
图 1.
磷光软盐配合物的合成
Figure 1.
The syntheses of phosphorescent soft salt complexes

过渡金属配合物是一类重要的光电材料, 它们具有丰富的氧化还原性质、光物理性质、强磁性等, 因此在学术研究和工业生产中占据举足轻重的地位[1~5].尤其是, 过渡金属配合物具有较高的发光量子效率、显著的斯托克斯位移、易调节的发光颜色等优异的光物理性质, 在有机光电子领域具有巨大的应用前景[6~10].近年来, 它们在各种光电领域, 如有机发光二极管、太阳能电池、化学传感中引起了广泛的关注并得到了迅速发展[11, 12].其中, 磷光离子型金属配合物因其具有分子设计简单、制备容易等特点, 引起了研究人员极大的研究兴趣[13~15].目前, 磷光离子型金属配合物已成功应用于发光电化学电池、环境监测、生物成像等领域[16~22].
磷光软盐配合物是不含碱金属和卤素等反离子, 而是仅由金属配合物型阴阳离子组成的一种新型配合物.之所以称这类材料为软盐, 是因为它们的离子半径较大, 离子间键合作用主要为弱静电或范德华作用力, 使得其晶格能较低.这类材料可将不同性质的金属配合物进行组合, 具有结构和光谱性质易调控的优势.然而, 尽管目前磷光阳离子与阴离子配合物已经研究得较为深入, 磷光软盐配合物的研究仍处于起步阶段.本文主要介绍磷光软盐配合物的光物理性质及其在各种光电应用研究领域的进展.首先, 我们详细介绍了磷光软盐配合物的合成与光物理性质.其次, 阐述了它们在光电领域的应用研究.最后, 我们总结并展望了磷光软盐配合物在光电应用研究领域的前景和面临的挑战.
2009年, Thompson课题组[23]报道了首例基于铱(Ⅲ)配合物的磷光软盐分子.他们将黄光和橙光发射的阳离子铱(Ⅲ)配合物分别与蓝光和绿光发射的阴离子铱(Ⅲ)配合物在水溶液中通过盐复分解反应进行制备.随后, 通过萃取与重结晶方法提纯得到了磷光软盐配合物.如图 1所示, 磷光软盐配合物S1由绿光阴离子铱(Ⅲ)配合物Na+[Ir(mppyH)2(CN)2]-(A1, mppyH=2-(对甲苯基)吡啶)与黄光阳离子铱(Ⅲ)配合物[Ir(mppyH)2(CNdt)2]+Cl-(C1, CNdt=2-甲基-N-甲基丙烷-2-氨基)反应制备获得; 磷光软盐配合物S2由蓝光阴离子铱(Ⅲ)配合物Na+[Ir(dFppy)2(CN)2]-(A2, dFppyH=4, 6-二氟苯基吡啶)与橙光阳离子铱(Ⅲ)配合物[Ir(mppyH)2(dtbubpy)]+Cl-(C2, dtbubpy=4, 4'-二叔丁基-2, 2'-联吡啶)制备而成.目前, 大多磷光软盐配合物的制备都基于该方法, 即根据阴阳离子自身的溶解性选择不同的溶剂通过盐复分解反应制备而得.
磷光软盐配合物的发射光谱由阴阳离子配合物两部分光谱叠加组成.与单组分发光材料相比, 磷光软盐配合物具有更丰富的激发态性质, 不同组分之间还有能量效应.因此, 详细研究这类材料的光物理性质对设计、制备具有特殊要求的发光分子并将它们应用于不同的光电领域具有重要的意义. Thompson教授报道的配合物A1和C1在乙腈溶液中发射波长分别为472 nm(量子效率=70%, 磷光寿命=4.0 μs)与458 nm(量子效率=38%, 磷光寿命=36.7 μs).类似地, 配合物A2在448 nm处显示磷光发射, 量子产率为70%, 发光寿命为4.1 μs, 而配合物C2在波长586 nm处具有发射峰, 量子产率为21%, 发光寿命为0.4 μs.进一步, 他们研究了由这些阴阳离子配合物制备的磷光软盐配合物S1和S2的光物理性质.结果表明, 它们的发射光谱表现出浓度依赖性, 说明阴离子配合物的发射通过Dexter能量转移被阳离子配合物淬灭.以S2为例, 由于A2发光量子效率高于C2, 在相对较低浓度下(10-5 mol•L-1), S2发射峰以蓝光阴离子配合物为主导.随着S2浓度从10-5 mol•L-1增加到10-3 mol•L-1, A2发射强度逐渐降低, 并且当浓度大于10-3 mol•L-1时, 只能观察到C2在586 nm处所显示的橙光发射.基于双分子淬灭模型, 计算出淬灭速率常数kq为1.71×1010 L•mol-1•s-1, 该值接近在乙腈中的扩散极限(2×1010 L•mol-1•s-1).
同年, De Cola课题组[24]利用绿光阴离子配合物NBu4+[Ir(ppy)2(CN)2]-(A3, ppy=2-苯基吡啶)与黄光阳离子铱(Ⅲ)配合物[Ir(dFppy)2(bpy)]+Cl-(C3, bpy=4, 4′-联吡啶), 以及蓝光阴离子铱(Ⅲ)配合物A2与橙光阳离子铱(Ⅲ)配合物[Ir(ppy)2(bpy)]+Cl-(C4)制备磷光软盐配合物(S3和S4, 图 2a). S3的X射线单晶结构表明, 这种软盐配合物形成了一个三维多孔网络.在该网络中, 二氯甲烷、乙酸戊酯、乙酸乙酯、二乙醚、甲苯或蒽醌等小溶剂分子可以很容易地插入(图 2b).随后, 他们研究了S3和S4晶体真空条件下是否存在客体分子包合情况下发射光谱的性质.在无氧二氯甲烷溶液中, 配合物C3发射的黄光波长为564 nm, 而A3发射的蓝绿光波长为502 nm.干燥的S3晶体的发射在591 nm处表现出红移现象.同样, 干燥的S4晶体在596 nm处表现出发光红移的现象, 而A2和C4发射波长则分别为460 nm和554 nm.与相对应的单核配合物相比, S3和S4的发射波长的红移归因于晶体网络中存在的相反电荷铱(Ⅲ)配合物的C^N配体之间的强π-π相互作用, 促进了Dexter能量从高能态阴离子铱(Ⅲ)供体A3和A2转移到低能态阳离子铱(Ⅲ)受体C3和C4, 并且促进了激基复合物的形成和相应低能激发态的发射.重要的是, 通过在其多孔网络内嵌入客体分子, 可以有效地调节晶体S3和S4的磷光发射性质.例如, 与干燥晶体相比, 甲苯或蒽醌分子插入S4晶体后, 其在590 nm和580 nm处的发射峰分别发生蓝移, 该现象可归因于分子间π-π相互作用被破坏.此外, 当甲苯分子被吸收到S4中后软盐的发光强度增强, 而当蒽醌分子插入S4晶体中后, 由于光诱导电子从S4给体转移到蒽醌受体, 其发射强度显著降低.

Godbert课题组[25]设计、合成了以双齿型阴离子作为辅助配体的绿光阴离子铱(Ⅲ)配合物(图 3中的A4a和A4b), 它们的发射峰分别位于530 nm和536 nm处, 其磷光量子效率分别为69%和58%.随后, 他们分别用配合物A4a和A4b与阳离子配合物[Ir(ppy)2(pyam)]+Cl-(C5, pyam=2-吡啶甲基胺)合成了具有高磷光量子效率的软盐配合物S5a和S5b.与之前报道的软盐配合物不同, S5a和S5b中阳离子配合物为供体单元, 而阴离子配合物则成为受体. S5a和S5b的发射分别来自于发射波长在480 nm, 525 nm和486 nm, 532 nm处的两种离子配合物, 量子产率分别为81%和83%.

Stagni课题组[26]用dFppy为环金属配体, 4-苯甲腈四唑酸盐作为辅助配体的蓝光发射阴离子铱(Ⅲ)配合物A5(图 3), 与红光发射的[Ir(ppy)2(ptz-Me)]+Cl-阳离子配合物(ptz-Me=5-甲基-2-苯基四唑)(C6), 制备了白光软盐配合物.在二氯甲烷溶液中, 配合物A5表现出典型的3LC/3MLCT态发射, 发射波长分别为462 nm和492 nm, 量子产率为3.3%.相比之下, 配合物C6的发射谱较宽, 波长为686 nm, 量子产率为2.7%.在二氯甲烷溶液中, 由于配合物S6中阴离子和阳离子配合物之间能量传递效率较低, 并且它们的发射强度几乎相同, 因此软盐配合物显示出量子效率为2.8%的纯白光(CIE: x=0.3288, y=0.3284).同时, A5中大量4-苯甲腈四唑酸盐的存在可能限制了它与阳离子C6的相互作用, 进一步减弱了其能量传递效率.
为了研究软盐配合物中两个互补发光体之间能量传递过程及其调控手段, Zysman-Colman等[27]报道了首个基于三组分杂金属软盐配合物S7(图 3), 其中包括了红色发光的阳离子钌(Ⅱ)配合物[Ru(dtBubby)3]2+Cl2-(C7, dtBubby=4, 4'-二叔丁基-2, 2'-联吡啶)以及蓝绿色发光的阴离子铱(Ⅲ)配合物NBu4+[Ir(ppy)2(CN)2]-(A3, ppy=2-苯基吡啶).对其光物理性质进行详细研究后发现, 改变溶剂或配合物浓度可以很容易地调控两种组分的相对发射强度.以这种方式, 可以观察到蓝绿色到橙红色的发射, 包括近白色的发射.因此, 他们认为这种杂金属软盐配合物是制备白光材料的潜在候选.
相对于单一组分的发光材料, 磷光软盐配合物的光物理性质更容易调节.首先, 软盐配合物具有两个发光中心, 只需修饰阴阳离子配合物中的一个便可实现光电性质的调控.其次, 软盐配合物中的基于金属配合物的阴阳离子组分通常存在能量传递过程, 通过外界刺激而调控两个组分之间的静电作用力便可影响它们之间的能量传递效率, 从而实现对光物理性质的调控.
基于软盐配合物优异的光物理性质与调控简单的特点, 目前研究人员已成功将它们用于不同光电领域, 包括有机发光二极管、生物成像、光动力治疗、电致发光变色、共晶、自组装等.
在电致发光器件领域, 中性铱(Ⅲ)配合物在有机发光二极管中得到了广泛的应用[28~33], 而阳离子铱(Ⅲ)配合物作为发光电化学电池中的发光材料得到了更为详尽的探索.然而, 研究表明, 以软盐配合物作为发光材料制备的有机发光二极管器件同样具有良好的性能[21].在发光器件中使用软盐配合物的主要优势在于能够在一个复合离子中引入两个磷光中心, 同时通过非共价相互作用控制两个金属配合物分离.
Thompson课题组[23]率先将基于铱(Ⅲ)配合物的磷光软盐分子用于有机发光二极管器件中.该有机发光二极管器件的外量子效率EQE为4.7%, 亮度超过7428 cd•m-2(发射波长586 nm处).研究表明有机发光二极管的良好性能直接与软盐中两种离子组分HOMO-LUMO水平的合理分布有关.然而, 固态下S2表现出的相对较低的量子产率(18%)极大地限制了器件的外量子效率.
Mayer课题组[34]设计、制备了由阴离子配合物NBu4+[Ir(dFppy)2(NCS)2]-(A6)和阳离子配合物[Ir(ppy)2-(bpy-non)]+PF6-(C8, bpy-non=4, 4'-二壬基-2, 2'-联吡啶)组成的软盐配合物并将其应用于有机发光二极管领域(图 4).将壬基链引入阳离子配合物C8中, 有助于提高所制备软盐配合物S8在有机溶剂中的溶解度, 从而改善溶液法制备下器件的薄膜形貌.软盐配合物S8在溶液状态、固体状态和电致发光器件中均表现为550 nm左右的黄色发光.由其制备所得的有机发光二极管的外量子效率为0.66%, 亮度为1114 cd•m-2.

此外, Mayer课题组[35]报道了两种基于双核阳离子铱(Ⅲ)配合物和单核阴离子铱(Ⅲ)配合物的软盐配合物S9和S10, 并研究了它们作为发光材料在有机发光二极管中的应用(图 4).桥接双核阳离子配合物中两个铱中心的单元由C9的咔唑衍生物或C10的苯乙烯基组成.双核配合物C9和C10在二氯甲烷溶液中均表现出较低的量子产率, 分别为9.1%和15.0%, 这是两种铱发光团之间的猝灭所导致的.由于苯乙烯桥有利于两个金属铱中心之间的连通, 软盐配合物S10的电致发光性能表现不如S9, 它们最大峰值电流效率分别为0.06 cd•A-1和0.44 cd•A-1, 最大亮度分别为101 cd•m-2和1022 cd• m-2.
基于荧光探针的生物成像为表征亚细胞分辨率下细胞内的形态细节提供了可靠的方法[36~40].然而, 大多数传统荧光探针会由于自发荧光和散射光而受到干扰, 这增加了背景噪声并降低了检测信噪比.最近, 磷光寿命成像作为一种新兴成像技术, 为扣除背景荧光干扰提供了一种有效方法[41~44].此外, 以寿命作为检测信号, 它与激发激光强度、目标分子浓度和光漂白无关, 这非常有利于生物成像应用.因此, 发展基于磷光过渡金属配合物的生物成像探针至关重要.
尽管基于磷光金属配合物的探针具有诸多优势, 然而目前报道的探针大多基于单一发射强度变化.细胞内不同区域形态的多样性可能会影响发射信号的准确性, 当研究细胞内生物分子的动态变化过程时可能会导致较大的误差.因此, 定量地测量细胞内生物分子的实际浓度或浓度的相对变化较为困难.目前, 比率成像通常被用于解决此问题, 它可以同时记录两个不同波长的相对变化, 从而能够提供对环境影响的内置校正, 降低细胞内环境对信号影响的程度[45~48].然而, 最近发展的用于生物分子成像的大多数比率探针是基于有机染料或纳米颗粒, 由于磷光金属配合物复杂的激发态性质, 设计并制备基于这类材料的探针难度较大, 目前仅有少数基于磷光金属配合物的生物探针被报道.因此, 设计基于磷光金属配合物的比率探针仍然是当前的挑战.
考虑到基于过渡金属配合物的软盐的两个发射波长可以通过对阴阳离子配合物的环金属化配体进行化学修饰而容易地被调节, 软盐将是用于设计磷光比率探针的良好且通用的平台. 2016年, 我们课题组[49]首次发展了基于磷光软盐配合物的探针用于监测活细胞内pH值的变化.细胞内pH是与细胞增殖、凋亡、耐药、酶活性和离子转运等细胞行为和病理条件相关的重要参数[50~52].因此, 监测生物细胞和组织内pH的变化以了解生理和病理过程是至关重要的.我们设计了以qpy (qpy=4, 4':2', 2'':4'', 4'''-四吡啶)作为辅助配体的阳离子配合物C11与阴离子配合物A2构建的软盐配合物S11(图 5).其中, 阳离子配合物C11辅助配体上吡啶单元的质子化或去质子化能够引起S11的发射波长的变化, 该变化可以通过稳态和瞬态光谱很容易地检测到.在乙腈溶液中, 阳离子配合物C11发射625 nm的橙红光, 其强度随pH值的降低而显著降低(图 5).相比之下, 阴离子配合物在451 nm处的蓝光发射几乎不受pH变化的影响.

随着pH值的增加, S11的发光颜色从蓝色变为红色.更为重要的是, 软盐配合物S11可用于细胞成像并且表现出较低细胞毒性, 并通过共聚焦显微镜成功实现了比率成像用于监测细胞内pH值的实时变化.此外, 磷光寿命成像实验可以通过发光寿命测量来检测细胞内pH变化, 极大地消除了背景荧光信号对目标信号的影响.基于pH校准曲线, 我们成功利用S11实现了由氧化应激引起的细胞内pH波动的定量测量.
半胱氨酸/高半胱氨酸是生物体内仅有的两个含硫醇的氨基酸, 它们在维系生命系统中的生理平衡起着举足轻重的作用[53~55].缺乏半胱氨酸可能导致如造血减少, 白细胞损失以及牛皮癣等疾病, 而缺少高半胱氨酸会与心血管病、阿尔茨海默氏病密切相关.为了解决目前基于磷光金属配合物的半胱氨酸/高半胱氨酸的比率探针缺失的问题.由α, β-不饱和酮基官能化的阳离子配合物(C12)和阴离子配合物A3组成制备了软盐配合物S12(图 6), 以用于比率和时间分辨的发光检测细胞内半胱氨酸/高半胱氨酸含量的变化[56].其后, 研究了S12的光物理性质和传感性能.在CH3CN/H2O (3/2, V/V)的混合溶液中, 配合物S12表现出绿光发射, 在485 nm和505 nm处具有两个发射峰.通过在含有配合物S12溶液中加入半胱氨酸, 阳离子配合物在560 nm的黄色磷光发射显著增强, 而仅观察到A3蓝绿色发光的微弱变化. S12对巯基分析物的检测能力来自于它们与C12 α, β-不饱和酮基发生了高效而快速的迈克加成反应, 从而使得阳离子铱(Ⅲ)配合物的发光增强(图 6).因此, 配合物S12实现了对半胱氨酸/高半胱氨酸的比率检测.更重要的是, 该探针通过比率成像与时间分辨成像技术, 可以实现细胞内生物硫醇变化的实时监测.
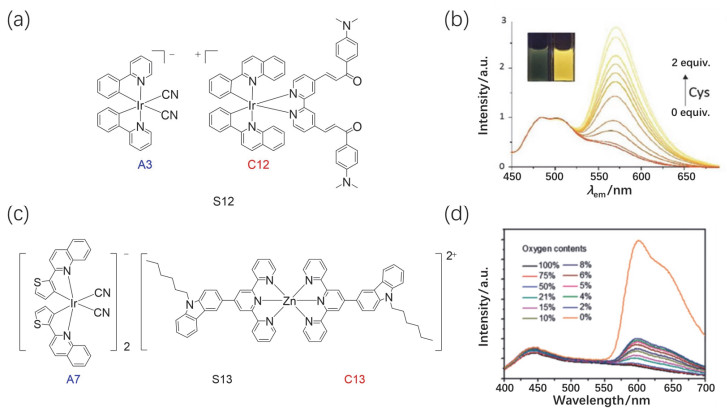
氧气是生物体中重要的分子之一, 在许多生理过程中氧气都发挥着关键作用[57, 58], 例如ATP的产生、离子流的调节、氧化磷酸化过程、信号传导、酶反应的诱导等生理过程都缺少不了氧气分子的参与.因此, 氧的正常利用对于人体的健康十分重要.而当机体中氧的利用异常时, 将会引起很多的病理变化, 比如机体内氧含量过低即乏氧状态是肿瘤、心血管病、中风、帕金森综合症的临床症状, 所以氧气含量作为这些疾病早期诊断的一种重要的标志物已经被很多的科学家所关注[59, 60].选择对氧气不敏感的阳离子锌(Ⅱ)配合物[Zn(tpy)2]2+(CH3COO)2-(C13, tpy=3-(2, 2′:6′, 2′三联吡啶)-4′-基]-9-己基-9H-咔唑)为阳离子组分, 以及对氧气敏感的阴离子铱(Ⅲ)配合物[Ir(tpq)2CN2]-Bu4N+(A7, tpq=2-(噻吩- 2-基)喹啉)制备了能够检测细胞内氧气比例的比率探针S13[61](图 6).软盐配合物S13在溶液中显示出两个发光颜色显著不同的发射峰(446 nm和600 nm), 随着氧气含量的增加, A7在600 nm的红光发射强度逐渐降低, 而C13在446 nm的蓝光强度几乎保持不变.随着氧含量降低, 分子发光颜色从蓝色变为红色的过程可以通过肉眼容易地观察到.随后, 配合物S13被用来监测活细胞中的氧含量变化.如图 6所示, HeLa细胞中红色通道(570~630 nm)的发光随着氧浓度从21%降低到2.5%而增强, 而来自蓝色通道(420~480 nm)的发光几乎没有变化.基于上述实验, 我们可以清楚地观察到比率通道的光强度随细胞的氧气含量变化的动态过程.
光动力学治疗是一种无创或微创性, 利用光化学反应对肿瘤组织或肿瘤细胞进行靶向治疗的方法[62~65].首先肿瘤组织选择性摄取光敏剂, 随后选择适当波长的激光对局部进行照射, 光敏剂能够吸收该光源光子的能量而达到单重激发态, 然后经过隙间穿越到三重激发态.由于光敏剂在三重激发态与氧气的能级匹配而发生能量转移, 基态氧分子发生光氧化反应生成单线态氧或氧自由基等, 从而实现治疗肿瘤组织的目的[66~68].近年来, 研究人员利用分子间能量传递可提高光敏剂的单线态氧产生速率.然而, 制备这类能量传递体系需要的合成步骤往往非常复杂.由于阴阳两种离子配合物的环金属配体可以很容易地被修饰, 这有助于调节离子对的阳离子和阴离子组分的吸收峰和发射峰.因此, 我们发展了基于软盐配合物的光敏剂并将其应用于光动力治疗领域.软盐配合物S14(图 7)由阳离子铱(Ⅲ)配合物[Ir(L)(L')]3+(PF6-)3(C14, L=4'-苯基-2, 2':6', 2'-三联吡啶, L'=3-([2, 2':6', 2'-三联吡啶]-4'-基)-9-己基-9H-咔唑), 和阴离子铱(Ⅲ)配合物[Ir(dFppy)2CN2]-Bu4N+(A2)组成[69].通过对其光物理性质的研究, 阳离子型铱(Ⅲ)配合物C14的吸收峰与阴离子型铱配合物A2的发射峰相重合, 并通过Stern-Volmer猝灭法证实了阴阳离子间高效的能量传递效率.通过单线态氧产生效率测试发现, 软盐配合物S14相对于A2和C14具有较高的单线态氧产生效率, 且在阴阳离子配合物比例在n(A2):n(C14)=3:1时效果最佳.最终, 通过细胞实验证实了软盐配合物S14具有较低的暗毒性和较高的光毒性, 较A2和C14相比具有更高的光动力治疗效果.

电致发光变色, 即电刺激诱导下的材料发光性能变化, 在逻辑门、光学信息记录、信息安全存储等领域具有潜在应用前景, 吸引了人们的研究兴趣[70~72].最近, 利用磷光金属配合物的优异光电性能, 电致发光变色材料逐渐引起了学者的极大关注.由于软盐配合物的正负电组分分别都发光, 且两组分之间存在静电作用力, 当两组分体积较大时, 二者的静电力减弱, 较小的电场便可以促使不同发光组分分离.因此, 磷光软盐配合物是一类潜在的电致发光变色材料.鉴于此, 赵强课题组[73]设计、合成了一系列磷光软盐配合物(S15~S20)用于实现电致发光变色应用(图 8a).在软盐配合物的乙腈溶液中施加3 V的电压, 发现在阴极附近, 溶液的发光颜色在短时间内由橙光变为绿光, 该颜色与阴离子配合物组分相同.而在阳极附近, 溶液的发光颜色由橙光变为红光, 该颜色与阳离子配合物组分相同.当电压施加一段时间后, 两种不同的发光颜色在比色皿中形成一条分明的界线(图 8b), 而当去除电压并搅拌比色皿中的溶液可以使其发光颜色恢复为橙色, 表明这种电致发光变色行为具有可逆性.随后, 他们进一步探索了用软盐配合物制备的准固态器件的电致发光变色现象.器件具有典型的三明治结构, 在两个铂电极之间添加一层由二氧化硅、无水二甲基亚砜、软盐配合物混合组成的准固态凝胶.当对器件施加3 V电压后, 我们可以观察到阴极部分的发光从橙光变成了绿光, 而阳极部分则变为了红光, 这与溶液中所观察到的现象一致.有趣的是, 改变电压方向, 阴阳两极的发光颜色逐渐互换, 并且该现象可以多次重复.

陈勇等[74]利用不同的阳离子配合物[Au(NHC)2]+(NHC=N-杂环卡宾)和阴离子配合物[MX2]-(M=Au或Cu; X=卤化物、氰化物或芳基乙酰基)制备了一系列的软盐配合物[Au(NHC)2]+[MX2]-.通过配体修饰和三组分共结晶或外延生长方法实现了整个可见光区域发射颜色可调的多色磷光.其激发态的性质的变化可归因于d10∙∙∙d10亲金属性辅助的配体(阴离子)-配体(阳离子)的电荷转移跃迁.具体的, 他们利用C19, A12和A15, n(C19):n(A12):n(A15)=2:1:1共结晶, 并成功获得C19-A12/A15三重盐的单晶[n(A12):n(A15)=1:1]. X射线单晶衍射结构确定二氰酸盐和二碘金酸盐阴离子各自在晶格中占50%, 并且将二碘金酸(A15)阴离子掺入C19-A12双盐中不会改变结晶相.使用荧光显微镜可以观察到每个晶体的发光颜色并记录其发射光谱. C19-A12/A15 [n(A12):n(A15)=1:1]单晶显示在586 nm与652 nm处具有双发射带.更为有趣的是, 通过在共结晶时调节n(A12):n(A15)的成分比或通过外延晶体生长成核-壳微晶, 可以进一步调节三重盐C19-A12/A15的发光颜色.
共晶:向阳离子C19前体的甲醇溶液中注入具有各种物质的量比的混合[Au(CN)2]-和[AuI2]-的甲醇溶液(图 9a).沉淀物含有片状的微晶, 其尺寸小于30 μm(图 9b).

外延生长:向悬浮在乙腈中的预先制备的C19-A12微晶样品中加入[AuI2]-(A15)的乙腈溶液(图 9c).在荧光显微镜下观察沉淀物为板状核壳微晶, 其中蓝色发光晶体被具有清晰边界的橙色发光层包围.
由分子组装而成的超分子纳米结构由于其丰富的聚集结构和独特的光学性质引起了科研工作者极大的研究兴趣[75~77].目前, 大量研究已经证明铂(Ⅱ)多吡啶配合物可以通过金属-金属相互作用显示构建超分子结构的潜力[78~85].然而, 它们主要局限于用单体的铂(Ⅱ)配合物进行超分子自组装, 利用双组分铂(Ⅱ)多吡啶配合物组装不论在结构表征方面还是光物理性质研究方面都不够深入.
任咏华课题组[86]参与设计、制备了一系列基于烷基铂(Ⅱ)衍生物的阳离子和阴离子配合物.其中, 阳离子配合物中含有亲水性的三甘醇链(TEG), 阴离子配合物中含有带负电荷的亲水性磺酸盐基团(图 10a).他们发现这些水溶性的软盐配合物的光物理性质在溶剂组成变化时会发生显著的变化, 并且进一步证实了这些变化与所形成的纳米形貌相关.

从软盐配合物S21a的二甲亚砜溶液(10-4 mol•L-1)观察到的透射电镜(TEM)图像表明未形成统一纳米尺寸的聚集体, 而相似浓度的纯水溶液中组装而成的纳米结构具有均匀直径的纳米纤维, 其直径约为60 nm, 长度范围在5至20 μm之间(图 10b).类似的组装结构可以从扫描电子显微镜(SEM)和原子力显微镜(AFM)中观察到.在扫描电镜和原子力显微镜图像中, 发现纳米线结构的高度约为20 nm, 宽度约为100 nm, 表明其具有扁平结构.相比之下, 软盐配合物S21b在水溶液中不形成大而竖直的纳米纤维, 而是形成直径更小的更柔软的纳米纤维, 而软盐配合物S21c只形成更短的纳米棒.放大后的透射电子显微镜图像显示, 纳米纤维是由一束直径约4 nm的小纳米纤维形成的.可以看出, 用苯基炔基配体取代氯配体会阻碍纳米纤维的形成, 这可能是由于苯基炔基配体对空间的要求相对较高, 从而避免了溶液状态下软盐分子的大量聚集.将软盐配合物应用于自组装领域证明了使用非共价分子间作用力协同引导分子以一维各向异性方式组装, 为开发一类在一维电荷传输中具有潜在应用前景的新型功能性超分子材料提供了可能.
近年来, 磷光软盐配合物作为一类新兴的光电材料, 由于其具有光谱易调控且易将具有不同性质的金属配合物组合等优势, 吸引了越来越多关注.由于磷光软盐配合物相比于单组分磷光金属配合物具有更丰富的激发态性质, 对其光物理性质的深入研究尤为重要, 本文详细介绍了由不同金属配合物组成的软盐配合物的发光性质及调控手段.在深入了解它们光物理性质的基础上, 目前磷光软盐配合物已经在包括有机发光二极管、生物成像、光动力治疗、电致发光变色等不同光电应用研究领域中开始崭露头角.
然而, 迄今为止, 磷光软盐配合物在光电应用研究方面仍没有得到足够的关注.进一步利用磷光软盐配合物光物理性质丰富且易调节的优点, 提高它们在各个光电领域的性能是今后研究的重点之一.此外, 进一步拓展磷光软盐配合物在不同光电领域的应用同样是一个重要的方向.我们相信, 随着更多的磷光软盐配合物的开发, 它们必将在有机光电子领域发挥越来越重要的作用.
Abd-El-Aziz, A. S.; Agatemor, C.; Wong, W. Y. Macromolecules Incorporating Transition Metals, Vol. 27, Royal Society of Chemistry, London, United Kingdom, 2018. doi: 10.1039/9781788010368
Zhou, G. J.; Wong, W. Y. Chem. Soc. Rev. 2011, 40, 2541. doi: 10.1039/c0cs00094a
Köhler, A.; Wilson, J. S.; Friend, R. H. Adv. Mater. 2002, 14, 701. doi: 10.1002/1521-4095(20020517)14:10<701::AID-ADMA701>3.0.CO;2-4
徐广涛, 李佳, 陈忠宁, 化学学报, 2014, 72, 667. doi: 10.6023/A14030217Xu, G. T.; Li, J.; Chen, Z. N. Acta Chim. Sinica 2014, 72, 667. doi: 10.6023/A14030217
Wang, L. H.; Guo, J. F.; Li, Y. J.; Su, Y. R.; Liu, J. W.; Li, Y. H.; Wang, S.; Shimada, S.; Huang, W. Chinese J. Chem. 2017, 35, 507. doi: 10.1002/cjoc.201600666
Chen, Z. Q.; Bian, Z. Q.; Huang, C. H. Adv. Mater. 2010, 22, 1534. doi: 10.1002/adma.200903233
Huang, T.; Jiang, W.; Duan, L. J. Mater. Chem. C 2018, 6, 5577. doi: 10.1039/C8TC01139G
Chou, P. T.; Chi, Y. Chem. Eur. J. 2007, 13, 380. doi: 10.1002/chem.200601272
Chow, P. K.; Cheng, G.; Tong, G. S. M.; To, W. P.; Kwong, W. L.; Low, K. H.; Kwok, C. C.; Ma, C. S.; Che, C. M. Angew. Chem., Int. Ed. 2015, 54, 2084. doi: 10.1002/anie.201408940
Ma, Y.; Shen, L.; She, P. F.; Hou, Y. Q.; Yu, Y. X.; Zhao, J. Z. Adv. Optical Mater. 2019, 1801657.
Zhang, K. Y.; Yu, Q.; Wei, H. J.; Liu, S. J.; Zhao, Q.; Huang, W. Chem. Rev. 2018, 118, 1770. doi: 10.1021/acs.chemrev.7b00425
Martir, D. R.; Zysman-Colman, E. Coord. Chem. Rev. 2018, 364, 86. doi: 10.1016/j.ccr.2018.03.016
Ma, D.; Duan, L.; Wei, Y.; He, L.; Wang, L.; Qiu, Y. Chem. Commun. 2014, 50, 530. doi: 10.1039/C3CC47362G
Zhang, K. Y.; Chen, X.; Sun, G.; Zhang, T.; Liu, S.; Zhao, Q.; Huang, W. Adv. Mater. 2016, 28, 7137. doi: 10.1002/adma.201601978
Zhao, Q.; Li, F.; Huang, C. Chem. Soc. Rev. 2010, 39, 3007. doi: 10.1039/b915340c
Chen, X. L.; Yu, R.; Zhang, Q. K.; Zhou, L. J.; Wu, X. Y.; Zhang, Q.; Lu, C. Z. Chem. Mater. 2013, 25, 3910 doi: 10.1021/cm4024309
Zhang, K. Y.; Liu, H. W.; Tang, M. C.; Choi, A. W. T.; Zhu, N.; Wei, X. G.; Lo, K. K. W. Inorg. Chem. 2015, 54, 6582. doi: 10.1021/acs.inorgchem.5b00944
Liu, J.; Yee, K. K.; Lo, K. K. W.; Zhang, K. Y.; To, W. P.; Che, C. M.; Xu, Z. J. Am. Chem. Soc. 2014, 136, 2818. doi: 10.1021/ja411067a
Housecroft, C. E.; Constable, E. C. Coord. Chem. Rev. 2017, 350, 155. doi: 10.1016/j.ccr.2017.06.016
Chen, G. Y.; Chang, B. R.; Shih, T. A.; Lin, C. H.; Lo, C. L.; Chen, Y. Z.; Yang, Z. P. Chem. Eur. J. 2019, 25, 5489. doi: 10.1002/chem.201805902
Xie, Z.; Ma, L.; de Krafft, K. E.; Jin, A.; Lin, W. J. Am. Chem. Soc. 2010, 132, 922. doi: 10.1021/ja909629f
You, Y.; Lee, S.; Kim, T.; Ohkubo, K.; Chae, W. S.; Fukuzumi, S.; Lippard, S. J. Am. Chem. Soc. 2011, 133, 18328. doi: 10.1021/ja207163r
Wu, C.; Chen, H. F.; Wong, K. T.; Thompson, M. E. J. Am. Chem. Soc. 2009, 132, 3133.
Mauro, M.; Schuermann, K. C.; Prétôt, R.; Hafner, A.; Mercandelli, P.; Sironi, A.; De Cola, L. Angew. Chem., Int. Ed. 2010, 49, 1222. doi: 10.1002/anie.200905713
Ionescu, A.; Szerb, E. I.; Yadav, Y. J.; Talarico, A. M.; Ghedini, M.; Godbert, N. Dalton Trans. 2014, 43, 784. doi: 10.1039/C3DT52077C
Fiorini, V.; D'Ignazio, A.; Magee, K. D.; Ogden, M. I.; Massi, M.; Stagni, S. Dalton Trans. 2016, 45, 3256. doi: 10.1039/C5DT04958J
Sandroni, M.; Zysman-Colman, E. Dalton Trans. 2014, 43, 3676. doi: 10.1039/c3dt53170h
Ho, C. L.; Wong, W. Y. Coord. Chem. Rev. 2013, 257, 1614. doi: 10.1016/j.ccr.2012.08.023
Fan, C.; Yang, C. Chem. Soc. Rev. 2014, 43, 6439. doi: 10.1039/C4CS00110A
Lee, J.; Chen, H. F.; Batagoda, T.; Coburn, C.; Djurovich, P. I.; Thompson, M. E.; Forrest, S. R. Nat. Mater. 2016, 15, 92. doi: 10.1038/nmat4446
Kim, J. B.; Han, S. H.; Yang, K.; Kwon, S. K.; Kim, J. J.; Kim, Y. H. Chem. Commun. 2015, 51, 58. doi: 10.1039/C4CC07768G
Kesarkar, S.; Mróz, W.; Penconi, M.; Pasini, M.; Destri, S.; Cazzaniga, M.; Bossi, A. Angew. Chem., Int. Ed. 2016, 55, 2714. doi: 10.1002/anie.201509798
陈仕琦, 代军, 周凯峰, 罗艳菊, 苏仕健, 蒲雪梅, 黄艳, 卢志云, 化学学报, 2017, 75, 367. doi: 10.6023/A17010015Chen, S. Q.; Dai, J.; Zhou, K. F.; Luo, Y. J.; Su, S. J.; Pu, X. M.; Huang, Y.; Lu, Z. Y. Acta Chim. Sinica 2017, 75, 367. doi: 10.6023/A17010015
Dumur, F.; Nasr, G.; Wantz, G.; Mayer, C. R.; Dumas, E.; Guerlin, A.; Miomandre, F.; Clavier, G.; Bertin, D.; Gigmes, D. Org. Electron. 2011, 12, 1683. doi: 10.1016/j.orgel.2011.06.014
Nasr, G.; Guerlin, A.; Dumur, F.; Beouch, L.; Dumas, E.; Clavier, G.; Miomandre, F.; Goubard, F.; Gigmes, D.; Bertin, D.; Wantze, G.; Mayer, C. R. Chem. Commun. 2011, 47, 10698. doi: 10.1039/c1cc13733f
Stephens, D. J.; Allan, V. J. Science 2003, 300, 82. doi: 10.1126/science.1082160
Dmitriev, R. I.; Papkovsky, D. B. Cell. Mol. Life Sci. 2012, 69, 2025. doi: 10.1007/s00018-011-0914-0
Wang, X. D.; Wolfbeis, O. S. Chem. Soc. Rev. 2014, 43, 3666. doi: 10.1039/C4CS00039K
Knox, H. J.; Hedhli, J.; Kim, T. W.; Khalili, K.; Dobrucki, L. W.; Chan, J. Nat. Commun. 2017, 8, 1794. doi: 10.1038/s41467-017-01951-0
Papkovsky, D. B.; Dmitriev, R. I. Chem. Soc. Rev. 2013, 42, 8700. doi: 10.1039/c3cs60131e
Dmitriev, R. I.; Borisov, S. M.; Dussmann, H.; Sun, S. W.; Muller, B. J.; Prehn, J.; Baklaushev, V. P.; Klimant, I.; Papkovsky, D. B. ACS Nano 2015, 9, 5275. doi: 10.1021/acsnano.5b00771
Aigner, D.; Dmitriev, R. I.; Borisov, S. M.; Papkovsky, D. B.; Klimant, I. J. Mater. Chem. B 2014, 2, 6792. doi: 10.1039/C4TB01006J
Baggaley, E.; Botchway, S. W.; Haycock, J. W.; Morris, H.; Sazanovich, I. V.; Williams, J. G.; Weinstein, J. A. Chem. Sci. 2014, 5, 879. doi: 10.1039/C3SC51875B
Wang, J. Q.; Hou, X. J.; Jin, C. Z.; Chao, H. Chinese J. Chem. 2016, 34, 583. doi: 10.1002/cjoc.201500769
Lee, M. H.; Han, J. H.; Lee, J. H.; Park, N.; Kumar, R.; Kang, C.; Kim, J. S. Angew. Chem., Int. Ed. 2013, 52, 6206. doi: 10.1002/anie.201301894
Sapsford, K. E.; Berti, L.; Medintz, I. L. Angew. Chem., Int. Ed. 2006, 45, 4562. doi: 10.1002/anie.200503873
Jares-Erijman, E. A.; Jovin, T. M. Nat. Biotechnol. 2003, 21, 1387. doi: 10.1038/nbt896
Xiong, L.; Zhao, Q.; Chen, H.; Wu, Y.; Dong, Z.; Zhou, Z.; Li, F. Inorg. Chem. 2010, 49, 6402. doi: 10.1021/ic902266x
Ma, Y.; Liang, H.; Zeng, Y.; Yang, H.; Ho, C. L.; Xu, W. J.; Zhao, Q.; Huang, W.; Wong, W. Y. Chem. Sci. 2016, 7, 3338. doi: 10.1039/C5SC04624F
Gottlieb, R. A.; Nordberg, J.; Skowronski, E.; Babior, B. M. Proc. Natl. Acad. Sci. 1996, 93, 654. doi: 10.1073/pnas.93.2.654
Hoyt, K. R.; Reynolds, I. J. J. Neurochem. 1998, 71, 1051.
Casey, J. R.; Grinstein, S.; Orlowski, J. Nat. Rev. Mol. Cell Biol. 2010, 11, 50. doi: 10.1038/nrm2820
Shahrokhian, S. Anal. Chem. 2001, 73, 5972. doi: 10.1021/ac010541m
Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P. F.; Rosenberg, I. H.; D'Agostino, R. B.; Wilson, P. W. F.; Wolf, P. A. N. Engl. J. Med. 2002, 346, 476. doi: 10.1056/NEJMoa011613
Ma, Y.; Liu, S. J.; Yang, H. R.; Wu, Y. Q.; Yang, C. J.; Liu, X. M.; Zhao, Q.; Wu, H. Z.; Liang, J. C.; Li, F. Y.; Huang, W. J. Mater. Chem. 2011, 21, 18974. doi: 10.1039/c1jm13513a
Liu, S.; Xu, A.; Chen, Z.; Ma, Y.; Yang, H.; Shi, Z.; Zhao, Q. Opt. Express. 2016, 24, 28247. doi: 10.1364/OE.24.028247
Acker, T.; Acker, H. J. Exp. Biol. 2004, 207, 3171. doi: 10.1242/jeb.01075
Tobita, S.; Yoshihara, T. Curr. Opin. Chem. Biol. 2016, 33, 39. doi: 10.1016/j.cbpa.2016.05.017
Denko, N. C. Nat. Rev. Cancer. 2008, 8, 705. doi: 10.1038/nrc2468
Simon, M. C.; Liu, L.; Barnhart, B. C.; Young, R. M. Annu. Rev. Physiol. 2008, 70, 51. doi: 10.1146/annurev.physiol.70.113006.100526
Ma, Y.; Dong, Y. F.; Zou, L.; Shen, L.; Liu, S. Y.; Liu, S. J.; Huang, W.; Zhao, Q.; Wong, W. Y. Eur. J. Inorg. Chem. 2018, 20, 345.
Ogilby, P. R. Chem. Soc. Rev. 2010, 39, 3181. doi: 10.1039/b926014p
Apel, K.; Hirt, H. Annu. Rev. Plant Biol. 2004, 55, 373. doi: 10.1146/annurev.arplant.55.031903.141701
Greer, A. Acc. Chem. Res. 2006, 39, 797. doi: 10.1021/ar050191g
周前雄, 王雪松, 化学学报, 2017, 75, 49. doi: 10.11862/CJIC.2017.013Zhou, Q. X.; Wang, X. S. Acta Chim. Sinica 2017, 75, 49. doi: 10.11862/CJIC.2017.013
Nam, J. S.; Kang, M. G.; Kang, J.; Park, S. Y.; Lee, S. J. C.; Kim, H. T.; Seo, J. K.; Kwon, O. H.; Lim, M. H.; Rhee, H. W.; Kwon, T. H.; J. Am. Chem. Soc. 2016, 138, 10968. doi: 10.1021/jacs.6b05302
Du, E.; Hu, X.; Roy, S.; Wang, P.; Deasy, K.; Mochizuki, T.; Zhang, Y. Chem. Commun. 2017, 53, 6033. doi: 10.1039/C7CC03337K
Lv, W.; Zhang, Z.; Zhang, K. Y.; Yang, H.; Liu, S.; Xu, A.; Guo, S.; Zhao, Q.; Huang, W. Angew. Chem., Int. Ed. 2016, 55, 9947. doi: 10.1002/anie.201604130
Ma, Y.; Zhang, S. J.; Wei, H. J.; Dong, Y. F.; Shen, L.; Liu, S. J.; Zhao, Q.; Liu, L.; Wong, W. Y. Dalton Trans. 2018, 47, 5582. doi: 10.1039/C8DT00720A
Sun, H. B.; Liu, S. J.; Lin, W. P.; Zhang, K. Y.; Lv, W.; Huang X.; Huo, F. W.; Yang, H. R.; Jenkins, G.; Zhao, Q.; Huang, W. Nat. Commun. 2014, 5, 3601. doi: 10.1038/ncomms4601
Ma, Y.; Yang, J.; Liu, S. J.; Xia, H. T.; She, P. F.; Jiang, R.; Zhao, Q. Adv. Optical Mater. 2017, 1700587.
Ma, Y.; Liu, S. J.; Yang, H. R.; Zeng, Y.; She, P. F.; Zhu, N. Y.; Ho, C. L.; Zhao, Q.; Huang, W.; Wong, W. Y. Inorg. Chem. 2017, 56, 2409. doi: 10.1021/acs.inorgchem.6b02319
Guo, S.; Huang, T. C.; Liu, S. J.; Zhang, K. Y.; Yang, H. R.; Han, J. M.; Zhao, Q.; Huang, W. Chem. Sci. 2017, 8, 348. doi: 10.1039/C6SC02837C
Liu, Q.; Xie, M.; Chang, X. Y.; Cao, S.; Zou, C.; Fu, W. F.; Che, C. M.; Chen, Y.; Lu, W. Angew. Chem., Int. Ed. 2018, 57, 6279. doi: 10.1002/anie.201803965
Lehn, J. M. Science 2002, 295, 2400. doi: 10.1126/science.1071063
Aida, T.; Meijer, E. W.; Stupp, S. I. Science 2012, 335, 813. doi: 10.1126/science.1205962
Aliprandi, A.; Mauro, M.; De Cola, L. Nat. Chem. 2016, 8, 10. doi: 10.1038/nchem.2383
Ma, Y.; Zhao, W. W.; She, P. F.; Liu, S. Y.; Shen, L.; Li, X. L.; Liu, S. J.; Zhao, Q.; Huang, W.; Wong, W. Y. Small Methods 2019, 1900142.
Po, C.; Tam, A. Y. Y.; Wong, K. M. C.; Yam, V. W. W. J. Am. Chem. Soc. 2011, 133, 12136. doi: 10.1021/ja203920w
Po, C.; Yam, V. W. W. Chem. Sci. 2014, 5, 4868. doi: 10.1039/C4SC01588F
Aliprandi, A.; Genovese, D.; Mauro, M.; De Cola, L. Chem. Lett. 2015, 44, 1152. doi: 10.1246/cl.150592
Chow, P.; Cheng, G.; Tong, G. S. M.; To, W.; Kwong, W.; Low, K.; Kwok, C.; Ma, C. S.; Che, C. M. Angew. Chem., Int. Ed. 2015, 54, 2084. doi: 10.1002/anie.201408940
Camerel, F.; Ziessel, R.; Donnio, B.; Bourgogne, C.; Guillon, D.; Schmutz, M.; Iacovita, C.; Bucher, J. Angew. Chem., Int. Ed. 2007, 46, 2659. doi: 10.1002/anie.200604012
Chan, M. H. Y.; Ng, M.; Leung, S. Y. L.; Lam, W. H.; Yam, V. W. W. J. Am. Chem. Soc. 2017, 139, 8639. doi: 10.1021/jacs.7b03635
Li, Y. H.; Zeng, W. J.; Lai, W. Y.; Shimada, S.; Wang, S.; Wang, L. H.; Huang, W. Chinese J. Chem. 2015, 33, 1206. doi: 10.1002/cjoc.201500418
Wong, V. C. H.; Po, C.; Leung, S. Y. L.; Chan, A. K. W.; Yang, S. Y.; Zhu, B. R.; Cui, X. D.; Yam, V. W. W. J. Am. Chem. Soc. 2018, 140, 657. doi: 10.1021/jacs.7b09770

图 2 (a) 磷光软盐配合物S3和S4的分子结构. (b) S3的单晶结构. (c)左图:归一化发射光谱图, S4(黑线)、嵌入甲苯分子的S4(蓝线)、A2(红线)、C4(绿线); 右图: S4(黑线)与嵌入蒽醌分子S4(红线)的归一化发射光谱图
Figure 2 (a) The chemical structures of phosphorescent soft salt complexes S3 and S4. (b) The single crystal structure of S3. (c) left: normalized PL spectra of S4 (black line), S4 intercalating toluene (blue line), complexes A2 (red line) and C4 (green line); right: normalized PL spectra of S4 (black line) and S4 with anthraquinone intercalated (red line)

图 3 磷光软盐配合物S5, S6和S7的化学结构
Figure 3 The chemical structures of phosphorescent soft salt complexes S5, S6 and S7

图 4 应用于有机发光二极管的磷光软盐配合物S8, S9和S10
Figure 4 Phosphorescent soft salt complexes S8, S9 and S10 for organic light emitting diodes

图 5 (a) 应用于细胞内pH检测的磷光软盐配合物S11. (b) S11在不同pH值中的发射光谱. (c) S11在不同pH值的活细胞内的磷光寿命成像
Figure 5 (a) Phosphorescent soft salt complex S11 for intracellular pH detection. (b) Changes in the PL spectra of S11 at different pH values. (c) Phosphorescence lifetime images of S11 in living cells at different pH values

图 6 (a) 应用于半胱氨酸检测的磷光软盐配合物S12. (b) S12在乙腈与水的混合溶剂中在不同浓度半胱氨酸下的发射光谱. (c)用于氧气含量检测的磷光软盐配合物S13. (d) S13在二甲基亚砜溶剂中在不同氧气含量下的发射光谱
Figure 6 (a) Phosphorescent soft salt complex S12 for detecting cysteine. (b) Changes in the PL spectra of S12 in CH3CN/H2O (3/2, V/V) upon addition of various amount of cysteine. (c) Phosphorescent soft salt complex S13 for detecting oxygen contents. (d) PL spectra of S13 at different oxygen concentrations in DMSO solution

图 7 (a) 应用于光动力治疗的磷光软盐分子S14. (b)配合物A2, C14和S14的光毒性测试
Figure 7 (a) Phosphorescent soft salt complex S14 for photodynamic therapy. (b) The phototoxicity of A2, C14 and S14

图 8 (a) 应用于电致发光变色的磷光软盐配合物S15~S20. (b) S15~S20乙腈溶液在施加电压前后发光颜色的变化
Figure 8 (a) Phosphorescent soft salt complexes S15~S20 for electrochromic luminescence. (b) The emission color of S15~S20 before and after application of a voltage

图 9 (a) 阴离子配合物A12~A25和阳离子配合物C18~C21的化学结构. (b)荧光显微镜下, 共晶法制备下的微晶图片. (c)荧光显微镜下, 外延生长法制备下的微晶图片
Figure 9 (a) Chemical structures of anionic complexes of A12~A25 and cationic complexes C18~C21. (b) Fluorescence micrographs of microcrystals prepared via co-crystallization. (c) Fluorescence micrographs of core-shell microcrystals prepared via epitaxial growth

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

 下载:
下载: