
引用本文:
郭金秋, 杜亚平, 张洪波. 稀土材料在多相催化中的应用研究进展概述[J]. 化学学报,
2020, 78(7): 625-633.
doi:
10.6023/A20030053

Citation: Guo Jinqiu, Du Yaping, Zhang Hongbo. A Brief Summary of Research Progress on the Application of Rare Earth Materials in Heterogeneous Catalysis[J]. Acta Chimica Sinica, 2020, 78(7): 625-633. doi: 10.6023/A20030053
Citation: Guo Jinqiu, Du Yaping, Zhang Hongbo. A Brief Summary of Research Progress on the Application of Rare Earth Materials in Heterogeneous Catalysis[J]. Acta Chimica Sinica, 2020, 78(7): 625-633. doi: 10.6023/A20030053
稀土材料在多相催化中的应用研究进展概述
摘要:
稀土在中国储量丰富,尤其在徐光宪等老一辈科学家不懈努力下开发了高效提纯稀土元素的有效方法,为当今的稀土性能应用研究打下了坚实的基础.稀土包括钪、钇以及镧系金属在内的17种元素,由于镧系元素最外层电子和次外层电子的结构相似([Xe]4fn-15d0~16s2(n=1~15)),尤其是其独特的4f电子层结构,使稀土元素具有化学性质稳定、化学价态可变、配位形式多样、总体具有Lewis酸性等特点,使其在催化领域具有广泛的应用.但稀土元素电子层结构与催化反应效果之间的关系尚未有明确的总结,因此本文从稀土材料的电子结构特点出发,综述了其在多相(热)催化领域中作为载体、Lewis酸(碱)中心以及催化助剂等方面的应用.
English
A Brief Summary of Research Progress on the Application of Rare Earth Materials in Heterogeneous Catalysis
Abstract:
Rare earth (RE) resources are in big amount in China, which can be effectively purified based on the strategies developed by Prof. Guangxian Xu et al. last century, which sets up solid fundamentals for applied research on rare earth materials nowadays. Rare earth elements, including scandium, yttrium and lanthanides, feature stable overall chemical properties, variable valence states and coordination form as well as special Lewis acidity due to the unique electron configuration in the outermost and secondary outer orbitals of the lanthanide elements ([Xe] 4fn-15d0~16s2 (n=1~15)), especially on their 4f electron shell structure, having been extensively used in catalysis. However, the efficiency and selectivity to the desired products are always the major challenges due to the complexity of catalysis, in particular, the mechanism by which rare earth metals affect catalytic reactions through structural or electronic effects has not been clarified. Therefore, this mini-review summarizes the research progress on the application of rare earth materials in heterogeneous catalysis (specifically on thermal catalysis). Firstly, a brief summary of rare earth materials' structural properties is provided with emphasis on the unique distribution of the 4f electron. Afterward, the application of RE elements in thermal catalysis was discussed in detail. For example:(1) as a support to promote catalytic reaction, such as CeO2, which has variable chemical valence and can be used as an active support to participate in the redox reaction; (2) as moderate Lewis acid (base) center to catalyze the aldol condensation of acetaldehyde/ethanol mixture and effectively control the C-C bond coupling; (3) as electronic and structural promoters to improve catalytic activity and stability. Hence, the structure-function relationship is illustrated in accordance with the studies of the rare earth materials as the supports, Lewis acid (base) active center and catalytic promoters, suggesting great potential of rare earth materials in catalysis.
-
Key words:
- rare earth material
- / support
- / Lewis acid (base)
- / catalytic promoters
-
1. 引言
钪、钇以及镧系(镧、铈、镨、钕、钷、钐、铕、钆、铽、镝、钬、饵、铥、镱、镥)构成稀土元素.自18世纪以来, 稀土元素在生物学、军事、农业以及化学工程等领域具有非常广泛的应用[1].我国稀土资源丰富, 种类齐全, 且储存总量位居世界第一位, 是世界上最大的稀土生产、应用和出口国[2].
近年来, 基于稀土元素自身的特点, 如:结构相对稳定、具有中等强度的Lewis酸、具有独特的4f电子层结构, 使其在光催化[3-6]、电催化[7, 8]、热催化[9-11]领域具有广泛的应用.但由于催化反应的复杂性使得催化反应产物分布广泛, 不能有效地合成目标产物.究其主要原因在于人们对于稀土元素通过结构、电子效应来影响催化反应的机理尚不明确.本综述依据稀土独特的电子层排布, 选取典型实例(集中讨论热催化), 概述了其作为催化载体、Lewis酸(碱)中心、催化助剂的应用, 并对稀土材料的发展前景进行了展望.
2. 稀土材料概述
2.1 稀土元素的电子构型特点
稀土金属具有独特的电子构型, 以镧系元素为例, 其电子构型为[Xe]4fn-15d0~16s2(n=1~15), 最外层都填充到6s2轨道, 5d轨道无电子填充或仅有一个电子填充, 且容易失去外层两个6s轨道电子和一个次外层5d轨道上的电子,
无d轨道电子时则失去一个4f轨道电子, 从而实现4fn→4fn-15d1的转变, 使稀土离子通常表现为+3价.根据洪特规则可知, 在原子的电子结构中, 当同一层的原子处于全空、半满或全满时原子相对稳定, 对于稀土元素, 具有[Xe]4f0、[Xe]4f7和[Xe]4f14电子构型的La3+、Gd3+、Lu3+比较稳定, 而对于比它们原子序数稍大的Ce3+、Pr3+、Tb3+, 比稳定态离子多1~2个电子, 所以易被氧化为+4价; 比它们原子序数小的Sm3+、Eu3+、Yb3+离子, 比稳定态离子少1~2个电子, 因此易被还原成+2价[12].
2.2 稀土材料在催化领域的适用性
部分稀土材料的化学价态可变, 可以将其应用于催化氧化(还原)反应, 比如, CeO2中铈离子可以实现从+3价到+4价的可逆转变, 同时释放出氧空位, 从而使CeO2具有独特的储存/释放氧的能力[13-19].而La元素由于4f轨道全空而十分稳定, 利用这一特性, 可以将La2O3引入催化体系, 以提高催化材料的稳定性[20, 21].另外, 由于稀土元素存在大量的4f空轨道, 使其在与其他材料复合以及掺杂作为催化助剂时, 可以与载体产生较强的相互作用, 从而表现出优良的催化效果[22-35].
3. 稀土材料在催化中的应用
基于稀土元素独特的电子层结构, 稀土材料被广泛地应用于各种催化转化, 包括光催化[3-6]、电催化[7, 8]和热催化[9-11], 本综述主要从稀土材料作为载体、Lewis酸(碱)催化剂, 以及作为催化助剂三个方面, 介绍稀土材料用于(热)催化领域时表现出的特异性.
3.1 作为载体
3.1.1 CeO2
在催化反应中, 载体不仅影响负载金属粒子的形貌和结构, 还会通过与金属粒子的相互作用来影响催化反应活性、选择性和稳定性[13], 因此选用合适的载体有助于催化反应的顺利进行.与SiO2等惰性载体不同, 以CeO2为典型代表的化学价态可变的部分稀土金属氧化物为载体时, 可作为活性载体, 参与到氧化还原反应之中.例如, Mattos等[14]根据CeO2可参与到氧化还原反应中的特性, 在CeO2载体上负载Pt, 将其用作乙醇选择性氧化催化剂, 分析了乙醇转化机理.发现其反应过程明显有CeO2参与, 具体反应路径如下:
$ \begin{array}{*{20}{l}} {{\rm{C}}{{\rm{H}}_{\rm{3}}}{\rm{C}}{{\rm{H}}_{\rm{2}}}{\rm{OH}}} \end{array}\begin{array}{*{20}{l}} {\left( {\rm{g}} \right){\rm{ + C}}{{\rm{e}}^{{\rm{4 + }}}} \to {\rm{C}}{{\rm{H}}_{\rm{3}}}{\rm{C}}{{\rm{H}}_{\rm{2}}}{\rm{O - C}}{{\rm{e}}^{{\rm{4 + }}}}{\rm{ + }}\left( {{\rm{1/2}}} \right){{\rm{H}}_{\rm{2}}}} \end{array} $
(1) $ \begin{array}{*{20}{l}} {{\rm{C}}{{\rm{H}}_{\rm{3}}}{\rm{C}}{{\rm{H}}_{\rm{2}}}{\rm{O - C}}{{\rm{e}}^{{\rm{4 + }}}} \to {\rm{C}}{{\rm{H}}_{\rm{3}}}{\rm{CHO - C}}{{\rm{e}}^{{\rm{4}} + }} + \left( {{\rm{1/2}}} \right){{\rm{H}}_{\rm{2}}}} \end{array} $
(2) $ \begin{array}{l} {\rm{C}}{{\rm{H}}_{\rm{3}}}{\rm{CHO - C}}{{\rm{e}}^{{\rm{4}} + }} + {\rm{C}}{{\rm{e}}^{{\rm{4}} + }}{\rm{ - }}{{\rm{O}}^{{\rm{2}} - }} \to {\rm{C}}{{\rm{e}}^{{\rm{3}} + }}{\rm{ - C}}{{\rm{H}}_{\rm{3}}}{\rm{COO - C}}{{\rm{e}}^{{\rm{3}} + }} + \\ \left( {{\rm{1/2}}} \right){{\rm{H}}_{\rm{2}}} \end{array} $
(3) 当温度进一步升高时, 部分氧化的产物则会进一步分解, 产生CH4, CO和H2等副产物.
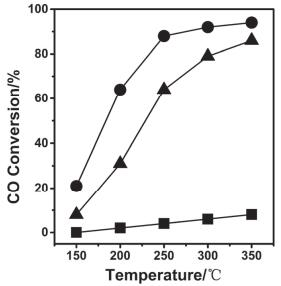
CeO2是商用汽车三效催化剂的载体, 三效催化剂是用来催化转化汽油燃烧过程中产生的不完全燃烧的碳氢化合物CHx, 氮氧化合物NOx和CO等对环境有害的污染物[15-17].由于无法控制汽车尾气中的氧含量, 当氧含量不足时, 会导致尾气的不完全燃烧, 从而使污染物残留.研究表明, 采用Rh、Pt等贵金属和二氧化铈形成的复合材料作为三效催化剂来储存氧气, 可获得接近化学计量比的污染物转化率, 但是当燃料十分充足或极其匮乏时, 这种三效催化剂的催化效率会显著降低.当CeO2载体含量增加时, 催化剂储存/释放氧的能力也相应提高, 因此增加CeO2载体含量会显著改善这一不足[18], 在燃料充足的条件下, Ce4+被还原成Ce3+, 同时释放氧; 然而当缺少燃料时, 铈的化学价则从+3价转变成+4价, 并储存氧.利用CeO2化学价态可变的特性, 提高了汽车三效催化剂的转化效率和抗老化能力[19]. CeO2作为载体时, 可以通过改变CeO2的形貌以获得不同的催化性能[36-39].贾春江等[36]将质量分数为1%的铜分别沉积到CeO2纳米立方体和纳米棒中, 用于催化CO氧化.结果发现Cu可在暴露{100}面的CeO2纳米立方体上产生Cu(I)活性位点, 这种活性位点可以在CO氧化的过程中产生Cu(I)-CO活性物种, 从而提高催化CO氧化的能力. Flytzani-Stephanopoulos等[38, 39]将Au负载在CeO2上, 用于低温催化水煤气变换反应.其采用两步法, 分别将Au负载在CeO2纳米棒、纳米立方体和纳米多面体上, 得到了不同的水煤气变换反应活性.透射电镜(TEM)和高分辨透射电镜(HRTEM)表征表明(如图 1所示)Au在CeO2纳米立方体上暴露{111}面.三种形貌的Au/CeO2催化剂(图 2)的活性顺序为: Au/CeO2纳米棒>Au/CeO2纳米多面体>Au/CeO2纳米立方体, 且在350 ℃的反应温度下, Au/CeO2纳米棒的转化率接近100%, 而Au/CeO2纳米立方体的转化率在350 ℃时只有20%.这是由于: (1) CeO2的纳米尺寸效应, 这种纳米尺寸效应可以提高CeO2固有的还原性, 将Ce4+还原成Ce3+, 同时产生氧空位[40]; (2) CeO2的形貌会显著影响Au的活性, CeO2纳米棒暴露{110}晶面, 氧空位在{110}晶面上的生成能低于{100}和{111}晶面, 这些氧空位可以稳定Au金属离子, 从而有利于提高Au/CeO2纳米棒的活性; (3)其活性的提高也与晶格应力有关.这一手段同样可以拓展到研究其它氧化物形貌对催化活性的影响.
图 1
图 2

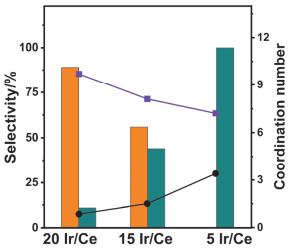
CeO2作为载体时, 还可与负载在其上的金属(Pd, Ru, Rh等)产生较强的相互作用(Strong Metal-Support Interaction, SMSI), 这种强相互作用可以提高一些电子敏感型催化转化速率, 并可以有效地控制反应产物的选择性, 如CeO2为载体用于催化CO氧化[41-47]、水煤气变换[48-50]和CO2加氢[51]等反应.乔波涛等[45]使用单原子金和金纳米颗粒对CeO2载体进行修饰, 并对比了其催化CO氧化的活性, 结果发现CeO2和单原子金之间的相互作用有利于形成氧化态的金, 在水存在的条件下可作为电子受体, 为CO+OH→COOH→CO2+H提供有效的反应通道, 增强了催化CO氧化的反应活性.通过金属与载体之间的强相互作用(SMSI), 载体不仅可以影响负载金属的分散性, 而且可以影响贵金属纳米颗粒的化学状态[52].例如:马丁课题组[53]用无配体的方法在CeO2纳米片上合成了高度分散、Ir含量不同的纳米颗粒, 并将其用于CO2加氢反应.其研究发现, 可以通过改变负载在CeO2上Ir纳米颗粒的尺寸来改变产物的选择性. 图 3显示不同Ir负载量的Ir/Ce催化剂中, CO和CH4选择性和Ir-Ir和Ir-O配位数(Coordination Number, CN)之间的关系, 当Ir加入量从20%(质量分数)减少到5%(质量分数)时, Ir的尺寸降低, CNIr-Ir从9.7降低为7.2, 但CNIr-O逐渐升高, 这表明当Ir占比小时, 氧原子会对Ir物种进行修饰, 这种结构修饰来源于CeO2载体与Ir之间的强相互作用, 使更多的氧原子富集在金属Ir表面, 从而使CNIr-O升高.部分氧化的Ir/CeO2有利于CO2的还原并促进CO的脱附, 使CO的选择性提高, 导致5% Ir/CeO2上CO选择性接近100%.
图 3
 图 3. 在不同Ir负载量的Ir/Ce催化剂上, Ir-Ir(■)和Ir-O(●)配位数(数据, 右轴)与催化选择性(棒, 左轴) (CO(橙色), CH4(深青色))的关系[53]Figure 3. The coordination number (CN) of Ir-Ir (■) and Ir-O (●) shells (data, right axis) correlate to catalytic selectivity (bars, left axis) (CO (orange), CH4 (dark cyan)) with Ir/Ce catalysts at various Ir loadings[53]
图 3. 在不同Ir负载量的Ir/Ce催化剂上, Ir-Ir(■)和Ir-O(●)配位数(数据, 右轴)与催化选择性(棒, 左轴) (CO(橙色), CH4(深青色))的关系[53]Figure 3. The coordination number (CN) of Ir-Ir (■) and Ir-O (●) shells (data, right axis) correlate to catalytic selectivity (bars, left axis) (CO (orange), CH4 (dark cyan)) with Ir/Ce catalysts at various Ir loadings[53]类似的, 严纯华、张亚文等[54]近期通过改变Ru在Ru/CeO2中的尺寸(单原子、纳米团簇1.2 nm、纳米颗粒4.0 nm), 揭示了金属与载体之间的强相互作用随粒径尺寸的变化规律, 同时讨论了H溢流效应与水脱除反应之间的竞争关系.具体体现为:当Ru是单原子时, CeO2与Ru之间的相互作用最强, 可以抑制金属羰基的活化; 而当Ru是纳米颗粒时, H溢流效应最强, 能够抑制表面水的脱除; 当Ru是纳米团簇时, 金属与载体间的相互作用以及H溢流效应在Ru/CeO2纳米团簇催化剂上达到平衡, 因此该复合材料表现出最佳的低温CO2甲烷化的活性, 甲烷的选择性可达98%~100%.
3.1.2 RE2O3
稀土金属氧化物RE2O3(Rare Earth, RE)可作为催化剂的载体[55], 以La2O3作为典型代表, 来说明RE2O3作为载体的作用.镧元素的4f轨道含有0个电子, 相对稳定, 因此可利用La2O3作为载体来提高催化体系的稳定性[20, 21]. La2O3作为载体时, 可以通过与金属粒子之间的相互作用[56, 57], 尤其是静电相互作用来促进金属粒子的分散. Flytzani-Stephanopoulos等[58]用阴离子吸附法制备了Au/La2O3催化剂, Au前驱体和镧基载体界面之间具有静电相互作用, 这种静电相互作用可在不降低Au担载量的前提下, 使Au高度分散在La2O3载体中.这种独特的静电相互作用还可以消除体系中残留的氯离子, 有利于在低温下进行水煤气转化, 转化率可达70%. La2O3还可以作为活化烷烃制低碳烯烃的载体.烷烃裂解制备低碳烯烃时, C—H键的活化往往是反应的决速步[59], 选用高活性的活化C—H键的材料作为催化剂对整体反应有利.但是C—H键的活化能垒较高, 从而导致反应体系能耗相对较高, 且产物分布多样.根据这一问题, Hardacre等[60]对不同氧化物载体制备烯烃的效果进行了测试, 催化性能如表 1所示, 其中La2O3展现了相对较高的总体反应速率(0.84 mmol•min-1•g-1)及最高的烯烃选择性(28.8%).之后将Au负载在La2O3载体上, 并将其用于丁烷裂解制烯烃中, 结果发现Au/La2O3复合材料的催化活性优于单相材料, 且在反应48 h之后仍可保持较高活性(转化率稳定在38%, 烯烃选择性为62.5%左右).
表 1

 下载:
导出CSV
下载:
导出CSV
催化剂 反应速率/(mmol·min-1·g-1 选择性/% 产率/% 烯烃 烷烃 COx C2H4 C3H6 La2O3 0.84 28.8 3.2 23.8 1.7 1.7 CeZrO4 0.86 17.9 2.2 69.5 1 0.3 ZrO2 0.38 16.3 3.8 24.5 0.4 0.5 TiO2 0.51 17.3 2.1 26.3 1 0.3 Nb2O5 0.25 20.5 5.3 27.2 0.2 0.3 这是由于La2O3有利于稳定离子态的金(Au+), 而不是将金还原成零价(Au0), Au+与La2O3之间的相互作用可以促进C—H键的解离产生有效自由基, 而未提供氧化的位点, 使碳氧化合物的选择性降低, 碳氢化合物的选择性升高.龚学庆等[61]和常春然等[62]用密度泛函理论(Density Functional Theory, DFT)对稀土金属氧化物活化甲烷过程中C—H键的解离进行了研究, 其中龚学庆等[61]发现调节La2O3暴露的晶面或在La2O3基体中掺杂Sr/Ce离子(Sr/Ce-La2O3)均可以促进C—H键的解离.其通过DFT理论计算进行分析, 发现La2O3中高密勒指数的(210)晶面可暴露出低配位数的氧(尤其是O2c), Sr/Ce-La2O3可提供Ce 4f空轨道, 这两种方法均使La2O3基材料具有高的H吸附能、低的氧空位形成能, 从而有效解离甲烷形成甲基中间物种, 提高碳氢化合物的选择性.
3.2 Lewis酸(碱)催化
Lewis酸、碱往往成对出现, 对于不同的催化反应, 起作用的形式各异.稀土元素具有独特的Lewis酸性, 与之结合的阴离子具有Lewis碱性, 因此可以将稀土材料用做Lewis酸(碱)催化的催化剂.
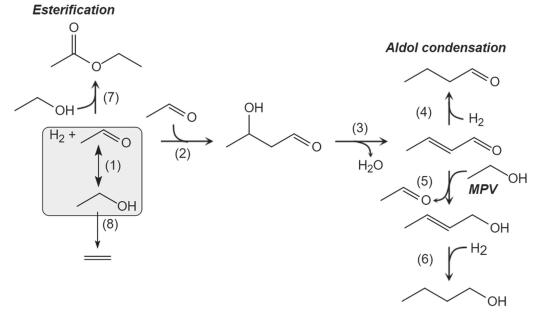
近年来, 由于化石能源储量日益降低, 使能源短缺问题更加凸显, 人们发现该能源危机有望通过生物质平台小分子催化转化为高附加值的产物来解决.生物质乙醇可以通过ABE (Acetone-Butanol-Ethanol)发酵而大量获得, 且乙醇作为典型的生物质平台小分子, 可以通过aldol加成反应生成异丁烯[63]、丁醇[64]、巴豆醛[65]等高附加值的化学品, 乙醛/乙醇混合物aldol加成的反应路径如图式 1.
图式 1
 图式 1. 乙醛/乙醇混合物aldol加成的反应路径Scheme 1. Reaction network of aldol condensation from acetaldehyde/ethanol mixtures
图式 1. 乙醛/乙醇混合物aldol加成的反应路径Scheme 1. Reaction network of aldol condensation from acetaldehyde/ethanol mixtures(1) 乙醇脱氢生成乙醛; (2)两分子乙醛经过aldol加成生成aldol产物——3-羟基丁醛; (3)3-羟基丁醛脱水生成巴豆醛; (4)巴豆醛的碳碳键加氢生成丁醛; (5)巴豆醛经过氢转移反应(Meerwein-Ponndorf-Verley, MPV反应), 使碳氧键加氢生成巴豆醇; (6)巴豆醇进一步加氢生成丁醇.在反应过程中通常伴随着许多副反应的发生, 如(7)乙醇与乙醛脱水生成乙酸乙酯、(8)乙醇直接脱水生成乙烯等, 而且可能会进一步生成C—C键从而得到高碳的产物[66].其中, C—C键的生成是aldol加成反应中的关键步骤[67], 然而, 经过一次C—C键生成所获得的一级aldol产物之间会发生复杂的C—C键交联反应, 使目标产物的选择性不受控制.大量的研究表明调控材料的Lewis酸(碱)强度可以显著影响C—C键的生成[68].稀土元素具有独特的Lewis酸性, 且构成稀土金属氧化物时存在Lewis酸、碱对[69](稀土元素作为Lewis酸, 氧原子作为Lewis碱), 因此可以利用稀土材料独特的Lewis酸(碱)特性, 用来催化aldol加成反应.
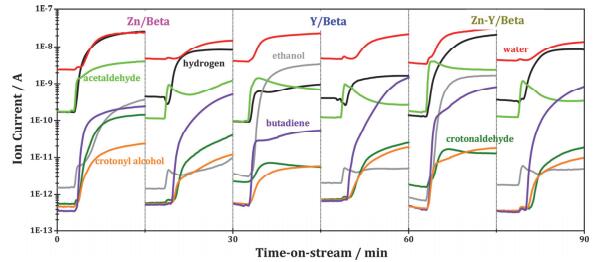
例如李兰冬等[70]借助稀土独特的Lewis酸性构筑了高效的aldol加成催化剂.该课题组利用金属锌(Zn)和稀土元素钇(Y)对Beta分子筛进行共修饰, 并将其作为乙醇aldol加成反应的催化剂, 高选择性地制备了1, 3-丁二烯(选择性约60%).并通过1H和13C的核磁共振波谱, 对[Si]Beta, Zn/Beta, Y/Beta以及Zn-Y/Beta进行表征.结果表明, Y和Zn的加入可以在Beta分子筛中引入Lewis酸性位点, 使催化剂aldol加成活性提高.采用不同金属修饰的Beta分子筛具有不同的催化反应活性及对应产物选择性.例如: Zn/Beta的脱氢活性相对较高, 而Y/Beta具有较高的aldol加成反应活性.为了进一步证明Y是aldol加成反应的主要活性位点, 其在反应体系中分别加入乙醇、巴豆醛以及乙醇与巴豆醛的混合物, 并用质谱在线检测不同催化剂所表现出的反应情况, 结果如图 4所示.分析表明, aldol加成反应主要在具有高aldol加成反应活性的Y活性位点上进行, 而Zn主要参与乙醇脱氢生成乙醛, 进而进行aldol加成反应.
图 4
 图 4. 在Zn/Beta、Y/Beta、Zn-Y-Beta催化剂上, 分别用乙醇(左)、乙醇和巴豆醛(右)作为反应物的催化结果.反应条件:重量时空速度(WHSV)=1 h-1, 反应温度T=623 K[70]Figure 4. Modulation experiments about crotonaldehyde and ethanol over Zn/Beta, Y/Beta and Zn-Y-Beta catalysts (left: ethanol; right: ethanol and crotonaldehyde cofeeding). Reaction conditions: weight hourly space velocity (WHSV)=1.0 h-1, reaction temperature of T=623 K[70]
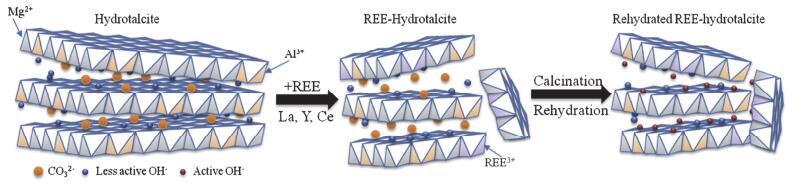
图 4. 在Zn/Beta、Y/Beta、Zn-Y-Beta催化剂上, 分别用乙醇(左)、乙醇和巴豆醛(右)作为反应物的催化结果.反应条件:重量时空速度(WHSV)=1 h-1, 反应温度T=623 K[70]Figure 4. Modulation experiments about crotonaldehyde and ethanol over Zn/Beta, Y/Beta and Zn-Y-Beta catalysts (left: ethanol; right: ethanol and crotonaldehyde cofeeding). Reaction conditions: weight hourly space velocity (WHSV)=1.0 h-1, reaction temperature of T=623 K[70]Wang等[71]的研究发现, 由于稀土元素有较低的电负性, 当用具有Lewis碱性的稀土金属氧化物(La2O3, Y2O3)对镁铝水滑石进行修饰时, 可以减小水滑石的晶粒尺寸、增大比表面积.未加修饰、稀土修饰以及加水修饰的镁铝水滑石的结构模型如图式 2表示.位于晶体棱边和转角处的氧(低配位的氧)的Lewis碱性强于其他位置处氧的碱性, 当水滑石的晶粒尺寸减小时, 低配位氧数量增加, 从而使水滑石的碱性增强[72, 73].将稀土金属Y、La修饰后的镁铝水滑石用作催化丙酮的aldol缩合反应, 表现出了很好的aldol加成活性.如:当用水对稀土修饰的水滑石材料进一步修饰之后, 在273 K的反应温度下, 丙酮大多转化为亚异丙基丙酮, 丙酮的转化率可达20%, 接近反应的平衡转化(23.1%).
图式 2

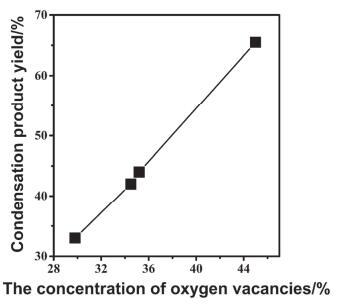
李小年等[74]研究发现, 具有Lewis酸、碱对的二氧化铈可以用于乙醛的aldol加成反应, 其中稀土金属铈作为路易斯酸中心, 晶格氧作为路易斯碱中心.其根据氧空位浓度的不同合成了四种CeO2, 并对其aldol加成效果进行了测定, 测试结果如图 5所示.从图中可以看出, 随着CeO2氧空位浓度的增大, aldol加成产物的产率也逐渐升高, 基本成线性增加的趋势, 其中在最高的氧空位浓度(43%)下, aldol加成产物的产率可达65.7%, 巴豆醛选择性高达88.3%.这是由于氧空位浓度最大的CeO2, 在表面形成大量的氧空位, 增加了材料总体的Lewis碱性, 从而有效催化乙醛的aldol加成反应.这一发现为设计高效的aldol加成反应催化剂提供了新思路.
图 5

3.3 助剂
催化助剂是指与催化活性组分产生特定作用从而使得催化剂活性、选择性和稳定性得到明显改善的一类物质.根据助剂所起的不同作用, 又可以将其分为防止活性组分烧结、长大的结构型助剂和可以改变催化剂表观电子态的电子性助剂[75].由于稀土元素具有独特的外层电子分布特性, 其4f轨道未完全充满, 有多个能级、多个亚稳态, 可改变催化剂的酸碱性、活性组分的电子态等诸多特性, 因此可以作为电子促进剂, 对催化剂的电子结构进行调控[76].同时, 还可以将稳定性优良的稀土元素引入反应体系充当结构助剂[77], 对载体的结构和形貌进行调控, 起到增加体系稳定性的作用[22-24].例如, Cunha等[76]将La2O3引入Fe/Al合金, 使La2O3同时作为电子性助剂和结构助剂.其采用原位热处理法制备了Fe/Al合金同La2O3的混合物.从X射线光电子能谱(XPS)表征可以看出, La2O3的加入使得催化剂中Fe 2p3/2的结合能降低, 这说明La2O3可充当电子性助剂, La2O3的电子转移到了Fe上.
另外, 从表 2可以看出, 加入适量的La2O3可以避免Fe/Al合金的烧结, 从而增加该复合催化剂的比表面积, 提高复合材料催化分解甲烷的活性和稳定性.如图 6所示, 在Fe/Al为50/50的合金中, 当La2O3的质量分数为20%时, 催化甲烷分解的活性最好.当La2O3的质量分数增加到50%时, 催化性能反而有所降低.这是由于, La2O3加入量过多, 阻碍了活性位点的迁移, 从而导致催化反应活性有所降低.
表 2
表 2 未加修饰、用20% La2O3修饰的Fe/Al合金催化剂的平均颗粒尺寸和比表面积[76]Table 2. Average particle sizes (d) and BET surface areas (SBET) of 20% La2O3 modified and unmodified Fe/Al alloy catalysts下载:
导出CSV
催化剂 平均颗粒尺寸d/μm SBET/(m2·g-1) 50Fe 13 5 50FeLa(20) 13 11 35Fe 29 6 35FeLa(20) 12 11 图 6

Lercher团队[25]研究了在3%(质量分数)的La修饰的CeO2-ZrO2上负载0.5%(质量分数)的Rh, 利用该催化剂实现了乙酸向富氢气体的高效转化(重整反应), H2的选择性高达98%.反应过程如图式 3.
图式 3

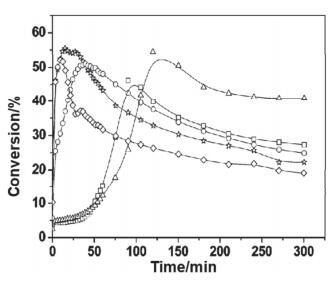
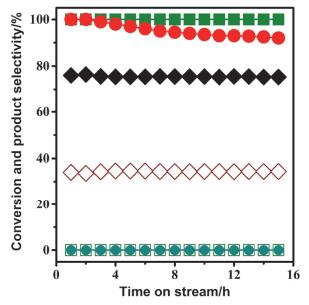
其研究发现在CeO2-ZrO2上负载Rh可以有效催化乙酸的重整, 但由于催化剂会烧结, 在反应15 h后会有20%的失活.为了解决催化剂烧结的问题, Lercher等[25]在催化剂中加入了少量La2O3.从图 7中可以看出, 反应15 h后, Rh/La2O3/CeO2-ZrO2催化剂的反应活性几乎没有下降, 这是因为La2O3的加入提高了晶格氧的迁移速率, 从而限制了积炭前驱体的浓度, 避免了大量积碳的产生, 增加了载体的稳定性, 有效地抑制了催化剂的失活, 使催化剂的稳定性提高.
图 7
 图 7. 在Rh/La2O3/CeO2-ZrO2催化剂上的乙酸水蒸气重整, 反应时间与转化率(红色●)、产物选择性之间的关系(H2(橄榄绿■); CO2 (黑色◆); CO (酒红色◇); (CH3)2CO (橄榄绿□); CH4 (深青色●)).反应条件: T=650 ℃, S/C=3, 体积时空速度(GHSV)=28000 h-1[25]Figure 7. Steam reforming of acetic acid over Rh/La2O3/CeO2-ZrO2 catalyst. The conversion (red ●) and product selectivities (H2(olive ■), CO2 (black ◆), CO(wine ◇), (CH3)2CO (olive □), CH4 (dark cyan ●)) as a function of time (T=650 ℃, S/C=3, gas hourly space velocity (GHSV)=28000 h-1)[25]
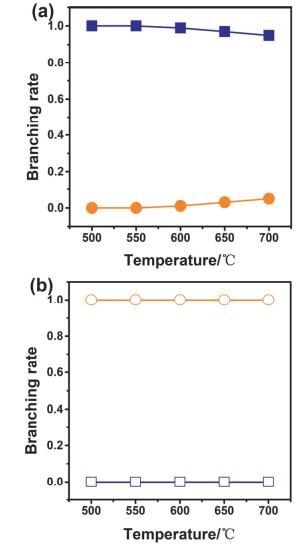
图 7. 在Rh/La2O3/CeO2-ZrO2催化剂上的乙酸水蒸气重整, 反应时间与转化率(红色●)、产物选择性之间的关系(H2(橄榄绿■); CO2 (黑色◆); CO (酒红色◇); (CH3)2CO (橄榄绿□); CH4 (深青色●)).反应条件: T=650 ℃, S/C=3, 体积时空速度(GHSV)=28000 h-1[25]Figure 7. Steam reforming of acetic acid over Rh/La2O3/CeO2-ZrO2 catalyst. The conversion (red ●) and product selectivities (H2(olive ■), CO2 (black ◆), CO(wine ◇), (CH3)2CO (olive □), CH4 (dark cyan ●)) as a function of time (T=650 ℃, S/C=3, gas hourly space velocity (GHSV)=28000 h-1)[25]镍在甲烷重整过程中有利于CO2的活化, 且价格相对便宜, 因此广泛用于甲烷干法重整反应[26-29](CH4+CO2→2CO+2H2), 但由于镍表面容易积碳, 导致其很容易失活, 因此需要找到合适的助催化剂来抑制表面积碳的产生[30].大多数稀土金属和氧化物都可以用作镍基催化剂的助剂, 钆、铈和镧等稀土元素被广泛用于修饰镍基催化剂[31-33], 其中借助镧氧化物来修饰镍基催化剂最为常见[32-34].综合之前的科研成果, 武志坚等[35]采用La或者La2O3对镍的(111)晶面进行修饰, 很可能改变了整体的反应路径, 在两种材料上不同反应路径的反应速率在图 8中表示.可以看出在La/Ni(111)表面, 反应被证实以P2反应(CH4+CO2→CH+3H+CO+O→CHO+3H+CO→2CO+2H2)为主, 而在La2O3/Ni(111)表面, 则以P4反应(CH4+CO2+La2O3→CH+CO2(La2O3-O)+3H→2CO+2H2+La2O3)为主. La2O3/Ni(111)在高温下的催化活性和稳定性优于La/Ni(111), 与La/Ni(111)相比, La2O3对Ni的(111)晶面进行修饰的主要区别是: La2O3/Ni(111)上CH→C+H转化速率较低, 且Ni在La2O3上的分散性良好, 有利于对CO2的吸附, 可以通过CO2+C→2CO途径来抑制焦炭的产生; 且在La2O3/Ni(111)上形成的La2O2CO3中间体(CO2(La2O2-O))可有效抑制积碳的产生, 从而提高了催化反应稳定性.
图 8
 图 8. 在La/Ni(111)、La2O3/Ni(111)催化剂上发生C—H键解离(深蓝色■、□)的速率, 以及(a)在La/Ni(111)上最容易发生的P2反应(橙色●)、在La2O3/Ni(111)上最容易发生的P4反应(橙色○)速率[35]Figure 8. The branching reaction rate of C—H dissociation (navy ■, □) and the most favorable pathway (a) P2 for La-Ni (orange ●) and (b) P4 for La2O3-Ni (orange ○) over La/Ni(111) and La2O3/Ni(111)[35]
图 8. 在La/Ni(111)、La2O3/Ni(111)催化剂上发生C—H键解离(深蓝色■、□)的速率, 以及(a)在La/Ni(111)上最容易发生的P2反应(橙色●)、在La2O3/Ni(111)上最容易发生的P4反应(橙色○)速率[35]Figure 8. The branching reaction rate of C—H dissociation (navy ■, □) and the most favorable pathway (a) P2 for La-Ni (orange ●) and (b) P4 for La2O3-Ni (orange ○) over La/Ni(111) and La2O3/Ni(111)[35]4. 总结与展望
稀土元素独特的外层电子结构特征导致其具有总体化学性质稳定、元素化学价态可变、多种配位形式、具有Lewis酸性等特点, 且可作为催化助剂调变载体的形貌、结构、酸碱性等, 因而受到研究者的广泛关注.本综述对稀土材料用于(热)催化领域的构效关系做了总结, 从稀土材料作为载体、Lewis酸(碱)活性中心、催化助剂三个不同的方面举例说明了稀土材料电子层结构与催化反应效果之间的关系, 说明稀土材料在催化领域中有着广泛的应用.
但是稀土材料用于催化领域也存在一些问题, 如合成方法不够环保、催化活性受载体限制较大、作为助剂的作用机理解释不明确等.因此需要研究人员的进一步研究, 以拓展稀土材料在催化领域的应用, 改善我国稀土资源利用率低的问题, 以期早日实现我国从稀土资源大国到稀土产业强国的转变.
-
-
[1]
Dong, H.; Du, S. R.; Zheng, X. Y.; Lyu, G. M.; Sun, L. D.; Li, L. D.; Zhang, P. Z.; Zhang, C.; Yan, C. H. Chem. Rev. 2015, 115, 10726. doi: 10.1021/acs.chemrev.5b00091
-
[2]
中华人民共和国国务院新闻办公室, 中国金属通报, 2012, 24, 20.Information Office of the State Council of the People's Republic of China. China Metal Bulletin 2012, 24, 20(in Chinese).
-
[3]
Wang, M.; Shen, M.; Jin, X. X.; Tian, J. J.; Li, M. L.; Zhou, Y. J.; Zhang, L. X.; Li, Y. S.; Shi, J. L. ACS Catal. 2019, 9, 4573.
-
[4]
Liang, M. F.; Borjigin, T.; Zhang, Y. H.; Liu, B. H.; Liu, H.; Guo, H. Appl. Catal. B: Environ. 2018, 243, 566.
-
[5]
Zhang, F.; Braun, G. B.; Shi, Y. F.; Sun, X. H.; Reich, N. O.; Zhao, D. Y.; Stucky, G. J. Am. Chem. Soc. 2010, 132, 2850.
-
[6]
孟双艳, 王明明, 吕柏霖, 薛群基, 杨志旺, 化学学报, 2018, 77, 1184. doi: 10.6023/A19070268Meng, S. Y.; Wang, M. M.; Lü, B. L.; Xue, Q. J.; Yang, Z. W. Acta Chim. Sinica 2019, 77, 1184(in Chinese). doi: 10.6023/A19070268
-
[7]
Xia, J. L.; Zhao, H. Y.; Pang, W. K.; Yin, Z. Y; Zhou, B.; He, G.; Guo, Z. P.; Du, Y. P. Chem. Sci. 2018, 9, 3421. doi: 10.1039/C7SC05185A
-
[8]
Ha, H. W.; Yun, N. J.; Kim, M. H.; Woo, M. H.; Kim, K. Electrochim. Acta 2006, 51, 3297.
-
[9]
Chen, P. L.; Chen, I. W. J. Am. Ceram. Soc. 1993, 76, 1577. doi: 10.1111/j.1151-2916.1996.tb07998.x
-
[10]
Frey, A. M.; Karmee, S. K.; de Jong, K. P.; Bitter, J. H.; Hanefeld, U. ChemCatChem 2013, 5, 594. doi: 10.1002/cctc.201200282
-
[11]
陈之尧, 刘捷威, 崔浩, 张利, 苏成勇, 化学学报, 2019, 77, 242. doi: 10.6023/A18100440Chen, Z. Y.; Liu, J. W.; Cui, H.; Zhang, L.; Su, C. Y. Acta Chim. Sinica 2019, 77, 242(in Chinese). doi: 10.6023/A18100440
-
[12]
宋天佑, 徐佳宁, 程功臻, 王莉, 无机化学(第三版), 高等教育出版社, 北京, 2014, p. 816.Song, T. Y.; Xu, J. N.; Cheng, G. Z.; Wang, L. Inorganic Chemistry (Third edition), Higher Education Press, Beijing, 2014, p. 816(in Chinese).
-
[13]
Risse, T.; Shaikhutdinov, S.; Nilius, N.; Sterrer, M.; Freund, H. J. Acc. Chem. Res. 2008, 41, 949. doi: 10.1021/ar800078m
-
[14]
Mattos, L. V.; Noronha, F. B. J. Catal. 2005, 233, 453. doi: 10.1016/j.apcata.2005.05.015
-
[15]
Summers, J. C.; Ausen, S. A. J. Catal. 1979, 58, 131. doi: 10.1016/0021-9517(79)90251-3
-
[16]
Yao, H. C.; Yao, Y. F. J. Catal. 1984, 86, 254.
-
[17]
Shyu, J. Z.; Otto, K.; Watkins, W. L. H.; Graham, G. W.; Belitz, R. K. J. Catal. 1988, 114, 23. doi: 10.1016/0021-9517(88)90005-x
-
[18]
Kašpar, J.; Fornasiero, P.; Graziani, M. Catal. Today 1999, 50, 285. doi: 10.1016/S0920-5861(98)00510-0
-
[19]
Fornasiero, P.; Monte, R. D.; Rao, G. R.; Kašpar, J.; Meriani, S.; Trovarelli, A.; Graziani, M. J. Catal. 1995, 151, 168. doi: 10.1006/jcat.1995.1019
-
[20]
Valsamakis, I.; Flytzani-Stephanopoulos, M. Appl. Catal. B: Environ. 2011, 106, 255.
-
[21]
Al-Sultan, F. S.; Basahel, S. N.; Narasimharao, K. Fuel 2018, 233, 796. doi: 10.1016/j.fuel.2018.06.130
-
[22]
Zheng, T. T.; He, J. J.; Zhao, Y. K.; Xia, W. Z.; He, J. L. J. Rare Earth 2014, 32, 97.
-
[23]
Korneeva, E. V.; Ivanova, A. S.; Bukhtiyarova, G. A.; Aleksandrov, P. V.; Zaikovskii, V. I.; Prosvirin, I. P.; Noskov, A. S. Kinet. Catal. 2011, 52, 579. doi: 10.1134/s0023158411030128
-
[24]
Mokhnachuk, O. V.; Soloviev, S. O.; Kapran, A. Y. Catal. Today 2007, 119, 145.
-
[25]
Lemonidou, A. A.; Vagia, E. C.; Lercher, J. A. ACS Catal. 2013, 3, 1919. doi: 10.1021/cs4003063
-
[26]
Li, X. Y.; Li, D.; Tian, H.; Zeng, L.; Zhao, Z. J.; Gong, J. L. Appl. Catal. B: Environ. 2017, 202, 683. doi: 10.1016/j.apcatb.2016.09.071
-
[27]
Sodesawa, T.; Dobashi, A.; Nozaki, F. Kinet. Catal. Lett. 1979, 12, 107. doi: 10.1007/bf02071433
-
[28]
Luo, J. Z.; Yu, Z. L.; Ng, C. F.; Au, C. T. J. Catal. 2000, 194, 198. doi: 10.1023/A:1019035220233
-
[29]
Singh, S.; Zubenko, D.; Rosen, B. A. ACS Catal. 2016, 6, 4199. doi: 10.1021/acscatal.6b00673
-
[30]
Pawar, V.; Appari, S.; Monder, D. S.; Janardhanan, V. M. Ind. Eng. Chem. Res. 2017, 56, 8448. doi: 10.1021/acs.iecr.7b01662
-
[31]
Dahdah, E.; Rached, J. A.; Aouad, S.; Gennequin, C.; Tidahy, H. L.; Estephane, J.; Aboukais, A.; Aad, E. A. Int. J. Hydrogen Energy 2017, 48, 12808. doi: 10.1007/s11356-016-7480-9
-
[32]
Liu, H. R.; Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Costa, P. D.; Gálvez, M. E. Fuel 2016, 182, 8. doi: 10.1016/j.fuel.2016.05.073
-
[33]
Tsipouriari, V. A.; Verykios, X. E. J. Catal. 1999, 187, 85. doi: 10.1006/jcat.1999.2565
-
[34]
Oemar, U.; Kathiraser, Y.; Mo, L.; Ho, X. K.; Kawi, S. Catal. Sci. Technol. 2016, 6, 1173. doi: 10.1039/C5CY00906E
-
[35]
Li, K.; He, F.; Yu, H. M.; Wang, Y.; Wu, Z. J. J. Catal. 2018, 364, 248.
-
[36]
May, Y. A.; Wang, W. W.; Yan, H.; Wei, S.; Jia, C. J. Chin. J. Catal. 2020, 41, 1017.
-
[37]
Zhou, Y.; Chen, A.; Ning, J.; Shen, W. J. Chin. J. Catal. 2020, 41, 928.
-
[38]
Si, R.; Flytzani-Stephanopoulos, M. Angew. Chem., Int. Ed. 2008, 47, 2884. doi: 10.1002/anie.200705828
-
[39]
Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Catal. Lett. 2001, 77, 87. doi: 10.1023/a:1012666128812
-
[40]
Zhang, F.; Chan, S. W.; Spanier, J. E.; Apak, E.; Jin, Q.; Robinson, R. D.; Herman, I. P. Appl. Phys. Lett. 2002, 80, 127. doi: 10.1063/1.1430502
-
[41]
Gatla, S.; Aubert, D.; Agostini, G.; Mathon, O.; Pascarelli, S.; Lunkenbein, T.; Willinger, M. G.; Kaper, H. ACS Catal. 2016, 6, 6151. doi: 10.1021/acscatal.6b00677
-
[42]
Nie, L.; Mei, D. H.; Xiong, H. F.; Peng, B.; Ren, Z. B.; Hernandez, X. I. P.; DeLaRiva, A.; Wang, M.; Engelhard, M. H.; Kovarik, L.; Datye, A. K.; Wang, Y. Science 2017, 358, 1419.
-
[43]
Spezzati, G.; Benavidez, A. D.; DeLaRiva, A. T.; Su, Y. Q.; Hofmann, J. P.; Asahina, S.; Olivier, E. J.; Neethling, J. H.; Miller, J. T.; Datye, A. K.; Hensen, E. J. M. Appl. Catal. B: Environ. 2019, 243, 36.
-
[44]
Liu, L. C.; Corma, A. Chem. Rev. 2018, 118, 4981.
-
[45]
Zhao, S.; Chen, F.; Duan, S. B.; Shao, B.; Li, T. B.; Tang, H. L.; Lin, Q. Q.; Zhang, J. Y.; Li, L.; Huang, J. H.; Bion, N.; Liu, W.; Sun, H.; Wang, A. Q.; Haruta, M.; Qiao, B. T.; Li, J.; Liu, J. Y.; Zhang, T. Nat. Commun. 2019, 10, 3824.
-
[46]
Liu, J. C.; Wang, Y. G.; Li, J. J. Am. Chem. Soc. 2017, 139, 6190.
-
[47]
Jones, J.; Xiong, H. F.; DeLaRiva, A. T.; Peterson, E. J.; Pham, H.; Challa, S. R.; Qi, G. S.; Oh, S.; Wiebenga, M. H.; Hernandez, X. I. P.; Wang, Y.; Datye, A. K. Science 2016, 353, 150. doi: 10.1126/science.aaf8800
-
[48]
Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Science 2003, 301, 935. doi: 10.1126/science.1085721
-
[49]
Dongil, A. B.; Pastor-Perez, L.; Escalona, N.; Sepulveda-Escribano, A. Carbon 2016, 101, 296. doi: 10.1016/j.apcata.2015.11.048
-
[50]
Reina, T. R.; Ivanova, S.; Centeno, M. A.; Odriozola, J. A. Appl. Catal. B: Environ. 2016, 187, 98. doi: 10.1016/j.apcatb.2016.01.031
-
[51]
Rodriguez, J. A.; Liu, P.; Stacchiola, D. J.; Senanayake, S. D.; White, M. G.; Chen, J. G. ACS Catal. 2015, 6, 6696. doi: 10.1021/acscatal.5b01755
-
[52]
Micoud, F.; Maillard, F.; Bonnefont, A.; Job, N.; Chatenet, M. Phys. Chem. Chem. Phys. 2010, 12, 1182. doi: 10.1039/b915244j
-
[53]
Li, S. W.; Xu, Y.; Chen, Y. F.; Li, W. Z.; Lin, L. L.; Li, M. Z.; Deng, Y. C.; Wang, X. P.; Ge, B. H.; Yang, C.; Yao, S. Y.; Xie, J. L.; Li, Y. W.; Liu, X.; Ma, D. Angew. Chem., Int. Ed. 2017, 56, 10761. doi: 10.1002/ange.201705002
-
[54]
Guo, Y.; Mei, S.; Yuan, K.; Wang, D. J.; Liu, H. C.; Yan, C. H.; Zhang, Y. W. ACS Catal. 2018, 8, 6203. doi: 10.1021/acscatal.7b04469
-
[55]
Xu, J.; Chen, X. Y.; Xu, Y. S.; Du, Y. P.; Yan, C. H. Adv. Mater. 2019, 1806461.
-
[56]
Pudukudy, M.; Yaakob, Z.; Takriff, M. S. Energy Convers. Manage. 2016, 126, 302. doi: 10.1016/j.enconman.2016.08.006
-
[57]
Pudukudy, M.; Yaakob, Z.; Jia, Q. M.; Takriff, M. S. Appl. Surf. Sci. 2019, 467~468, 236.
-
[58]
Lessard, J. D.; Valsamakis, I.; Flytzani-Stephanopoulos, M. Chem. Commun. 2012, 48, 4857. doi: 10.1039/c2cc31105d
-
[59]
Akhmedov, V. M.; Al-Khowaiter, S. H. Catal. Rev.-Sci. Eng. 2007, 44, 455.
-
[60]
Jacinto, S.; Ace, M.; Delgado, J. J.; Goguet, A.; Hardacre, C.; Morgan, K. ChemCatChem 2011, 3, 394. doi: 10.1002/ange.201102066
-
[61]
Wang, Z. Q.; Wang, D.; Gong, X. Q. ACS Catal. 2020, 10, 586.
-
[62]
Huang, Z. Q.; Zhang, T. Y.; Chang, C. R.; Li, J. ACS Catal. 2019, 9, 5523.
-
[63]
Sun, J. M.; Zhu, K. K.; Gao, F.; Wang, C. M.; Liu, J.; Peden, C. H. F.; Wang, Y. J. Am. Chem. Soc. 2011, 133, 11096. doi: 10.1021/ja204235v
-
[64]
Ogo, S.; Onda, A.; Iwasa, Y.; Hara, K.; Fukuoka, A.; Yanagisawa, K. J. Catal. 2012, 296, 24. doi: 10.1016/j.jcat.2012.08.019
-
[65]
Zhang, H. B.; Ibrahim, M. Y. S.; Flaherty, D. W. J. Catal. 2018, 361, 290. doi: 10.1016/j.jcat.2018.02.030
-
[66]
Pang, J. F.; Zheng, M. Y.; He, L.; Li, L.; Pan, X. L.; Wang, A. Q.; Wang, X. D.; Zhang, T. J. Catal. 2016, 344, 184.
-
[67]
Moteki, T.; Flaherty, D. W. ACS Catal. 2016, 6, 4170. doi: 10.1021/acscatal.6b00556
-
[68]
Dai, J. J.; Zhang, H. B. Sci. China Mater. 2019, 62, 1642.
-
[69]
Cota, I.; Ramírez, E.; Medina, F.; Layrac, C.; Tichit, D.; Gérardin, C. J. Mol. Catal. A: Chem. 2016, 412, 101.
-
[70]
Yan, T. T.; Dai, W. L.; Wu, G. J.; Lang, S.; Hunger, M.; Guan, N. J.; Li, L. D. ACS Catal. 2018, 8, 2760. doi: 10.1021/acscatal.8b00014
-
[71]
Wang, Z.; Fongarland, P.; Lu, G. Z.; Essayem, N. J. Catal. 2014, 318, 108. doi: 10.1016/j.jcat.2014.07.006
-
[72]
Meis, N. N. A. H.; Bitter, J. H.; de Jong, K. P. Ind. Eng. Chem. Res. 2010, 49, 1229. doi: 10.1021/ie901114d
-
[73]
Álvarez, M. G.; Plísková, M.; Segarra, A. M.; Medina, F.; Figueras, F. Appl. Catal. B: Environ. 2012, 113~114, 212.
-
[74]
Liang, Z.; Jiang, D. H.; Fang, G. Q.; Leng, W. H.; Tu, P. X.; Tong, Y. Q.; Liu, L.; Ni, J.; Li, X. N. ChemistrySelect 2019, 4, 4364. doi: 10.1002/slct.201900712
-
[75]
甄开吉, 李荣生, 王国甲, 毕颖丽, 阚秋斌, 催化作用基础(第三版), 科学出版社, 北京, 2004, p. 239Zhen, K. J.; Li, R. S.; Wang, G. J.; Bi, Y. L.; Kan, Q. B. Catalysis Basics (Third edition), Science Press, Beijing, 2004, p. 239. (in Chinese).
-
[76]
Cunha, A. F.; Mahata, N.; Órfão, J. J. M.; Figueiredo, J. L. Energy Fuels 2009, 23, 4047.
-
[77]
Gao, J.; Hou, Z. Y.; Guo, J. Z.; Zhu, Y. H.; Zheng, X. M. Catal. Today 2008, 131, 278. doi: 10.1016/j.cattod.2007.10.019
-
[1]
-

图 3 在不同Ir负载量的Ir/Ce催化剂上, Ir-Ir(■)和Ir-O(●)配位数(数据, 右轴)与催化选择性(棒, 左轴) (CO(橙色), CH4(深青色))的关系[53]
Figure 3 The coordination number (CN) of Ir-Ir (■) and Ir-O (●) shells (data, right axis) correlate to catalytic selectivity (bars, left axis) (CO (orange), CH4 (dark cyan)) with Ir/Ce catalysts at various Ir loadings[53]
图式 1 乙醛/乙醇混合物aldol加成的反应路径
Scheme 1 Reaction network of aldol condensation from acetaldehyde/ethanol mixtures
图 4 在Zn/Beta、Y/Beta、Zn-Y-Beta催化剂上, 分别用乙醇(左)、乙醇和巴豆醛(右)作为反应物的催化结果.反应条件:重量时空速度(WHSV)=1 h-1, 反应温度T=623 K[70]
Figure 4 Modulation experiments about crotonaldehyde and ethanol over Zn/Beta, Y/Beta and Zn-Y-Beta catalysts (left: ethanol; right: ethanol and crotonaldehyde cofeeding). Reaction conditions: weight hourly space velocity (WHSV)=1.0 h-1, reaction temperature of T=623 K[70]
图 7 在Rh/La2O3/CeO2-ZrO2催化剂上的乙酸水蒸气重整, 反应时间与转化率(红色●)、产物选择性之间的关系(H2(橄榄绿■); CO2 (黑色◆); CO (酒红色◇); (CH3)2CO (橄榄绿□); CH4 (深青色●)).反应条件: T=650 ℃, S/C=3, 体积时空速度(GHSV)=28000 h-1[25]
Figure 7 Steam reforming of acetic acid over Rh/La2O3/CeO2-ZrO2 catalyst. The conversion (red ●) and product selectivities (H2(olive ■), CO2 (black ◆), CO(wine ◇), (CH3)2CO (olive □), CH4 (dark cyan ●)) as a function of time (T=650 ℃, S/C=3, gas hourly space velocity (GHSV)=28000 h-1)[25]
图 8 在La/Ni(111)、La2O3/Ni(111)催化剂上发生C—H键解离(深蓝色■、□)的速率, 以及(a)在La/Ni(111)上最容易发生的P2反应(橙色●)、在La2O3/Ni(111)上最容易发生的P4反应(橙色○)速率[35]
Figure 8 The branching reaction rate of C—H dissociation (navy ■, □) and the most favorable pathway (a) P2 for La-Ni (orange ●) and (b) P4 for La2O3-Ni (orange ○) over La/Ni(111) and La2O3/Ni(111)[35]
表 1 在650 ℃下氧化物催化丁烷裂解的催化结果[60]
Table 1. Catalytic performance on the oxidative cracking of n-butane at 650 ℃[60]
催化剂 反应速率/(mmol·min-1·g-1 选择性/% 产率/% 烯烃 烷烃 COx C2H4 C3H6 La2O3 0.84 28.8 3.2 23.8 1.7 1.7 CeZrO4 0.86 17.9 2.2 69.5 1 0.3 ZrO2 0.38 16.3 3.8 24.5 0.4 0.5 TiO2 0.51 17.3 2.1 26.3 1 0.3 Nb2O5 0.25 20.5 5.3 27.2 0.2 0.3  下载: 导出CSV
下载: 导出CSV
-












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 119
- 文章访问数: 2882
- HTML全文浏览量: 933
 下载:
下载:
