二个问题有直接的联系.而P450甲烷羟化酶的开发, 一定程度上要依赖于这三个问题的解决.
有观点认为“利用生物氧化原理实现烃类化合物羟化理论上是可行的, 但用于大宗化学品的生产是有困难的”[95], 对此我们应该看到, 科学家们正通过不同的途径在努力解决这些问题.如通过酶的固定化[96]和生物矿化[97]技术来改善其稳定性; 通过蛋白质工程技术和化学活化策略来增强其催化活性; 通过建立通用性的还原伴侣[98]或开发辅酶NADPH再生系统[99]来完善其电子传递催化体系; 以及通过开发化学能如H2O2驱动的P450过加氧酶来解除P450酶对还原辅酶的依赖.另外, 多相微反应器的发展也为生物催化在实验室和工业级应用之间搭起了一个桥梁[100].我们相信随着化学、生物以及生物工程等技术的发展, 酶催化终将为烷烃的选择性羟化提供一个条件温和且环境友好的策略.
Luo, Y. R. Handbook of Bond Dissociation Energies in Organic Compounds, Routledge, New York, 2003.
Hashiguchi, B. G.; Konnick, M. M.; Bischof, S. M.; Gustafson, S. J.; Devarajan, D.; Gunsalus, N.; Daniel H.; Ess, D. H.; Periana, R. A. Science 2014, 343, 1232. doi: 10.1126/science.1249357
Soussan, L.; Pen, N.; Belleville, M. P.; Marcano, J. S.; Jeanjean, D. P. J. Biotechnol. 2016, 222, 117. doi: 10.1016/j.jbiotec.2016.02.007
Sirajuddin, S.; Rosenzweig, A. C. Biochemistry 2015, 54, 2283. doi: 10.1021/acs.biochem.5b00198
Cahalan, E.; Ernfors, M.; Müller, C.; Devaney, D.; Laughlin, R. J.; Watson, C. J.; Hennessy, D.; Grant, J.; Khalil, M. I.; McGeough, K. L.; Richards, K. G. Agric. Ecosyst. Environ. 2015, 199, 339. doi: 10.1016/j.agee.2014.09.008
Van Beilen, J. B.; Wubbolts, M. G.; Witholt, B. Biodegradation 1994, 5, 161. doi: 10.1007/BF00696457
Ortiz de Montellano, P. R. Cytochrome P450: Structure, Mechanism, and Biochemistry, Routledge, New York, 2005.
(a) Urlacher, V. B.; Girhard, M. Trends Biotechnol. 2019, 37, 882. (b) Wei, Y.; Ang, E. L.; Zhao, H. Curr. Opin. Chem. Biol. 2018, 43, 1. (c) Jiang, Y.; Li, S. Chin. J. Org. Chem. 2018, 38, 2307 (in Chinese). (蒋媛媛, 李盛英, 有机化学, 2018, 38, 2307). (d) Cheng, Z.; Chen, P.; Liu, G. Acta Chim. Sinica 2019, 77, 856 (in Chinese). (成忠明, 陈品红, 刘国生, 化学学报, 2019, 77, 856). (e) Liu, Q.; Zhou, D.; Li, Z.; Luo, W.; Guo, C. Chin. J. Chem. 2017, 35, 1063. (f) He, X.; Chen, L.; He, Q.; Xiao, H.; Zhou, X.; Ji, H. Chin. J. Chem. 2017, 35, 693. (g) Lu, W.; Chen, X.; Feng, J.; Bao, Y.-J.; Wang, Y.; Wu, Q.; Zhu, D. Appl. Environ. Microbiol. 2018, 84, e00503-18.
Paddon, C. J.; Westfall, P. J.; Pitera, D. J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M. D.; Tai, A.; Main, A.; Eng, D.; Polichuk, D. R.; Teoh, K. H.; Reed, D. W.; Treynor, T.; Lenihan, J.; Fleck, M.; Bajad, S.; Dang, G.; Dengrove, D.; Diola, D.; Dorin, G.; Ellens, K. W.; Fickes, S.; Galazzo, J.; Gaucher, S. P.; Geistlinger, T.; Henry, R.; Hepp, M.; Horning, T.; Iqbal, T.; Jiang, H.; Kizer, L.; Lieu, B.; Melis, D.; Moss, N.; Regentin, R.; Secrest, S.; Tsuruta, H.; Vazquez, R.; Westblade, L. F.; Xu, L.; Yu, M.; Zhang, Y.; Zhao, L.; Lievense, J.; Covello, P. S.; Keasling, J. D.; Reiling, K. K.; Renninger, N. S.; Newman, J. D. Nature 2013, 496, 528. doi: 10.1038/nature12051
(a) Zhou, Q.; Luo, G.; Zhang, H.; Tang, G. Chin. J. Org. Chem. 2019, 39, 1169 (in Chinese). (周强, 罗光彩, 张惠展, 唐功利, 有机化学, 2019, 39, 1169). (b) Bai, X.; Guo, H.; Chen, D.; Yang, Q.; Tao, J.; Liu, W. Org. Chem. Front. 2020, 7, 584. (c) Lv, J.-M.; Hu, D.; Gao, H.; Kushiro, T.; Awakawa, T.; Chen, G.-D.; Wang, C.-H.; Abe, I.; Yao, X.-S. Nat. Commun. 2017, 8, 1644.
(a) Qi, F.; Lei, C.; Li, F.; Zhang, X.; Wang, J.; Zhang, W.; Fan, Z.; Li, W.; Tang, G.; Xiao, Y.; Zhao, G.; Li, S. Nat. Commun. 2018, 9, 2342. (b) Sun, W.; Xue, H.; Liu, H.; Lv, B.; Yu, Y.; Wang, Y.; Huang, M.; Li, C. ACS Catal. 2020, 10, 4253. (c) Tian, X.; Ruana, J.-X.; Huang, J.-Q.; Yang, C.-Q.; Fang, X.; Chen, Z.-W.; Hong, H.; Wang, L.-J.; Mao, Y.-B.; Lu, S.; Zhang, T.-Z.; Chen, X.-Y. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5410. (d) Wang, W.-F.; Xiao, H.; Zhong, J.-J. Biotechnol. Bioeng. 2018, 115, 1842.
Coelho, P. S.; Brustad, E. M.; Kannan, A.; Arnold, F. H. Science 2013, 339, 307. doi: 10.1126/science.1231434
McIntosh, J. A.; Coelho, P. S.; Farwell, C. C.; Wang, Z. J.; Lewis, J. C.; Brown, T. R.; Arnold, F. H. Angew. Chem., Int. Ed. 2013, 52, 9309. doi: 10.1002/anie.201304401
Kan, S.; Huang, X.; Gumulya, Y. Nature 2017, 552, 132. doi: 10.1038/nature24996
Haynes, C. A.; Gonzalez, R. Nat. Chem. Biol. 2014, 10, 331. doi: 10.1038/nchembio.1509
Munz, D.; Strassner, T. Inorg. Chem. 2015, 54, 5043. doi: 10.1021/ic502515x
Bordeaux, M.; Galarneau, A.; Drone, J. Angew. Chem., Int. Ed. 2012, 51, 10712. doi: 10.1002/anie.201203280
Lawton, T. J.; Rosenzweig, A. C. J. Am. Chem. Soc. 2016, 138, 9327. doi: 10.1021/jacs.6b04568
Nelson, D. R. Biochim. Biophys. Acta. Proteins Proteom. 2018, 1866, 141. doi: 10.1016/j.bbapap.2017.05.003
(a) Poulos, T. L.; Finzel, B. C.; Howard, A. J. J. Mol. Biol. 1987, 195, 687. (b) Tripathi, S.; Li, H.; Poulos, T. L. Science 2013, 340, 1227.
Ravichandran, K. G.; Boddupalli, S. S.; Hasemann, C. A.; Peterson, J. A.; Deisenhofer, J. Science 1993, 261, 731. doi: 10.1126/science.8342039
Haines, D. C.; Tomchick, D. R.; Machius, M.; Peterson, J. A. Biochemistry 2001, 40, 13456. doi: 10.1021/bi011197q
(a) Rittle, J.; Green, M. T. Science 2010, 330, 933. (b) Li, X.-X.; Postils, V.; Sun, W.; Faponle, A. S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S. P. Chem. Eur. J. 2017, 23, 6406.
Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A. M.; Maves, S. A.; Benson, D. E.; Sweet, B. M.; Ringe, D.; Petsko, G. A.; Sligar, S. G. Science 2000, 287, 1615. doi: 10.1126/science.287.5458.1615
Whitehouse, C. J.; Bell, S. G.; Wong, L. L. Chem. Soc. Rev. 2012, 41, 1218. doi: 10.1039/C1CS15192D
Xu, F.; Bell, S. G.; Lednik, J.; Insley, A.; Rao, Z.; Wong, L. L. Angew. Chem., Int. Ed. 2005, 44, 4029. doi: 10.1002/anie.200462630
Fasan, R.; Chen, M. M.; Crook, N. C.; Arnold, F. H. Angew. Chem., Int. Ed. 2007, 46, 8414. doi: 10.1002/anie.200702616
Chen, J.; Kong, F.; Ma, N.; Zhao, P.; Liu, C.; Wang, X.; Cong, Z. ACS Catal. 2019, 9, 7350. doi: 10.1021/acscatal.9b02507
(a) Scheps, D.; Malca, S. H.; Hoffmann, H.; Nestl, B. M.; Hauer, B. Org. Biomol. Chem. 2011, 9, 6727. (b) Funhoff, E. G.; Bauer, U.; García-Rubio, I.; Witholt, B.; Van Beilen, J. B. J. Bacteriol. 2006, 5220. (c) Bordeaux, M.; Girval, D.; Rullaud, R.; Subileau, M.; Dubreucq, E.; Drone, J. Appl. Microbiol. Biot. 2014, 98, 6275.
(a) Hsieh, S.-C.; Wang, J.-H.; Lai, Y.-C.; Su, C.-Y.; Lee, K.-T. Appl. Environ. Microbiol. 2018, 84, e01806-17. (b) Kochius, S.; Marwijk, J.; Ebrecht, A. C.; Opperman, D. J.; Smit, M. S. Catalysts 2018, 8, 531.
Nie, Y.; Liang, J.-L.; Fang, H.; Tang, Y.-Q.; Wu, X.-L. Appl. Microbiol. Biotechnol. 2014, 98, 163. doi: 10.1007/s00253-013-4821-1
Zimmer, T.; Ohkuma, M.; Ohta, A.; Takagi, M.; Schunck, W. H. Biochem. Biophys. Res. Commun. 1996, 224, 784. doi: 10.1006/bbrc.1996.1100
Hanano, A.; Shaban, M.; Almousally, I.; Al-Ktaifani, M. Chemosphere 2015, 135, 418. doi: 10.1016/j.chemosphere.2014.11.011
Craft, D. L.; Madduri, K. M.; Eshoo, M.; Wilson, C. R. Appl. Environ. Microbiol. 2003, 5983. https://www.researchgate.net/publication/9060951_Identification_and_characterization_of_the_CYP52_family_of_Candida_tropicalis_ATCC_20336_important_for_the_conversion_of_fatty_acids_and_alkanes_to_alphaomega-dicarboxylic_acids
Van Bogaert, I. N. A.; Demey1, M.; Develter, D.; Soetaert, W.; Vandamme, E. J. FEMS Yeast Res. 2009, 9, 87. doi: 10.1111/j.1567-1364.2008.00454.x
(a) Lida, T.; Sumita, T.; Ohta, A.; Takagi, M. Yeast 2000, 16, 1077. (b) Panwar, S. L.; Krishnamurthy, S.; Gupta, V.; Alarco, A. M.; Raymond, M.; Sanglard, D.; Prasad, R. Yeast 2001, 18, 1117. (c) Carratore, R. D.; Gervasi, P. G.; Contini, M. P.; Beffy, P.; Maserti, B. E.; Giovannetti, G.; Brondolo, A.; Longo, V. Biotechnol. Lett. 2011, 33, 1201.
Trippe, K. M.; Wolpert, T. J.; Hyman, M. R.; Ciuffetti, L. M. Biodegradation 2014, 25, 137. doi: 10.1007/s10532-013-9646-1
Park, H.; Park, G.; Jeon, W.; Ahn, J.-O.; Yang, Y.-H.; Choi, K.-Y. Biotechnol. Adv. 2020, DOI: 10.1016/j.biotechadv.2020.107504.
(a) Von Bühler, C. J.; Urlacher, V. B. Chem. Commun. 2014, 50, 4089. (b) Tieves, F.; Erenburg, I. N.; Mahmoud, O.; Urlacher, V. B. Biotechnol. Bioeng. 2016, 113, 1845.
Syed, K.; Porollo, A.; Lam, Y. W. Appl. Environ. Microbiol. 2013, 79, 2692. doi: 10.1128/AEM.03767-12
(a) Greer, S.; Wen, M.; Bird, D.; Wu, X.; Samuels, L.; Kunst, L.; Jetter, R. Plant Physiol. 2007, 145, 653. (b) Zhang, D.; Yang, H.; Wang, X.; Qiu, Y.; Tian, L.; Qi, X.; Qu, L. Q. New Phytol. 2020, 225, 2094.
Minerdi, D.; Sadeghi, S. J.; Nardo, G. D.; Rua, F.; Castrignanò, S.; Allegra, P.; Gilardi, G. Mol. Microbiol. 2015, 95, 539. doi: 10.1111/mmi.12883
Fisher, M. B.; Zheng, Y. M.; Rettie, A. E. Biochem. Biophys. Res. Commun. 1998, 248, 352. doi: 10.1006/bbrc.1998.8842
(a) Maseme, M. J.; Pennec, A.; Marwijk, J.; Opperman, D. J.; Smit, M. S. Angew. Chem., Int. Ed. 2020, DOI: 10.1002/anie.202001055. (b) Manning, J.; Tavanti, M.; Porter, J.; Kress, N.; DeVisser, S.; Turner, N.; Flitsch, S. Angew. Chem., Int. Ed. 2019, 58, 5668. (c) Sakai, K.; Matsuzaki, F.; Wise, L.; Sakai, Y.; Jindou, S.; Ichinose, H.; Takaya, N.; Kato, M.; Wariishi, H.; Shimizu, M. Appl. Environ. Microbiol. 2018, 84, e01091-18.
(a) Johnston, J. B.; Kells, P. M.; Podust, L. M.; Ortiz de Montellano, P. R. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20687. (b) Salamanca, D.; Karande, R.; Schmid, A.; Dobslaw, D. Appl. Microbiol. Biotechnol. 2015, 99, 6889.
(a) Yin, Y.-C.; Yu, H.-L.; Luan, Z.-J.; Li, R.-J.; Ouyang, P.-F.; Liu, J.; Xu, J.-H. ChemBioChem 2014, 15, 2443. (b) Xie, L.; Chen, K.; Cui, H.; Wan, N.; Cui, B.; Han, W.; Chen, Y. ChemBioChem 2020, 20, DOI: 10.1002/cbic.201900691.
Bell, S. G.; Yang, W.; Dale, A.; Zhou, W.; Wong, L. L. Appl. Microbiol. Biotechnol. 2013, 97, 3979. doi: 10.1007/s00253-012-4278-7
Shoji, O.; Aiba, Y.; Watanabe, Y. Acc. Chem. Res. 2019, 52, 925. doi: 10.1021/acs.accounts.8b00651
Dus, K.; Katagiri, M.; Yu, C. A.; Erbes, D. L.; Gunsalus, I. C. Biochem. Biophys. Res. Commun. 1970, 40, 1423. doi: 10.1016/0006-291X(70)90026-4
Stevenson, J. A.; Westlake, A. C. G.; Whittock, C.; Wong, L. L. J. Am. Chem. Soc. 1996, 118, 12846. doi: 10.1021/ja963087q
Stevenson, J. A.; Bearpark, J. K.; Wong, L. L. New J. Chem. 1998, 22, 551. doi: 10.1039/a801637b
Bell, S. G.; Stevenson, J. A.; Boyd, H. D.; Campbell, S.; Riddle, A. D.; Orton, E. L.; Wong, L. L. Chem. Commun. 2002, 490.
Bell, S. G.; Orton, E. L.; Boyd, H. D.; Stevenson, J. A.; Riddle, A. D.; Campbell, S.; Wong, L. L. Dalton Trans. 2003, 2133.
Poulos, T. L.; Finzel, B. C.; Howard, A. J. Biochemistry 1986, 25, 5314. doi: 10.1021/bi00366a049
Miura, Y.; Fulco, A. J. Biochim. Biophys. Acta 1975, 388, 305. doi: 10.1016/0005-2760(75)90089-2
Adam, W.; Lukacs, Z.; Saha-Möller, C. R.; Weckerle, B.; Schreier, P. Eur. J. Org. Chem. 2000, 16, 2923.
Appel, D.; Lutz, S.; Fischer, P.; Schwaneberg, U.; Schmid, R. D. J. Biotechnol. 2001, 88, 167. doi: 10.1016/S0168-1656(01)00249-8
Glieder, A.; Farinas, E. T.; Arnold, F. H. Nat. Biotechnol. 2002, 20, 1135. doi: 10.1038/nbt744
Peters, M. W.; Meinhold, P.; Glieder, A.; Arnold, F. H. J. Am. Chem. Soc. 2003, 125, 13442. doi: 10.1021/ja0303790
Meinhold, P.; Peters, M. W.; Chen, M. M.; Takahashi, K.; Arnold, F. H. ChemBioChem 2005, 6, 1765. doi: 10.1002/cbic.200500261
Farinas, E. T.; Schwaneberg, U.; Gliede, A.; Arnold, F. H. Adv. Synth. Catal. 2001, 343, 601. doi: 10.1002/1615-4169(200108)343:6/7<601::AID-ADSC601>3.0.CO;2-9
Weber, E.; Seifert, A.; Antonovici, M.; Geinitz, C.; Pleiss, J.; Urlacher, V. B. Chem. Commun. 2011, 47, 944. doi: 10.1039/C0CC02924F
Staudt, S.; Burda, E.; Giese, C.; Müller, C. A.; Marienhagen, J.; Schwaneberg, U.; Hummel, W.; Drauz, K.; Gröger, H. Angew. Chem., Int. Ed. 2013, 52, 2359. doi: 10.1002/anie.201204464
Müller, C. A.; Akkapurathu, B.; Winkler, T.; Svenja Staudt, S.; Hummel, W.; Gröger, H.; Schwaneberg, U. Adv. Synth. Catal. 2013, 355, 1787. doi: 10.1002/adsc.201300143
Pennec, A.; Hoomann, F.; Smit, M. S.; Opperman, D. J. ChemCatChem 2015, 7, 236. doi: 10.1002/cctc.201402835
Roiban, G. D.; Reetz, M. T. Chem. Commun. 2015, 51, 2208. doi: 10.1039/C4CC09218J
Roiban, G. D.; Agudo, R.; Reetz, M. T. Angew. Chem., Int. Ed. 2014, 53, 8659. doi: 10.1002/anie.201310892
Zhang, W.; Tang, W.; Wang, Z.; Li, Z. Adv. Synth. Catal. 2010, 352, 3380. doi: 10.1002/adsc.201000266
Chang, D. L.; Feiten, H. J.; Witholt, B.; Li, Z. Tetrahedron: Asymmetry 2002, 13, 2141. doi: 10.1016/S0957-4166(02)00534-7
Chang, D. L.; Feiten, H. J.; Engesser, K. H.; Van Beilen, J. B.; Witholt, B.; Li, Z. Org. Lett. 2002, 4, 1859. doi: 10.1021/ol025829s
Reetz, M. T. Angew. Chem., Int. Ed. 2011, 50, 138. doi: 10.1002/anie.201000826
Yang, Y.; Liu, J.; Li, Z. Angew. Chem., Int. Ed. 2014, 53, 3120. doi: 10.1002/anie.201311091
Landwehr, M.; Hochrein, L.; Otey, C. R.; Kasrayan, A.; Backvall, J. E.; Arnold, F. H. J. Am. Chem. Soc. 2006, 128, 6058. doi: 10.1021/ja061261x
Li, S.; Chaulagain, M. R.; Knauff, A. R.; Podust, L. M.; Montgomery, J.; Sherman, D. H. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18463. doi: 10.1073/pnas.0907203106
Narayan, A. R. H.; Jiménez-Osés, G.; Liu, P.; Negretti, S.; Zhao, W.; Gilbert, M. M.; Ramabhadran, R. O., Yand, Y.-F.; Furan, L. R.; Li, Z.; Podust, L. M.; Montgomery, J.; Houk, K. N.; Sherman, D. H. Nat. Chem. 2015, 7, 653. doi: 10.1038/nchem.2285
Ma, N.; Chen, Z.; Chen, J.; Wang, C.; Zhou, H.; Yao, L.; Shoji, O.; Watanabe, Y.; Cong, Z. Angew. Chem., Int. Ed. 2018, 57, 7628. doi: 10.1002/anie.201801592
Xu, J.; Wang, C.; Cong, Z. Chem. Eur. J. 2019, 25, 6853. doi: 10.1002/chem.201806383
Shoji, O.; Yanagisawa, S.; Stanfield, J. K.; Suzuki, K.; Cong, Z.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Angew. Chem., Int. Ed. 2017, 56, 10324. doi: 10.1002/anie.201703461
Cong, Z.; Shoji, O.; Kasai, C.; Kawakami, N.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. ACS Catal. 2015, 5, 150. doi: 10.1021/cs501592f
Zhang, W.; Ma, M.; Hollmann, F. J. Am. Chem. Soc. 2019, 141, 3116. doi: 10.1021/jacs.8b12282
Demming, R. M.; Hammer, S. C.; Nestl, B. M.; Gergel, S.; Fademrecht, S.; Pleiss, J.; Hauer, B. Angew. Chem., Int. Ed. 2019, 58, 173. doi: 10.1002/anie.201810005
(a) Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Curr. Opin. Chem. Biol. 2017, 37, 1. (b) Piontek, K.; Strittmatter, E.; Ullrich, R.; Gröbe, G.; Pecyna, M. J.; Kluge, M.; Scheibner, K.; Hofrichter, M.; Plattner, D. A. J. Biol. Chem. 2013, 288, 34767.
王曦翎, 陈杰, 陈置丰, 周海峰, 丛志奇, 生物资源, 2017, 39, 75. http://www.cnki.com.cn/Article/CJFDTotal-AJSH201702001.htmWang, X.; Chen, J.; Chen, Z.; Zhou, H.; Cong, Z. Biotic Resources 2017, 39, 75 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-AJSH201702001.htm
Chen, Z.; Chen, J.; Ma, N.; Zhou, H.; Cong, Z. J. Porphyr. Phthalocya. 2018, 22, 831. doi: 10.1142/S108842461850061X
Jiang, Y.; Wang, C.; Ma, N.; Chen, J.; Liu, C.; Wang, F.; Xu, J.; Cong, Z. Catal. Sci. Technol. 2020, 10, 1219. doi: 10.1039/D0CY00241K
Kawakami, N.; Shoji, O.; Watanabe, Y. Angew. Chem., Int. Ed. 2011, 50, 5315. doi: 10.1002/anie.201007975
(a) Zilly, F. E.; Acevedo, J. P.; Augustyniak, W.; Deege, A.; Häusig, U. W.; Manfred, T.; Reetz, M. T. Angew. Chem., Int. Ed. 2011, 50, 2720. (b) Zilly, F. E.; Acevedo, J. P.; Augustyniak, W.; Deege, A.; Häusig, U. W.; Manfred, T.; Reetz, M. T. Angew. Chem., Int. Ed. 2013, 52, 13503.
Kawakami, N.; Shoji, O.; Watanabe, Y. Chem. Sci. 2013, 4, 2344. doi: 10.1039/c3sc50378j
Ariyasu, S.; Kodama, Y.; Kasai, C.; Cong, Z.; Stanfield, J. K.; Aiba, Y.; Watanabe, Y.; Shoji, O. ChemCatChem 2019, 11, 4709. doi: 10.1002/cctc.201901323
Kawakami, N.; Cong, Z.; Shoji, O.; Watanabe, Y. J. Porphyr. Phthalocya. 2015, 19, 329. doi: 10.1142/S1088424615500145
Munday, S. D.; Shoji, O.; Watanabe, Y.; Wong, L. L.; Bell, S. G. Chem. Commun. 2016, 52, 1036. doi: 10.1039/C5CC09247G
Peter, S.; Kinne, M.; Wang, X.; Ullrich, R.; Kayser, G.; Groves, J. T. FEBS J. 2011, 278, 3667. doi: 10.1111/j.1742-4658.2011.08285.x
Cooley, R. B.; Dubbels, B. L.; Sayavedra-Soto, L. A.; Bottomley, P. J.; Arp, D. Microbiology 2009, 155, 2086. doi: 10.1099/mic.0.028175-0
Chen, M.; Coelho, P. S.; Arnold, F. H. Adv. Synth. Catal. 2012, 354, 964. doi: 10.1002/adsc.201100833
Labinger, J. A.; Bercaw, J. E. Nature 2002, 417, 507. doi: 10.1038/417507a
Lee, J. H.; Nam, D. H.; Lee, S. H.; Park, J. H.; Park, S. J.; Lee, S. H.; Park, C. B.; Jeong, K. J. Bioconjugate Chem. 2014, 25, 2101. doi: 10.1021/bc500404j
Ge, J.; Lei, J.; Zare, R. N. Nat. Nanotechnol. 2012, 7, 428. doi: 10.1038/nnano.2012.80
Khatri, Y.; Hannemann, F.; Ewen, K. M.; Pistorius, D.; Perlova, O.; Kagawa, N.; Brachmann, A. O.; Müller, R.; Bernhardt, R. Chem. Biol. 2010, 17, 1295. doi: 10.1016/j.chembiol.2010.10.010
Lee, J. H.; Nam, D. H.; Lee, S. H.; Park, J. H.; Park, C. B.; Jeong, K. J. J. Ind. Eng. Chem. 2016, 33, 28. doi: 10.1016/j.jiec.2015.10.002
Karande, R.; Schmid, A.; Buehler, K. Org. Process Res. Dev. 2016, 20, 361. doi: 10.1021/acs.oprd.5b00352

图 1 细胞色素P450单加氧酶催化的烷烃羟化反应机理
Figure 1 The mechanism of alkanes hydroxylation catalyzed by Cytochrome P450 monooxygenases

图 2 天然P450单加氧酶的代表性底物
Figure 2 Representative substrates of natural P450 monooxygenases

图 3 部分P450单加氧酶催化烷烃选择性羟化反应
Figure 3 Selected reactions of alkane hydroxylation catalyzed by P450 monooxygenases

图 4 (A) P450cam的晶体结构(PDB code: 2CPP[20a]), (B) P450cam结合莰酮的底物口袋结构.
Figure 4 (A) Crystal structure of P450cam (PDB code: 2CPP). (B) Structure of substrate pocket of P450cam in complex with camphor.

图 5 天然P450BM3单加氧酶的晶体结构(PDB code: 1JPZ[22])及其催化的脂肪酸羟化反应
Figure 5 Crystallographic structure of native P450BM3 monooxygenase (PDB code: 1JPZ) and the catalytic hydroxylation of fatty acids.

图 6 组合细胞色素P450单加氧酶、醇脱氢酶以及Baeyer-Villiger单加氧酶的多酶串级催化环烷烃氧化制备内酯.
Figure 6 Oxidations of cycloalkanes to lactones catalyzed by a multienzyme cascade route consist of cytochrome P450 monooxygenase, alcohol dehydrogenase and Baeyer-Villiger monooxygenase.

图 8 P450单加氧酶催化天然底物氧化(上)与诱饵分子支持的P450单加氧酶催化的非天然底物氧化(下)催化循环机理图例.
Figure 8 Illustrated catalytic cycles for (top) oxidation of natural substrate catalyzed by P450 monooxygenase and (bottom) oxidation of non-native substrate catalyzed by P450 monooxygenase in assistance with decoy molecule.

图 9 (A) 天然过加氧酶Aae APO利用“酸-碱”性谷氨酸残基Glu196协助活化H2O2的催化循环机理[82], (B)过加氧酶Aae APO活性位点结构显示谷氨酸具有“酸-碱”催化功能的羧基侧链与催化中心铁离子的距离[82b], (C)双功能小分子协同P450BM3酶催化策略示意图[76].
Figure 9 (A) The "acid-basic" residue Glu196-assisted activation of H2O2 in the catalytic cycle of natural peroxygenase Aae APO; (B) Structure of Aae UPO active site showing the distance between the carboxyl group of Glu196 with the "acid-base" catalytic function and the catalytic iron ion center; (C) Dual-functional small-molecule co-catalysis strategy for generating artificial P450BM3 peroxygenase.

图 10 细胞色素P450BM3单加氧酶与诱饵分子(PFC9-L-Trp)的共结晶结构(左, PDB code: 3WSP), 活性中心与PFC9-L-Trp以及DMSO结合部分结构放大图(右)[79].
Figure 10 Crystallographic structure of cytochrome P450BM3 monooxygenase in complex with decoy molecule (PFC9-L-Trp) (Left, PDB code: 3WSP); Active site of P450BM3 bound with PFC9-L-Trp and DMSO (Right).
表 1 部分CYP153家族酶催化的烷烃选择性羟化反应的例子
Table 1. Selected examples for hydroxylation of alkanes catalyzed by some P450 monooxygenases from CYP153 family
| Enzyme | Alkanes | Final Product | [Final Product]a | PFRb | TTNc |
| CYP153AP.Sp.[29a] | Pentane (C5) | 1-Pentanol (100%d) | 30 | — | 20 |
| Hexane (C6) | 1-Hexanol (100%d) | 62 | — | 41 | |
| Heptane (C7) | 1-Heptanol (100%d) | 103 | — | 69 | |
| Octane (C8) | 1-Octanol (90%d) | 165 | — | 110 | |
| Nonane (C9) | 2-Nonanol (59%d) | 114 | — | 76 | |
| Decane (C10) | 2-Decanol (100%d) | 99 | — | 66 | |
| CYP153A16[29a] | Pentane (C5) | 1-Pentanol | 34 | — | 22 |
| Hexane (C6) | 1-Hexanol | 24 | — | 16 | |
| Heptane (C7) | 1-Heptanol | 81 | — | 54 | |
| Octane (C8) | 1-Octanol | 120 | — | 80 | |
| Nonane (C9) | Nonane-1, 9-diol | 113 | — | 75 | |
| Decane (C10) | Decane-1, 10-diol | 65 | — | 43 | |
| CYP153A6[29b] | Hexane (C6) | 1-Pentanol | — | 0.74 | — |
| Heptane (C7) | 1-Hexanol | — | 21.2 | — | |
| Octane (C8) | 1-Heptanol | — | 60.8 | — | |
| Nonane (C9) | 1-Octanol | — | 49.7 | — | |
| Decane (C10) | 1-Nonanol | — | 13.8 | — | |
| Undecane (C11) | 1-Undecanol | — | 0.4 | — | |
| 2-Methyloctane (C9) | 2-Methyl-1-octanol | — | 35 | — | |
| Limonene (C10) | Perillyl alcohol | — | 31.2 | — | |
| p-Cymene (C10) | p-Cumic alcohol | — | 38.9 | — | |
| CYP153A13[29c] | Octane (C8) | 1-Octanol | 116 | — | 233 |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. d Regioselectivity. | |||||
 下载: 导出CSV
下载: 导出CSV
表 2 部分CYP52家族酶催化的烷烃选择性羟化反应
Table 2. Selected examples for selective hydroxylation of alkanes catalyzed by some P450 monooxygenases from CYP52 family
| Enzyme | Alkanes | Final Product | PFRb | TTN c |
| CYP52A3, A4, A5, A9[32] | n-Dodecane (C12) | 1-Dodecanol | 3~44 | 6~88 |
| n-Hexadecane (C16) | 1-Hexadecanol | 12~49 | 24~98 | |
| CYP52A58[33] | n-Hexadecane (C16) | 1-Hexadecanol | 0.6 | — |
| CYP52A12, A13, A14, A17, A18[34] | n-Octadecane (C18) | α, ω-Dicarboxylic acid | — | — |
| CYP52L1[37] | Propane | 1-Propanol | — | — |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. | ||||
下载: 导出CSV
表 3 各种各样的细胞色素P450酶催化的烷烃选择性羟化反应
Table 3. Selected examples for selective hydroxylation of alkanes catalyzed by miscellaneous Cytochrome P450 enzymes
| Enzyme | Alkanes | Final Product | [Final Product]a | PFRb | TTNc |
| CYP154A8[39a] | Heptane(C7) | (S)-2-Heptanol (84%d) | 1900 | 1.13 | 2800 |
| Octane(C8) | (S)-2-Octanol (91%d) | 2200 | 4.60 | 3200 | |
| Nonane(C9) | (S)-2-Nonanol (84%d) | 3000 | 3.73 | 4400 | |
| Decane(C10) | (S)-2-Decanol (63%d) | 1200 | 0.37 | 1700 | |
| CYP63A2[40] | n-Alkane(C9~C12, C15~C19) | ω-Alkanol (9-30%e) | — | — | — |
| CYP96A15[41a] | n-Alkane (C27, C29, C31) | Secondary alcohol and ketone | — | — | — |
| CYP116B5[42] | C14, C16, C24, C36 | Primary alcohol | — | — | — |
| CYP4B1[43] | Heptane (C7) | 1-Heptanol (96%f) | — | 33 | — |
| Octane (C8) | 1-Octanol (86%f) | — | 26 | — | |
| Nonane (C9) | 1-Nonanol (71%f) | — | 31 | — | |
| Decane (C10) | 1-Decanol (62%f) | — | 11 | — | |
| CYP505E3[44a] | Decane (C10) | 7-Decanol (71%f) | — | — | 8013 |
| Dodecane (C12) | 7-Dodecanol (71%f) | — | — | 1969 | |
| Tetradecane (C14) | 7-Tetradecanol (74%f) | — | — | 138 | |
| Hexadecane (C16) | 7-Hexadecanol (100%f) | — | — | 138 | |
| CYP124[45a] | Phytane & Pristane | ω-Oxidation | — | — | — |
| CYPCHX[45b] | Cyclopentane | Cyclopentanol | 10239 | — | — |
| Cyclohexane | Cyclohexanol | 9834 | — | — | |
| Cycloheptane | Cycloheptanol | 1933 | — | — | |
| Cyclooctane | Cyclooctanol | 3069 | — | — | |
| CYP101D2[47] | Camphor | 5-exo-Hydroxycamphor (99%f) | — | 2008i (99%g) | — |
| Adamantane | 1-Adamantanol (98%f) | — | 84.1i (62%g) | — | |
| Cyclooctane | Cyclooctanol (99%h) | — | 167i (75%g) | — | |
| Hexane | 2-OH/3-OH (56%:44%f) | — | 3.0i (9%g) | — | |
| 2-Methylpentane | 2-methyl-2-pentanol (55%f) | — | 14.7i (28%g) | — | |
| a The concentration of final product in μmol·L-1. b PFR: product formation rate in μmol·(μmol P450)-1·min-1. c TTN: total turnover number. d ee: enantiomeric excess percent. e Alkanes oxidation ratio. f Regioselectivity. g NADPH coupling efficiency. h Chemoselectivity, cyclooctenone (1%). i PFR in nmol·(nmol P450)-1· min-1. | |||||
下载: 导出CSV
表 4 P450cam及其突变酶对烷烃底物的氧化活性[26, 50~53] a
Table 4. Oxidation activity of alkanes by P450cam and its mutants
| P450cam enzyme | PFRb of the yielded alcoholsc (CE%d) | ||||
| n-Hexane | 3-Methylpentane | n-Butane | Propane | Ethane | |
| Wild-Type | 0.4 (2.0) | 27.7 (21.3) | 0.4 (0.5) | 0.5 (0.5) | — |
| Y96A | 89.3 (23.0) | 99.1 (52.0) | — | — | — |
| Y96F | 109.8 (47.0) | 122.6 (54.7) | 42 (42) | 2.2 (12) | — |
| Y96A/V274A | 122.2 (30.2) | 199.0 (63.6) | — | — | — |
| Y96F/V274L | 176.1 (66.8) | 42.3 (50.6) | 186 (75) | 43 (39) | — |
| F87W/Y96F/T101L/V247L | — | — | 750 (95) | 110 (32) | — |
| F87W/Y96F/T101L/V247L/L244M (EB) | — | — | 520 (90) | 176 (66.2) | — |
| EB/L294M/T185M/L1358P/G248A | — | — | — | 505 (85.6) | 78.2 (10.5) |
| a Summarized result for P450cam-catalyzed hydroxylation of alkanes according to references 26, 50~53. b PFR: product formation rate in nmol·(nmol P450)-1· min-1. c The yielded alcohols are 2-hexanol and 3-hexanol for n-hexane, 3-methyl-2-pentanol and 3-methyl-3-pentanol for 3-methyl-pentane, 2-butanol for n-butane, 2-propanol for propane, and ethanol for ethane. d CE: Coupling efficiency of NADPH, CE%=PFR/(NADPH consuming rate)×100%. | |||||
下载: 导出CSV
表 5 P450BM3突变体对乙烷、丙烷及辛烷的氧化活性[27, 58~60] a
Table 5. Oxidation activity of ethane, propane and n-octane by P450BM3 mutants
| P450BM3 variants | Ethane | Propane | n-Octane | ||||||||
| Rateb | TTNc | CE%d | Rate | TTN | CE% | Rate | TTN | CE% | |||
| 139-3 | — | — | — | 12 | 500 | 12 | 480 | 1000 | 22 | ||
| J | — | — | — | 30 | 800 | 5 | 660 | 3000 | 23 | ||
| 9-10A | — | — | — | 23 | 1100 | 5 | 540 | 3000 | 21 | ||
| 1-12G | — | — | — | 160 | 6000 | 100 | 150 | 7500 | 37 | ||
| 53-5H | 0.4 | 50 | 0.06 | 370 | 5000 | — | 660 | 8000 | — | ||
| 35-E11 | 0.4 | 250 | 0.08 | 210 | 5650 | 17.4 | 420 | 8000 | — | ||
| P450PMOR1 | — | — | — | 455 | 35600 | 94.4 | — | — | — | ||
| P450PMOR2 | — | — | — | 370 | 45800 | 98.2 | — | — | — | ||
下载: 导出CSV
表 6 诱饵分子策略在P450BM3催化烷烃选择性羟化上的应用a, b
Table 6. Selective hydroxylation of alkanes catalyzed by P450BM3 using decoy molecule strategy
| Decoy molecule | Alkanes | Final product | PFRb | TTNc | Reference |
| PFC10 | Propane | 2-Propanol | 70 | 700 | [86] |
| PFC9 | n-Butane | 2-Butanol | 110 | 1100 | [86] |
| PFC9 | Cyclohexane | Cyclohexanol | 112 | 1120 | [86] |
| PFC10d | Cyclohexane | Cyclohexanol | 484 | — | [91] |
| PFC10e | Methane | Methanol | — | 2472 | [87] |
| PFC11 | Propane | 2-Propanol | — | 1021 | [87] |
| PFC7 | n-Butane | 2-Butanol | — | 3632 | [87] |
| PFC11 | n-Hexane | 2-/3-Hexanol=77:23g | — | 525 | [87] |
| PFC9 | n-Octane | 2-/3-/4-Octanol=10:42:48g | — | 1184 | [87] |
| PFC10f | Ethane | Ethanol | 0.67 | — | [88] |
| PFC10f | Propane | 2-Propanol | 72 | — | [88] |
| PFC10f | Propane | 1-Propanol | 1.7 | — | [88] |
| PFC9-L-Leu | Ethane | Ethanol | 45 | — | [79] |
| PFC9-L-Leu | Propane | 2-Propanol | 256 | — | [79] |
| a Wild type P450BM3 enzyme is used unless otherwise stated. b PFR: Product formation rates in μmol·(μmol P450)-1·min-1. c TTN: Total turnover numbers. d A P450BM3 mutant KT2 is used. e The authors made a corrigendum about the system being unavailable for methane oxidation but available for other alkanes hydroxylation, see ref. 87b. f The reactions are carried out under high pressure conditions. g Molar ratio of alcohol products. | |||||
下载: 导出CSV
表 7 人工P450过加氧酶催化的小分子烷烃选择性羟化[28]
Table 7. Selective hydroxylation of small alkanes catalyzed by artificial P450 peroxygenasea, b
| P450BM3 variants | 2-Propanol | 2-Butanol | 1-/2-/3-Pentanol | 2-Hexanol | |||||||
| PFRc | TONd | PFRc | TONd | PFRc | TONd | PFRc | TONd | ||||
| F87A/T268I | 635 | 1582 | 902 | 1882 | 318 | 1056 | 297 | 775 | |||
| F87V/T268I | 499 | 1264 | 913 | 2009 | 233 | 1092 | 171 | 370 | |||
| F87A/T268I/A82T | 473 | 1353 | 1042 | 2253 | 442 | 1341 | 464 | 1003 | |||
| F87A/T268I/A184V | 619 | 1626 | 992 | 2262 | 391 | 1319 | 450 | 1203 | |||
| F87A/T268I/A184I | 630 | 1775 | 979 | 2289 | 383 | 1463 | 439 | 1286 | |||
| F87A/T268I/A82T/A184V | 481 | 1246 | — | — | 516 | 1640 | 600 | 1322 | |||
| a Reaction conditions: P450BM3 (0.5 μmol·L-1), H2O2 (60 mmol·L-1), Im-C6-Phe (500 μmol·L-1), in pH 8.0 phosphate buffer at 25 ℃. b All the control reactions did not show obvious activity of hydroxylation in the absence of Im-C6-Phe. c PFR: Product formation rates were estimated as μmol·(μmol P450)-1·min-1 over a 1-min reaction. d TON: Turnover numbers were estimated over a 30-min reaction. | |||||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: