Received Date:
16 January 2020 Available Online:
15 April 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21433013, 21303129), Outstanding Youth Fund of Jiangxi Province (No. 20192BCB23028) and the Science and Technology Project of Jiangxi Province (No. 20192BCD40017)
Abstract:
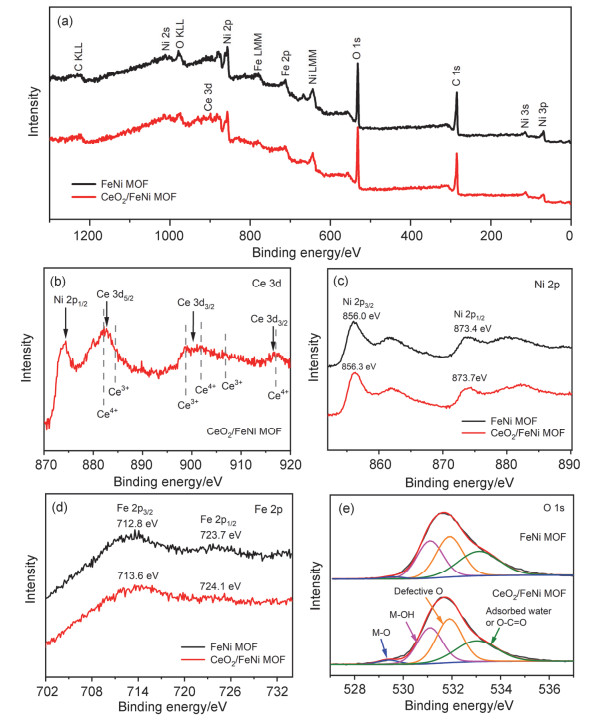
Oxygen evolution reaction (OER) is a crucial half reaction of electrochemical water splitting and metal-air batteries. But its sluggish four-electron reaction leads to a high overpotential. Current commercial OER catalysts are mainly noble metal-based materials, but their high cost restricts their broad application. Therefore, extensive efforts have been devoted to exploring low-cost and efficient OER catalysts. Nonprecious metal-based materials have been regarded as promising OER catalyst candidates, due to their abundancy on the earth, controllable morphologies and tunable chemical states. Among various nonprecious metal-based materials, metal-organic frameworks (MOFs) have attracted much attention, because of their large specific surface area and rich metal centers. However, their poor electrochemical activities, stabilities and conductivities severely affect their application in OER catalysis. To improve the activities of MOFs, several methods have been adopted, such as synthesizing ultrathin nanosheets, growing MOFs on nickel foam or carbon cloth, doping heteroatoms, and introducing synergistic interactions between two materials. In 1970, Wagner proposed a space-charge theory, which indicates that the carrier property can be tuned through adjusting interface. Inspired by this theory, constructing metal oxide-catalyst interface seems to be a promising strategy to improve activities of catalysts. CeO2 is a well-known cocatalyst due to its reversible Ce3+/Ce4+ redox. Previous works have demonstrated that OER performance can be effectively improved through introducing CeO2 since it can speed up the electron mobility and induce strong interaction between CeO2 and metal sites. In this work, an efficient OER catalyst was achieved through introducing CeO2 into FeNi MOF catalyst. FeNi MOF nanosheet arrays grown on nickel foam was firstly prepared via a solvothermal process. Then CeO2 nanoclusters (5 nm) were coated onto FeNi MOF surface by electrodeposition. A series of characterizations were employed to study the morphology, structure and surface electronic state information of the as-obtained CeO2/FeNi MOF. From X-ray photoelectron spectroscopic analysis, the doping of CeO2 clusters and the strong electronic interaction between CeO2 clusters and FeNi MOF induce the formation of Fe/Ni-O-Ce bonds and optimize the electronic structures of Fe/Ni sites, which will enhance OER activities. The OER performance tests confirm that CeO2/FeNi MOF indeed exhibits a superior OER activity than FeNi MOF alone. The hybrid catalyst delivers a higher mass activity (235.4 A·g-1) and a faster turnover frequency (0.065 s-1) than those of FeNi MOF (43.8 A·g-1, 0.018 s-1). Compared with FeNi MOF, CeO2/FeNi MOF also shows better OER kinetics, as evidenced by a decreased Tafel slope, a reduced charge transfer resistance. Besides, CeO2/FeNi MOF presents an outstanding stability (50 h, 50 mA·cm-2). All these features make our CeO2/FeNi MOF a potential catalyst in the future application. The interfacial strategy through introducing CeO2 to modulate Fe and Ni active sites may open a door for developing high-performance OER catalysts in future.
Figure 2.
(a) TEM image of CeO2/FeNi MOF. (b) High-resolution TEM image of CeO2/FeNi MOF. (c) Energy disperse X-ray spectrum of CeO2/FeNi MOF. (d) XRD patterns of FeNi MOF and CeO2/FeNi MOF. Inset is the magnification of XRD patterns
Figure 4.
(a) OER polarization curves of NF, RuO2/C, CeO2, FeNi MOF and CeO2/FeNi MOF in 1 mol•L-1 KOH solution. The inset is the magnification of the part marked by dash line. (b) Comparisons of overpotential η at 50 and 100 mA•cm-2. (c) Tafel plots of NF, RuO2/C, CeO2, FeNi MOF and CeO2/FeNi MOF. (d) Nyquist plots at the η of 256 mV of these above catalysts
图 5.
(a) FeNi MOF和CeO2/FeNi MOF在η为240 mV下的质量活性和TOF. (b) FeNi MOF和CeO2/FeNi MOF在1.28 V vs. RHE电压下电流密度-扫描速率图
Figure 5.
(a) Mass activities and TOFs of these prepared samples at η of 240 mV. (b) The plot of the capacitive currents versus scan rates for FeNi MOF and CeO2/FeNi MOF at 1.28 V vs. RHE
Figure 6.
(a) Chronopotentiometric curves of FeNi MOF and CeO2/FeNi MOF at 50 mA•cm-2 in 1 mol•L-1 KOH. (b) XRD patterns of CeO2/FeNi MOF before and after OER stability test
Figure 2
(a) TEM image of CeO2/FeNi MOF. (b) High-resolution TEM image of CeO2/FeNi MOF. (c) Energy disperse X-ray spectrum of CeO2/FeNi MOF. (d) XRD patterns of FeNi MOF and CeO2/FeNi MOF. Inset is the magnification of XRD patterns
Figure 4
(a) OER polarization curves of NF, RuO2/C, CeO2, FeNi MOF and CeO2/FeNi MOF in 1 mol•L-1 KOH solution. The inset is the magnification of the part marked by dash line. (b) Comparisons of overpotential η at 50 and 100 mA•cm-2. (c) Tafel plots of NF, RuO2/C, CeO2, FeNi MOF and CeO2/FeNi MOF. (d) Nyquist plots at the η of 256 mV of these above catalysts
图 5
(a) FeNi MOF和CeO2/FeNi MOF在η为240 mV下的质量活性和TOF. (b) FeNi MOF和CeO2/FeNi MOF在1.28 V vs. RHE电压下电流密度-扫描速率图
Figure 5
(a) Mass activities and TOFs of these prepared samples at η of 240 mV. (b) The plot of the capacitive currents versus scan rates for FeNi MOF and CeO2/FeNi MOF at 1.28 V vs. RHE
Figure 6
(a) Chronopotentiometric curves of FeNi MOF and CeO2/FeNi MOF at 50 mA•cm-2 in 1 mol•L-1 KOH. (b) XRD patterns of CeO2/FeNi MOF before and after OER stability test


 下载:
下载:





 下载:
下载:







 下载:
下载:
