State Key Laboratory of Chemical Resource Engineering, College of Chemistry, Institute of Computational Chemistry, Beijing University of Chemical Technology, Beijing 100029, China
Received Date:
26 November 2019 Available Online:
15 May 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21672018), the State Key Laboratory of Physical Chemistry of Solid Surfaces (Xiamen University) (No. 201811) and the Fundamental Research Funds for the Central Universities (No. XK1802-6)
Abstract:
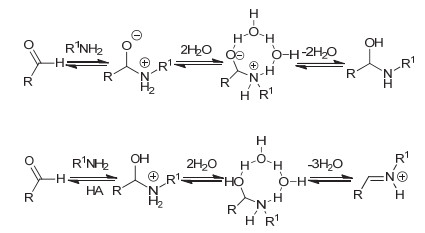
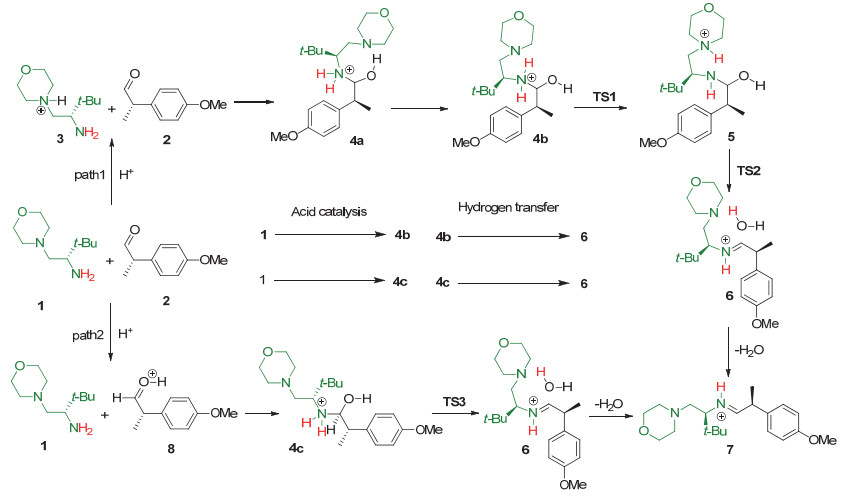
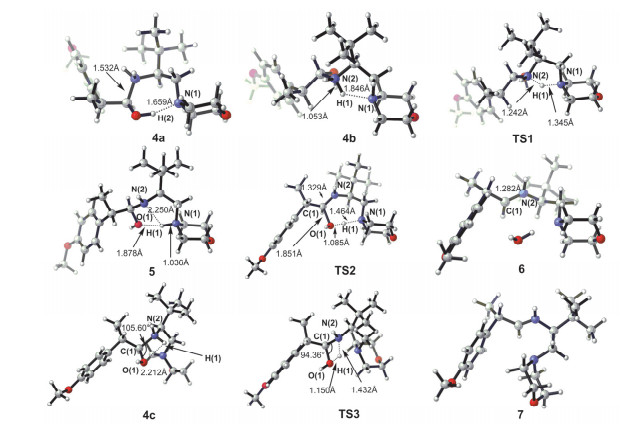
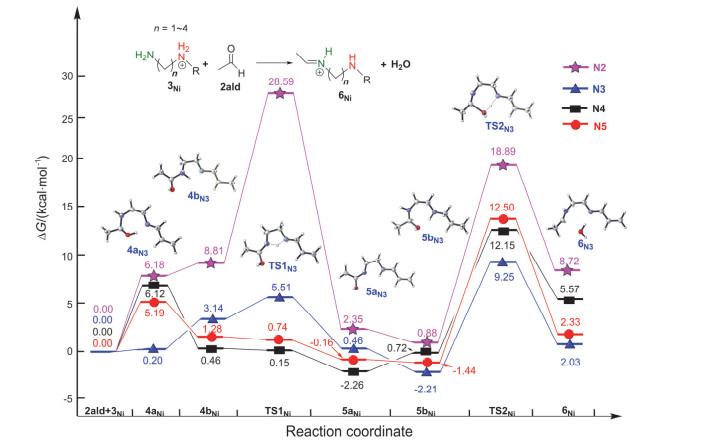
Imines and the intermediate methylamine by the aldimine condensation of primary amines with aldehydes have a potential application in the field of pharmacy, life science, catalysis, material science, etc. In this reaction, the hydrogen transfer in the dehydration step normally prefers the pathway via a water bridge in aqueous solution or a directly dehydration in organic solvent. It is a different mechanism for the aldimine condensation of amine owning neighbouring nitrogenous heterocycle. Herein we investigated the mechanism of aldimine condensation of primary amine containing nitrogenous heterocycle with aldehyde in dichloromethane under acidic conditions using density functional theory (DFT) at ωB97X-D/6-31++G(d, p) level, the calculated results show that compared with specific acid catalysis, the heterocyclic nitrogen with stronger basicity is easier to be protonated than the oxygen of carbonyl group. The whole reaction proceeds two hydrogen transfer steps via nitrogen bridge owning an energy span of 13.08 kcal/mol. The rate-determing step is the second hydrogen transfer step. In each step the heterocyclic nitrogen is a bridge to assist the hydrogen transfer, which could reduce the free energy barrier of the aldimine condensation. It is unfavorable for the reaction pathway via directly hydrogen transfer with a four-membered ring transition state owning a free energy barrier of 32.73 kcal/mol, and the reaction pathway via a water bridge is not located. Meanwhile, the energy barriers increased for systems in which the N atom in heterocycle of primary amine is replaced by P/As atoms. The rate-determining step changes from the second hydrogen transfer step for N system to the first hydrogen transfer step for As system. The position effect of adjacent nitrogen atom is also investigated. The γ position owns the highest reactivity of the aldimine condensation, which implies that the ring strain plays an important role in the aldimine condensation of primary amine containing nitrogenous heterocycle with aldehyde. This theoretical study may provide insights to unveil the nature of aldimine condensation of aldehyde and primary amine owning nitrogeneous heterocycle.
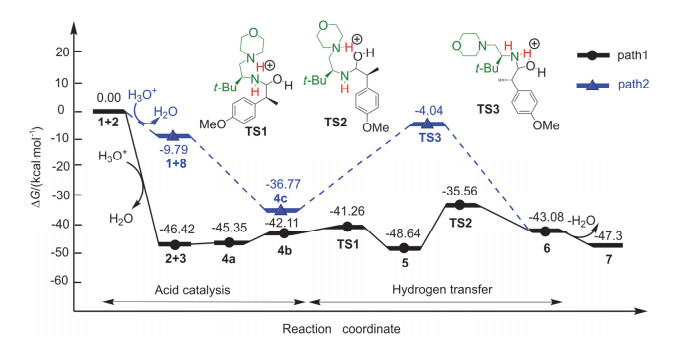
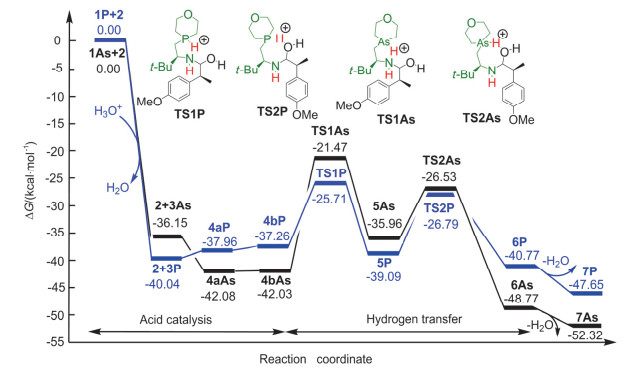
Figure 3.
Free energy profiles for the aldimine condensation of amine with nitrogenous heterocycle and aldehyde in acidic conditions (all energies are denoted in kcal/mol)
Leth, L. A.; Naesborg, L.; Reyes-Rodriguez, G. J.; Tobiesen, H. N.; Iversen, M. V.; Jorgensen, K. A. J. Am. Chem. Soc. 2018, 140, 12687. doi: 10.1021/jacs.8b07394
[3]
Rezayee, N. M.; Lauridsen, V. H.; Naesborg, L.; Nguyen, T. V. Q.; Tobiesen, H. N.; Jorgensen, K. A. Chem. Sci. 2019, 10, 3586. doi: 10.1039/C9SC00196D
[4]
Liu, Y.; Yue, X.; Luo, C.; Zhang, L.; Lei, M. Energy Environ. Mater. 2019, 2, 292. doi: 10.1002/eem2.12050
Lu, T.; Chen, F. J. Comput. Chem. 2012, 33, 580. doi: 10.1002/jcc.22885
[38]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision B. 01. Gaussian, Inc., Wallingford, 2010.
Figure 3
Free energy profiles for the aldimine condensation of amine with nitrogenous heterocycle and aldehyde in acidic conditions (all energies are denoted in kcal/mol)

 下载:
下载:

 下载:
下载:








 下载:
下载:
