引用本文:
熊麟, 凡勇, 张凡. 稀土纳米晶用于近红外区活体成像和传感研究进展[J]. 化学学报,
2019, 77(12): 1239-1249.
doi:
10.6023/A19080305 Citation:
Xiong Lin, Fan Yong, Zhang Fan. Research Progress on Rare Earth Nanocrystals for In Vivo Imaging and Sensing in Near Infrared Region[J]. Acta Chimica Sinica,
2019, 77(12): 1239-1249.
doi:
10.6023/A19080305
Received Date:
16 August 2019 Available Online:
15 December 2019
Fund Project:
Project supported by National Science Fund for Distinguished Young Scholars (No. 21725502), the National Key R & D Program of China (No. 2017YFA0207303), the Key Basic Research Program of Science and Technology Commission of Shanghai Municipality (No. 17JC1400100)
Abstract:In vivo imaging and sensing play a critical role in modern biological and medical research. Compared with other techniques such as computed tomography (CT), positron emission tomography (PET) and nuclear magnetic resonance (NMR), fluorescence imaging and analysis are featured by fast feedback, high sensitivity, and high spatiotemporal resolution. Especially, the application of near infrared (NIR) light as both excitation and emission signals provides increased tissue penetration and improved imaging quality and sensitivity due to reduced light scattering and auto-fluorescence. Among various materials investigated for in vivo imaging and bio-sensing, lanthanide-based nanocrystals display rich excitation/emission wavelengths in the NIR range, good photo and chemical stability, large Stokes shifts. In recent years, the research on lanthanide-based nanocrystals for in vivo imaging and sensing has seen rapid progress. Through nanoscale material design and synthesis, it is possible to fine tune the optical properties of lanthanide-based nanocrystals. By properly choosing different lanthanide ions as activators and sensitizers, multiple excitation/emission wavelengths can be obtained. The careful design of core-shell structure of nanocrystals enables improved fluorescence efficiency and tailorable fluorescence life time through controlled energy transfer. On the other side, the surface of lanthanide-based nanocrystals can be modified through coating, absorption or ligand exchange to enhance the biocompatibility, targeting capability, and bio-responsiveness. Taking advantage of this high flexibility and versatility, there are great opportunities for these lanthanide-based nanocrystals in various in vivo imaging and sensing applications. This review first outlines the general technique requirements for in vivo imaging and sensing. Then, the composition, synthesis and basic properties of lanthanide-based nanocrystals are briefly introduced. Subsequently, the routes for tailoring the optical and biochemical properties of lanthanide-based nanocrystals are discussed in detail, with an emphasis on the material designs and surface modifications for in vivo imaging and analysis. It is expected that this work will inspire new ideas for accelerating the clinic translation of rare earth nanocrystals-based imaging and sensing techniques.
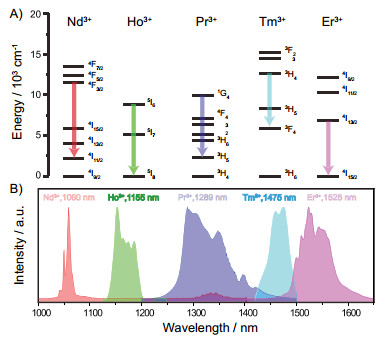
Figure 2.
(A) Structure of the C/S1/S2/S3 nanocrystals for 1525 nm luminescence. (B) Proposed energy transfer mechanisms in the multi-layer core/shell nanocrystals[68]
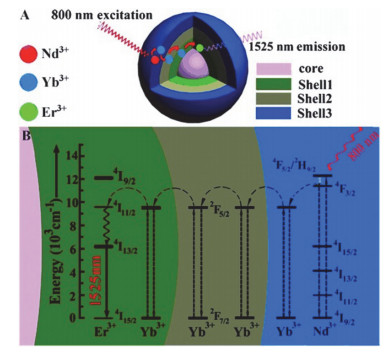
Figure 3.
(A) Energy transfer mechanism in the NaErF4:2 %Ho@NaYF4 core-shell UCNPs. (B) TEM, HAADF-STEM, HRTEM images, and (C) upconversion emission spectrum of the obtained NIR-Ⅱ UCNPs[70]
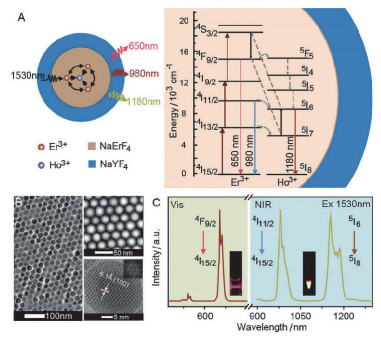
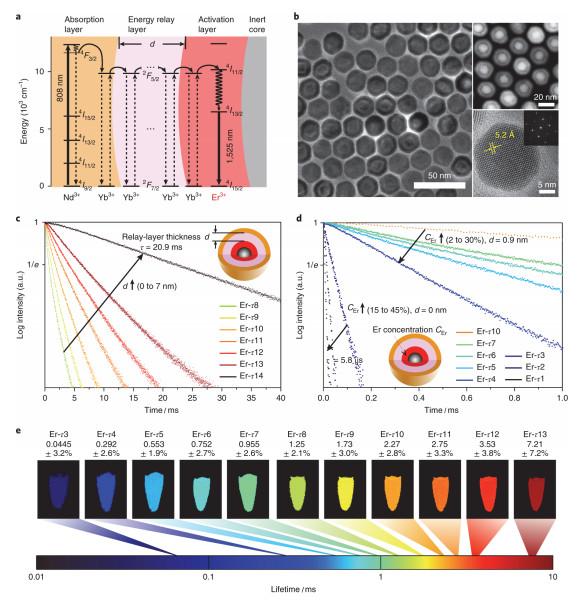
Figure 4.
(a) Energy level diagram of the core–multi-shell nanoparticles. (b) TEM, HAADF-STEM, HRTEM images of NaGdF4@NaGdF4:Yb, Er@ NaYF4:Yb@NaNdF4:Yb nanocrystals. (c) Luminescence decay curves of nanoparticles with energy relay shells of increasing thickness d from 0 to 7 nm. (d) Luminescence decay curves of the nanoparticles with incremental Er3+ doping concentration CEr from 2% to 30%. (e) Pseudocolour-mapped lifetime images of the Er nanoparticles[72]
Figure 5.
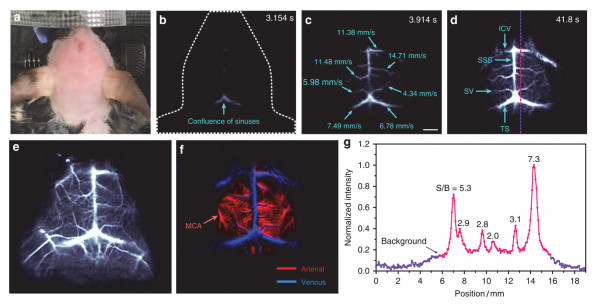
(a) Color photograph of a mouse preceding NIR-Ⅱ fluorescence imaging. (b~d) Time-course NIR-Ⅱ brain fluorescence images. (e, f) Cerebral vascular image in NIR-Ⅱ region with corresponding PCA overlaid image. (g) Signal/background ratio analysis of NIR-Ⅱ cerebrovascular image[61]
Figure 6.
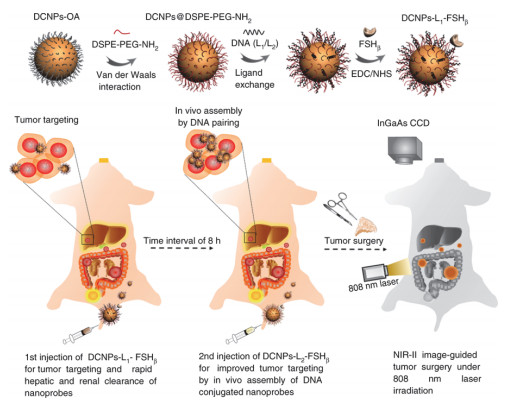
Schematic of stepwise fabrication of DNA and FSHβ modified DCNPs (DCNPs-L1-FSHβ) and in vivo assembly of DCNPs-L1-FSHβ (first injection) and DCNPs-L2-FSHβ (second injection) with improved tumor targeting and rapid hepatic and renal clearance after two-staged in sequence injections[77]
Figure 7.
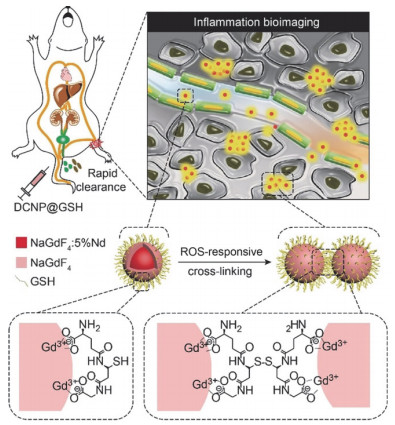
Illustration of bioimaging for acute local epidermal inflammation in mice utilizing ultra‐small DCNP@GSH nanoprobes. DCNP@GSH nanoprobes cross‐link at inflamed areas in response to ROS and they were rapidly excreted from the body[79]
Figure 9.
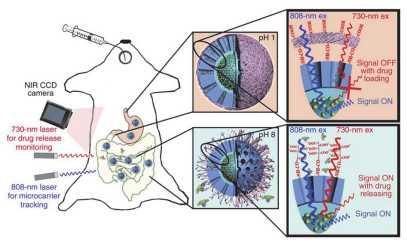
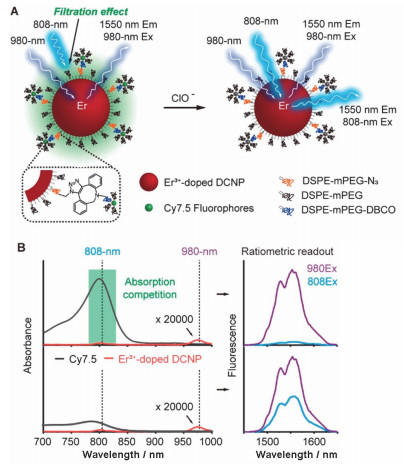
Schematic illustration showing the ratiometric response of DCNP@Cy7.5 to HOCl based on an absorption competition-induced emission (ACIE) mechanism[81]
Figure 10.
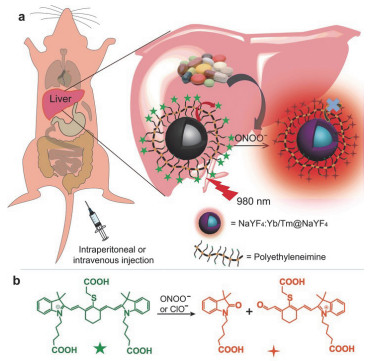
(a) Rational design of chromophore-assembled UCNPs for the detection of nitrosative hepatotoxicity in vivo. (b) Proposed reaction mechanism for the "turn-on" luminescence by which the energy acceptor Cy7 (marked with green star) degrades after oxidation by ONOO− or ClO−[82]
Naczynski, D. J.; Tan, M. C.; Zevon, M.; Wall, B.; Kohl, J.; Kulesa, A.; Chen, S.; Roth, C. M.; Riman, R. E.; Moghe, P. V. Nat. Commun.2013, 4, 2199. doi: 10.1038/ncomms3199
Dang, X.; Gu, L.; Qi, J.; Correa, S.; Zhang, G.; Belcher, A. M.; Hammond, P. T. Proc. Natl. Acad. Sci. U. S. A.2016, 113, 5179. doi: 10.1073/pnas.1521175113
Zhao, J.; Jin, D.; Schartner, E. P.; Lu, Y.; Liu, Y.; Zvyagin, A. V.; Zhang, L.; Dawes, J. M.; Xi, P.; Piper, J. A.; Goldys, E. M.; Monro, T. M. Nat. Nanotechnol.2013, 8, 729. doi: 10.1038/nnano.2013.171
Garfield, D. J.; Borys, N. J.; Hamed, S. M.; Torquato, N. A.; Tajon, C. A.; Tian, B.; Shevitski, B.; Barnard, E. S.; Suh, Y. D.; Aloni, S.; Neaton, J. B.; Chan, E. M.; Cohen, B. E.; Schuck, P. J. Nat. Photonics2018, 12, 402. doi: 10.1038/s41566-018-0156-x
[29]
Wei, W.; Chen, G.; Baev, A.; He, G. S.; Shao, W.; Damasco, J.; Prasad, P. N. J. Am. Chem. Soc.2016, 138, 15130. doi: 10.1021/jacs.6b09474
Xu, W.; Lee, T. K.; Moon, B. S.; Song, H. W.; Chen, X.; Chun, B.; Kim, Y. J.; Kwak, S. K.; Chen, P.; Kim, D. H. Adv. Opt. Mater.2018, 6, 1701119. doi: 10.1002/adom.201701119
[32]
Huang, B.; Sun, M.; Dougherty, A. W.; Dong, H.; Xu, Y.-J.; Sun, L.-D.; Yan, C.-H. Nanoscale2017, 9, 18490. doi: 10.1039/C7NR06729A
[33]
Zou, W.; Visser, C.; Maduro, J. A.; Pshenichnikov, M. S.; Hummelen, J. C. Nat. Photonics2012, 6, 560. doi: 10.1038/nphoton.2012.158
Wang, X.; Valiev, R. R.; Ohulchanskyy, T. Y.; Ågren, H.; Yang, C.; Chen, G. Chem. Soc. Rev.2017, 46, 4150. doi: 10.1039/C7CS00053G
[45]
Chen, G.; Damasco, J.; Qiu, H.; Shao, W.; Ohulchanskyy, T. Y.; Valiev, R. R.; Wu, X.; Han, G.; Wang, Y.; Yang, C.; Ågren, H.; Prasad, P. N. Nano Lett.2015, 15, 7400. doi: 10.1021/acs.nanolett.5b02830
[46]
Shao, W.; Chen, G.; Kuzmin, A.; Kutscher, H. L.; Pliss, A.; Ohulchanskyy, T. Y.; Prasad, P. N. J. Am. Chem. Soc.2016, 138, 16192. doi: 10.1021/jacs.6b08973
Gargas, D. J.; Chan, E. M.; Ostrowski, A. D.; Aloni, S.; Altoe, M. V.; Barnard, E. S.; Sanii, B.; Urban, J. J.; Milliron, D. J.; Cohen, B. E.; Schuck, P. J. Nat. Nanotechnol.2014, 9, 300. doi: 10.1038/nnano.2014.29
Tian, B.; Fernandez-Bravo, A.; Najafiaghdam, H.; Torquato, N. A.; Altoe, M. V. P.; Teitelboim, A.; Tajon, C. A.; Tian, Y.; Borys, N. J.; Barnard, E. S.; Anwar, M.; Chan, E. M.; Schuck, P. J.; Cohen, B. E. Nat. Commun.2018, 9, 3082. doi: 10.1038/s41467-018-05577-8
[58]
Johnson, N. J.; He, S.; Diao, S.; Chan, E. M.; Dai, H.; Almutairi, A. J. Am. Chem. Soc.2017, 139, 3275. doi: 10.1021/jacs.7b00223
Zhong, Y.; Ma, Z.; Zhu, S.; Yue, J.; Zhang, M.; Antaris, A. L.; Yuan, J.; Cui, R.; Wan, H.; Zhou, Y.; Wang, W.; Huang, N. F.; Luo, J.; Hu, Z.; Dai, H. Nat. Commun.2017, 8, 737. doi: 10.1038/s41467-017-00917-6
[62]
Chen, G.; Shen, J.; Ohulchanskyy, T. Y.; Patel, N. J.; Kutikov, A.; Li, Z.; Song, J.; Pandey, R. K.; Ågren, H.; Prasad, P. N.; Han, G. ACS Nano2012, 6, 8280. doi: 10.1021/nn302972r
[63]
Villa, I.; Vedda, A.; Cantarelli, I. X.; Pedroni, M.; Piccinelli, F.; Bettinelli, M.; Speghini, A.; Quintanilla, M.; Vetrone, F.; Rocha, U.; Jacinto, C.; Carrasco, E.; Rodríguez, F. S.; Juarranz, Á.; del Rosal, B.; Ortgies, D. H.; Gonzalez, P. H.; Solé, J. G.; García, D. J. Nano Research2014, 8, 649.
[64]
Diao, S.; Hong, G.; Antaris, A. L.; Blackburn, J. L.; Cheng, K.; Cheng, Z.; Dai, H. Nano Research2015, 8, 3027. doi: 10.1007/s12274-015-0808-9
[65]
Xie, X.; Li, Z.; Zhang, Y.; Guo, S.; Pendharkar, A. I.; Lu, M.; Huang, L.; Huang, W.; Han, G. Small2017, 13, 1602843. doi: 10.1002/smll.201602843
Levy, E. S.; Tajon, C. A.; Bischof, T. S.; Iafrati, J.; Fernandez-Bravo, A.; Garfield, D. J.; Chamanzar, M.; Maharbiz, M. M.; Sohal, V. S.; Schuck, P. J.; Cohen, B. E.; Chan, E. M. ACS Nano2016, 10, 8423. doi: 10.1021/acsnano.6b03288
Figure 2
(A) Structure of the C/S1/S2/S3 nanocrystals for 1525 nm luminescence. (B) Proposed energy transfer mechanisms in the multi-layer core/shell nanocrystals[68]
Figure 3
(A) Energy transfer mechanism in the NaErF4:2 %Ho@NaYF4 core-shell UCNPs. (B) TEM, HAADF-STEM, HRTEM images, and (C) upconversion emission spectrum of the obtained NIR-Ⅱ UCNPs[70]
Figure 4
(a) Energy level diagram of the core–multi-shell nanoparticles. (b) TEM, HAADF-STEM, HRTEM images of NaGdF4@NaGdF4:Yb, Er@ NaYF4:Yb@NaNdF4:Yb nanocrystals. (c) Luminescence decay curves of nanoparticles with energy relay shells of increasing thickness d from 0 to 7 nm. (d) Luminescence decay curves of the nanoparticles with incremental Er3+ doping concentration CEr from 2% to 30%. (e) Pseudocolour-mapped lifetime images of the Er nanoparticles[72]
Figure 5
(a) Color photograph of a mouse preceding NIR-Ⅱ fluorescence imaging. (b~d) Time-course NIR-Ⅱ brain fluorescence images. (e, f) Cerebral vascular image in NIR-Ⅱ region with corresponding PCA overlaid image. (g) Signal/background ratio analysis of NIR-Ⅱ cerebrovascular image[61]
Figure 6
Schematic of stepwise fabrication of DNA and FSHβ modified DCNPs (DCNPs-L1-FSHβ) and in vivo assembly of DCNPs-L1-FSHβ (first injection) and DCNPs-L2-FSHβ (second injection) with improved tumor targeting and rapid hepatic and renal clearance after two-staged in sequence injections[77]
Figure 7
Illustration of bioimaging for acute local epidermal inflammation in mice utilizing ultra‐small DCNP@GSH nanoprobes. DCNP@GSH nanoprobes cross‐link at inflamed areas in response to ROS and they were rapidly excreted from the body[79]
Figure 9
Schematic illustration showing the ratiometric response of DCNP@Cy7.5 to HOCl based on an absorption competition-induced emission (ACIE) mechanism[81]
Figure 10
(a) Rational design of chromophore-assembled UCNPs for the detection of nitrosative hepatotoxicity in vivo. (b) Proposed reaction mechanism for the "turn-on" luminescence by which the energy acceptor Cy7 (marked with green star) degrades after oxidation by ONOO− or ClO−[82]

 下载:
下载:

 下载:
下载:











 下载:
下载:
