引用本文:
余俊, 杨宇森, 卫敏. 水滑石基负载型催化剂的制备及其在催化反应中的应用[J]. 化学学报,
2019, 77(11): 1129-1139.
doi:
10.6023/A19070260

Citation: Yu Jun, Yang Yusen, Wei Min. Preparation and Catalytic Performance of Supported Catalysts Derived from Layered Double Hydroxides[J]. Acta Chimica Sinica, 2019, 77(11): 1129-1139. doi: 10.6023/A19070260
Citation: Yu Jun, Yang Yusen, Wei Min. Preparation and Catalytic Performance of Supported Catalysts Derived from Layered Double Hydroxides[J]. Acta Chimica Sinica, 2019, 77(11): 1129-1139. doi: 10.6023/A19070260
水滑石基负载型催化剂的制备及其在催化反应中的应用
摘要:
负载型催化剂作为一类重要工业催化剂,广泛应用于合成氨工业、能源化工和精细化工等重要的工业生产过程.水滑石(LDHs)是一类阴离子型二维层状无机功能材料,其具有层板元素比例可调、金属阳离子高分散和结构拓扑转变等特性,在多相催化中,其作为负载型催化剂的前体或者载体具有广阔的应用前景.总结了以LDHs或其拓扑转变得到的复合金属氧化物(MMO)作为催化剂载体,以LDHs作为催化剂前体,制备高性能的负载型单金属或双金属催化剂,聚焦于其在电催化、氧化脱氢、选择性加氢和合成气转化反应中的最新研究进展.最后,进一步讨论了LDHs基负载型催化剂未来的发展趋势以及面临的挑战,并提出了解决这些问题的有效方案.
English
Preparation and Catalytic Performance of Supported Catalysts Derived from Layered Double Hydroxides
Abstract:
Supported catalysts have been widely used in a large variety of industrial processes, including ammonia synthesis, energy conversion and fine chemical synthesis. Layered double hydroxides (LDHs) are a class of two-dimensional functional anionic materials. By virtue of the unique structural characteristics (e.g., tunability of host layers, high dispersion of metal cations and structure topological transformation), LDHs have shown potential applications in heterogeneous catalysis as precursors or supports. In this review, high-performance monometallic or bimetallic supported catalysts by using LDHs as supports/precursors, or by utilizing mixed metal oxides (MMO) as supports via topotactic transformation from LDHs is highlighted. Their recent progresses in electrocatalysis, oxidative dehydrogenation, selective hydrogenation and syngas conversion reaction are reviewed. In the final section, future opportunities and challenges in the preparation of LDHs-based catalysts are discussed, and some strategies to resolve these critical problems are further proposed.
-
1. 引言
负载型催化剂是一类重要的工业催化剂, 其在合成氨、费托合成、选择性加氢和水蒸气重整等多种重要的工业反应中发挥着不可替代的作用, 引起了国内外研究者的广泛关注.然而, 采用传统的浸渍法制备的负载型催化剂, 由于浸渍溶液的表面张力以及溶剂效应等因素的影响常常导致活性组分在载体上分散不均匀.此外, 载体和活性前体之间的弱相互作用会使得金属在高温焙烧过程中发生迁移而出现团聚现象, “特别是在反应过程中, 如在水蒸气重整反应、水煤气变换反应以及CO氧化等反应过程中, 活性金属的团聚致使催化剂的活性和稳定性显著降低, 最终导致催化剂失活[1].随着人们对工业生产的要求不断提高, 发展新型、高效、高稳定性的负载型催化剂成为科研工作者共同关心的重要课题和未来工业发展的必然趋势.
水滑石(layered double hydroxides, LDHs)是阴离子型层状双金属氢氧化物[2~7], 其主体层板与水镁石Mg(OH)2结构相似, 其化学组成可表示为
$[{\rm{M}}_{{\rm{1 - }}x}^{2 + }{\rm{M}}_x^{{\rm{3 + }}}{\rm{ - }}$ (OH)2][An−]x/n•mH2O; 其中M2+和M3+分别为主体层板中占据八面体中心的二价和三价金属阳离子, 而且层板中的二价金属阳离子M2+在一定的比例范围内可以被半径相似的三价金属阳离子M3+同晶取代, 从而使层板带有正电荷; An−代表层间阴离子, 起到电荷平衡的作用, 使得LDHs整体呈现电中性. LDHs材料具有层板金属元素呈原子级分散、层板组成比例可调、插层阴离子可交换[6, 8~12]、限域效应以及结构记忆效应[4, 13]等结构特点, 由此可构筑多种多样的无机功能材料.近年来, 随着纳米材料以及二维层状材料的飞速发展, LDHs由于自身结构的独特性, 在光催化、电催化、固体碱催化、催化氧化、选择性加氢以及合成气转化等诸多领域表现出广泛的应用前景[2~4, 8, 9, 13~22]. LDHs主体层板金属阳离子的高分散以及层间阴离子的取向排列使得LDHs焙烧/还原之后形成缺陷结构, 这极大地提高了LDHs基催化材料的催化活性[8, 23].同时, 焙烧/还原之后活性组分与载体之间的强相互作用防止了活性组分的团聚, 改善了催化剂的稳定性.通过各种合成策略可对LDHs基催化材料的粒径、形貌、载体性质、表面缺陷结构以及电子结构进行调控, 提供催化反应所需要的特定结构和活性中心[16, 24~28].本文总结了以LDHs或者其拓扑转变得到的复合金属氧化物(mixed metal oxides, MMO)作为催化剂载体制备高度分散的负载型催化剂, 并对LDHs的结构拓扑转变过程进行了探讨.特别地, 以LDHs为前体, 基于LDHs的结构拓扑转变效应, LDHs在不同气氛下(氢气、空气或者氩气)可诱导形成高分散、高稳定、高活性的负载型金属催化剂, 并聚焦于其在电催化、氧化脱氢、选择性加氢和合成气转化中等领域的最新研究进展.本文还详细讨论了催化剂活性结构的表征、确定以及高催化性能与催化剂结构的内在关系, 为新型LDHs基负载型催化剂的合理设计以及制备提供了一定的理论依据.最后, 从LDHs基负载型催化剂微观结构的精确调控以及反应机理的揭示等方面讨论了今后LDHs基负载型催化剂的机遇和挑战, 并提出了解决这些科学问题的可行策略.希望这篇综述能够吸引更多研究者关注LDHs基负载型催化剂, 鼓励人们在这一迅速发展的领域开展富有创新性的研究.2. 水滑石拓扑转变效应
LDHs作为催化剂的前体或者催化剂载体应用于非均相催化领域引起了人们的广泛关注[16, 29~35]. LDHs有两个独特的性质, 一是M2+和M3+金属阳离子在层板中呈现原子级分散, 层板元素具有可调变性; 二是LDHs的结构拓扑效应[4, 8], 即在焙烧和还原过程中LDHs层板元素保持均匀分散的结构特征, 显著提升金属催化剂的分散度和稳定性.大量研究表明[4, 6, 16, 36~38]通过调变拓扑转变过程的条件(焙烧温度、还原温度、升温速率、时间、气氛等), 可制备一系列尺寸、形貌、表面结构、电子结构可控的高分散负载型催化剂.所谓拓扑转变反应是指同一反应中反应物晶体或者产物晶体在晶体结构上至少存在一种等价关系[39].对于LDHs的拓扑转变过程, 前人已经进行广泛的研究. Garcia等[40]使用原位傅里叶变换红外光谱(FT-IR)研究MgAl-LDH热分解来跟踪化学组成变化, 并且使用程序升温脱附-质谱(TPD-MS)监测每个阶段中特定化学物质.发现MgAl- LDH热分解至少经历层间水脱除和层板脱羟基、层间阴离子分解、层板结构的收缩和坍塌以及新相的生成四个步骤. Ferreira等[41]通过实验手段[热重、X射线衍射(XRD)、FT-IR和穆斯堡尔光谱]研究了MgFe-LDH的热分解过程, 发现在300 ℃时, MgFe-LDH层状结构被破坏, 温度超过500 ℃时形成MgO和MgFe2O4尖晶石. Zhao等[42]在焙烧ZnAl-LDH前体时, 通过高倍透射电子显微镜(HRTEM)观察到ZnO相成核, 随后沿(1010)方向定向生长, 并在高于500 ℃的温度下形成ZnO和ZnAl2O4混合尖晶石结构.本课题组[43]报道了一种在无定型Al2O3微球表面原位生长的ZnAl-LDH纳米片, 该纳米片经拓扑还原后得到(0001)面高度暴露的六方ZnO纳米片. HRTEM和选取电子衍射(SAED)结果表明:在拓扑转变过程中, ZnAl-LDH的片层结构诱导ZnO沿(1010)方向优先生长并抑制其在c轴方向上的生长, 从而导致六方ZnO纳米片(0001)面的高度暴露.相对于ZnO纳米棒和ZnO纳米片对照样品, 基于LDH前体经拓扑转变得到的六方ZnO纳米片表现出更优异的可见光催化活性和稳定性. HRTEM和紫外-可见漫反射光谱结果表明ZnO纳米片存在多级结构的高度分散, 而且ZnO(0001)晶面具有丰富的氧缺陷与边缘位错, 提供了催化反应活性位点, 从而导致其优异的可见光催化活性.
虽然研究者采用实验方法对LDHs拓扑转变过程中的形貌、晶面以及晶体参数等进行了研究, 但是难以获得原子排布和电子结构等方面的信息; 而理论计算(DFT)弥补了实验上的不足[44~46]. Costa等[45]采用DFT计算结合热力学和动力学分析的方法对CO32-插层的MgAl-LDH在25~350 ℃内的热分解机理进行了研究, 发现在280 ℃时CO32-会以单齿配体的形式进行分解, 350 ℃变成双齿配体.本课题组[46]利用分子动力学模拟以及实验相结合的手段对MgAl-LDH热致拓扑转变过程进行了探究.发现在450 ℃时, 金属阳离子在LDHs二维层板平面上几乎维持着与原来相同的原子排布方式, 仅在垂直于层板平面的方向(c轴方向)发生移动, 揭示了LDHs结构拓扑转变特性来源于LDHs层板水平方向上的拓扑不变性, 为利用LDHs拓扑转变特性制备高性能负载型催化剂提供了一定的理论依据.
3. LDHs基负载型催化剂的制备
3.1 LDHs或MMO作为催化剂载体
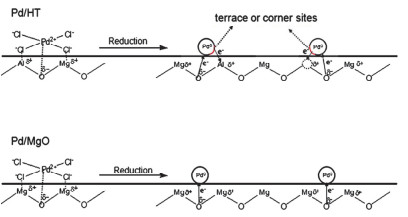
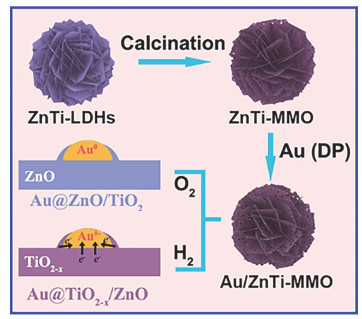
LDHs及其焙烧产物MMO由于具有优异的吸附能力、表面可调的酸碱性以及限域效应等优点可用作催化剂的载体[4, 16].根据LDHs层板构筑规则[47, 48], 对于难以进入层板的元素, 比如Ru、Pd、Pt、Au等, 通过层板的金属阳离子位点、阳离子空位以及碱性位点等对活性金属进行锚定, 经还原得到高分散、高稳定的LDHs基负载型催化剂.李殿卿等[49]采用水热法制备MgAl-LDH载体, 随后通过湿法浸渍将Pd负载在MgAl-LDH、MgO(图 1)和Al2O3表面发现MgAl-LDH载体上的碱性位点以及金属空位等不仅可以稳定并促进Pd金属的分散, 而且还可以调节Pd的电子密度, 使Pd/MgAl-LDH催化剂相对于Pd/MgO和Pd/Al2O3在乙炔选择性加氢反应中表现出更高的活性以及选择性. Ebitani等[50]使用可溶性淀粉作为绿色还原剂和稳定剂合成了新型LDHs负载型PtAu合金催化剂, 其在常温常压条件下可催化氧化甘油和1, 2-丙二醇选择性地生成甘油酸和乳酸. Tichit等[51]用浸渍、共沉淀以及阴离子交换三种不同的途径将Pd负载于MgAl-MMO表面并用于1-丁炔-1, 4-二醇的选择性加氢反应.所制备的三种催化剂都具有良好的分散性, 特别是浸渍法得到的负载型Pd催化剂(Pd的粒径约为2 nm), 其对于1-丁炔-1, 4-二醇的选择性加氢具有最优的活性以及选择性.本课题组[38]以ZnTi-LDH经结构拓扑转变得到的ZnTi-MMO为载体, 以尿素为沉淀剂, 采用沉淀沉积法将Au纳米颗粒固定在ZnTi-MMO上, 通过调节不同还原温度可得到电子结构可调的Au@TiO2-x/ZnO催化剂(图 2).该催化剂在水煤气变换反应中显示了高活性和良好的稳定性.此外, 本课题组[24]采用原位生长法合成了多级结构NiAl-LDHs, 再通过原位焙烧还原得到具有高表面缺陷浓度的负载型Ni基催化剂.随后, 以Ni表面缺陷位为成核位点, 诱导Ru团簇选择性地锚定在Ni表面缺陷位, 最终得到Ru-Ni双金属异质结构催化剂, 该催化剂对乙醇水蒸气重整反应表现出很高的活性和选择性, 极大提高了H2产率.肖丰收等[52]通过浸渍法直接将Au负载于MgAl-LDH表面, 随后在N2气氛中进行高温焙烧得到被载体部分包覆的Au/MgAl-MMO催化剂.该包覆结构有效抑制了Au纳米颗粒在高温下的聚集, 对于CO氧化和乙醇脱氢反应表现出较高的稳定性.利用LDHs层间阴离子可交换的特点, 将含有活性金属阴离子的配合物引入层间, 还原之后也可以得到相应负载型金属催化剂. LDHs层间限域效应可在一定程度上防止金属纳米粒子的聚集.李峰等[53]将PdCl42-离子通过离子交换的方法引入到MgAl-LDH层间制备了高度分散的负载型Pd基催化剂, 该催化剂可以促进均匀多壁碳纳米管的生长. Wei等[54]采用共沉淀法制备了FeMgAl-LDH, 随后将焙烧的材料在含MoO42-的盐溶液中恢复LDHs层状结构, MoO42-作为平衡电荷的阴离子嵌入层间, 得到高密度、高分散和高稳定的负载型Fe基催化剂, 该催化剂对单壁双螺旋碳纳米管的合成具有高催化活性.
图 1
图 2

3.2 LDHs作为负载型催化剂前体
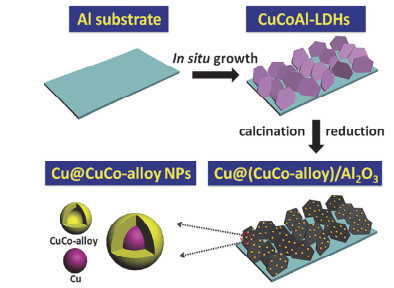
以LDHs为前驱体, 基于其结构拓扑转变效应, 经过焙烧还原可制备粒径、形貌、几何/电子结构可调的负载型单金属、双金属合金以及金属间化合物催化剂. LDHs中的金属离子在主体层板中呈现原子级均匀分散, 有利于在焙烧还原过程中形成稳定性高且分散性好的负载型金属催化剂[4, 15, 16, 47].此外, 活性金属与氧化物载体之间存在强相互作用, 可以有效地抑制纳米金属颗粒的团聚和烧结.本课题组[37]将Ni2+和Ti4+通过尿素法引入层板, 制得NiTi-LDH纳米片, 随后经过焙烧和不同还原温度处理得到TiO2-x部分包裹的负载型Ni基催化剂, 优化后的Ni@TiO2-x (450)催化剂在高温水煤气变换反应中表现出良好的催化活性.此外, 本课题组[55]以γ-Al2O3为载体, 采用水热法在γ-Al2O3上原位生长CuCoAl-LDH, 经过焙烧还原过程制备得到了具有核壳结构的负载型Cu@(CuCo-alloy)/Al2O3催化剂(图 3)用于合成气制高级醇.通过改变CuCoAl-LDH前体中的Cu/Co物质的量之比可以调节Cu@(CuCo-alloy)纳米粒子的组成、粒径以及壳的厚度.李福伟等[56]通过改进的共沉淀方法制备了多种比例的CuxNiyMgAl-LDH前体, 在不同温度下还原得到高度分散的负载型CuNi双金属合金催化剂, 研究发现, 催化剂前体的还原温度直接影响CuNi合金纳米粒子的分散性以及催化剂表面的酸碱度, 从而影响催化剂对于糠醛的加氢选择性.载体表面的酸碱性也可以通过改变焙烧和还原温度来调控. Coq等[57]采用共沉淀法制备了NiMgAl-LDHs, 再经不同焙烧和还原温度处理得到表面酸碱性各异的Ni/MgAl-MMO催化剂; 发现经623 K焙烧、723 K还原处理制备的Ni/MgAl-MMO催化剂, 其表面弱布朗斯特酸性位点与碱性位点的比率最低, 其对乙腈加氢生成乙胺的选择性最高.类似地, Tomishige等[58]采用共沉淀法合成了NiFeMgAl-LDHs前体, 经焙烧还原得到了均匀分散于载体MgAl-MMO表面的NiFe合金催化剂, 其在甲苯水蒸气重整反应中表现出很高的活性.此外, 通过调节LDHs前体中Ni/Fe比例可以实现对合金中Ni/Fe比例的调控.本课题组[59]将Ni和Ga元素引入LDHs层板制得三元NixMgyGaz-LDHs前体, 经拓扑还原得到负载型Ni-Ga金属间化合物催化剂, 通过调节LDHs层板的Ni/Ga比或者调节拓扑转变中的还原温度, 可以调节Ni-Ga金属间化合物的组成和粒径.
图 3

4. LDHs基负载型催化剂在催化反应中的应用
4.1 在电催化反应中的应用
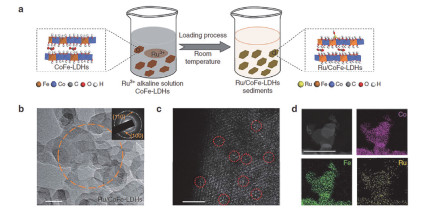
随着经济社会的快速发展, 煤、石油、天然气等不可再生能源不断被消耗, 加剧了能源危机与环境污染; 清洁、高效的电化学能源转化和储存技术在应对能源危机与环境污染中扮演着重要的角色[60, 61], 而发展高性能、低成本的电催化剂成为电化学能源转化和储存技术的关键问题[62, 63].单原子催化剂(Single-Atom Catalysts, SACs)是一种分散在载体上由互相隔离的金属原子组成的催化剂, 具有最大化的原子利用率及高度配位不饱和等优势, 在多相催化以及电催化方面显示出独特的催化活性和选择性, 成为解决这一关键问题的重要途径[64~66].近日, 孙晓明等[66]报道了一种锚定在CoFe-LDHs的单原子Ru催化剂(图 4), 该催化剂中的单原子Ru与载体CoFe-LDHs具有很强的电子偶合作用, 当钌的负载量达到0.45%时, Ru/CoFe-LDHs表现出优异的析氧反应(oxygen evolution reaction, OER)性能, 在10 mA•cm-2电流密度下, 催化剂的过电位仅198 mV, Tafel斜率为39 mV•dec-1.作者通过X射线吸收光谱(XAS)研究发现, 由于金属Ru与LDHs之间强电子偶合作用, 单原子Ru在高的过电位依旧保持+4价的氧化态, 而且*OOH有着最佳的吸附自由能, 这使得催化剂具有优异的电化学活性以及稳定性, 优于商业化的RuO2和IrO2催化剂.近年来, NiFe-LDH作为一类OER性能优异的非贵金属催化剂受到广泛研究.张兵等[67]通过电化学沉积法在NiFe-LDH上负载单原子Au (sAu/NiFe LDH), 在280 mV的过电位下, 其电流密度可达129.8 mA• cm-2, 比单独NiFe-LDH提高约6倍; sAu/NiFe LDH进行2000次连续循环后, 并没有发生明显的活性衰减.这表明sAu/NiFe LDH有着优异的OER活性以及稳定性.此外, LDHs不仅可以作为金属催化剂的载体, LDHs自身或者焙烧得到的MMO也可以直接作为电催化剂[63, 68].张铁锐等[69]采用简便的超声波剥离的方法合成了粒径约为3.5 nm、厚度约为0.5 nm超薄单层ZnCo- LDH纳米片(ZnCo-UF).所得到的ZnCo-UF表现出优异的OER性能, 这主要归因于ZnCo-UF表面具有丰富的氧空位和配位不饱和位点, 增强了对H2O的吸附.赵宇飞等[70]采用反相微乳液法制备单层NiTi-LDH纳米片, 经拓扑转变后得到粒径约为4.0 nm、厚度约为1.1 nm的超薄NiO纳米片.所得到单层NiTi-MMO催化剂表现出优异的OER性能:在1.55 V时, 电流密度高达10 mA• cm-2, 性能远优于对照样体相NiTi-MMO.实验表征结合DFT计算研究发现单层NiTi-MMO纳米片高度暴露(110)晶面, 其含有丰富Ni3+位点、界面位点以及Ti3+缺陷位点, Ti3+的存在改善了电荷传输效率, 促进了H2O的吸附, 从而显著提高了OER性能.
图 4
 图 4. (a) 水解沉积制备Ru/CoFe-LDHs的示意图、(b) Ru/CoFe-LDHs纳米片的透射电镜(TEM)图像、(c) Cs校正的Ru/CoFe-LDHs纳米片的STEM图像和(d) Ru/CoFe-LDHs的高角环形暗场扫描透射电镜(HAADF-STEM)图像及相应的Ni, Fe和Ru元素分布图[66]Figure 4. (a) Schematic illustration for the hydrolysis-deposition to form Ru/CoFe-LDHs, (b) transmission electron microscopy (TEM) images of as-prepared Ru/CoFe-LDHs nanosheets (inset shows the corresponding SAED pattern of Ru/CoFe-LDHs nanosheets marked in orange circle), (c) Cs-corrected STEM image of Ru/CoFe-LDHs nanosheets shows the monoatomic ruthenium dispersed on the surface of LDHs (some of the isolated Ru atoms are marked with red circles) and (d) HAADF-STEM images of the Ru/CoFe-LDHs and corresponding elemental distribution maps of Ni, Fe, and Ru in the Ru/CoFe-LDHs[66]
图 4. (a) 水解沉积制备Ru/CoFe-LDHs的示意图、(b) Ru/CoFe-LDHs纳米片的透射电镜(TEM)图像、(c) Cs校正的Ru/CoFe-LDHs纳米片的STEM图像和(d) Ru/CoFe-LDHs的高角环形暗场扫描透射电镜(HAADF-STEM)图像及相应的Ni, Fe和Ru元素分布图[66]Figure 4. (a) Schematic illustration for the hydrolysis-deposition to form Ru/CoFe-LDHs, (b) transmission electron microscopy (TEM) images of as-prepared Ru/CoFe-LDHs nanosheets (inset shows the corresponding SAED pattern of Ru/CoFe-LDHs nanosheets marked in orange circle), (c) Cs-corrected STEM image of Ru/CoFe-LDHs nanosheets shows the monoatomic ruthenium dispersed on the surface of LDHs (some of the isolated Ru atoms are marked with red circles) and (d) HAADF-STEM images of the Ru/CoFe-LDHs and corresponding elemental distribution maps of Ni, Fe, and Ru in the Ru/CoFe-LDHs[66]张涛课题组[71]以NiAl-LDH为前驱体制备了高度分散、高负载量的Ni/Al2O3催化剂, 该催化剂在室温条件下就可以催化水合肼完全分解, 选择性高达93%, 有效降低了副反应生成的氨对于Nafion膜以及燃料电池催化剂的毒化作用.通过CO2-TPD对碱性位的数量以及强度进行定量, 发现催化剂高的活性以及选择性主要归因于金属位点和强碱性位点的协同催化作用.本课题组[72]通过成核和老化两步法制备了NiFeMg-LDHs, 经焙烧还原制备含有NiFe合金活性中心以及固体碱的双功能NiFe/MgO合金催化剂.高倍透射电子显微镜及高角环形暗场扫描透射电子显微镜(HAADF-STEM)结果证实NiFe合金高度分散在基底MgO上, X射线吸收近边结构(XANES)表明NiFe合金中Ni和Fe之间存在电子相互作用, 使得Fe电子密度升高, 这种富电Fe的产生为氢原子从催化剂表面的分离提供了充足的电子, 并且Ni和Fe之间的协同效应也降低了N—H键断裂的能垒.这些结构特点使得催化剂在室温下高效催化水合肼制氢, 转化率达到100%, 对H2选择性达到99%.此外, 本课题组[73]在ITO基底上直接生长CoAl-LDH纳米阵列(LDH-NW), 随后利用LDH-NW与PdCl42-前体之间的原位氧化还原反应制备了锚定在LDH-NW上的Pd纳米粒子.由于Pd活性位点的有效暴露, 所得到的Pd/LDH- NW催化剂对乙醇的电氧化反应表现出高的催化活性以及耐受性. DFT计算研究表明, LDH载体通过Pd—HO键稳定Pd纳米颗粒, 并伴随着电子从LDH转移到Pd纳米颗粒上.因此, 金属载体之间的协同作用有助于增强乙醇电氧化性能.
4.2 在氧化脱氢反应中的应用
醇脱氢羰基化反应[74]以及烷烃氧化脱氢反应[75] (oxidative dehydrogenation, ODH)不仅可以制备高热值的清洁能源H2, 还可以合成醛/酮化合物以及重要的化工原料烯烃.肖丰收等[76]通过离子交换和NaBH4还原两步法在MgAl-LDH上制备了粒径为1~5 nm的小尺寸Au纳米粒子.在对醇类氧化反应性能评价中发现, Au/LDH在非常温和的条件下, 对多种伯醇和仲醇的氧化表现出优异的活性以及高的选择性. X射线光电子能谱(XPS)表明Au/LDH上Au带部分负电荷, 可能由于Au纳米粒子与LDH之间有强相互作用; 而且带负电的Au纳米粒子有利于醇有氧氧化中分子氧的活化.另一方面, 2-丙醇的程序升温表面反应(TPSR)实验证明载体对于2-丙醇具有优异的脱氢能力.李殿卿等[77]同样以MgAl-LDH为载体, 制备了负载型Pt/MgAl-LDH催化剂, 相对于TiO2和MgO载体负载的Pt基催化剂, Pt/MgAl-LDH催化剂在1-辛醇的氧化反应中表现出最高的活性以及选择性.张法智等[32]采用均相沉淀沉积法将Au负载在MgAl-LDH上制得Au/MgAl-LDH催化剂(图 5).有趣的是, 研究发现Au优先沉积于LDHs边缘具有高密度悬空键的(1010)晶面上, 形成小尺寸的Au纳米颗粒; 随着金属负载量的增加, LDHs的(0001)晶面表面的缺陷位点也成为金属离子的吸附中心, Au逐渐会在LDHs的(0001)晶面上沉积, 形成较大尺寸的Au纳米粒子; 不同粒径的Au/MgAl-LDH催化剂也直接导致了对苯乙烯环氧化反应催化性能的明显差异. LDH经过焙烧得到的复合金属氧化物具有高的表面积、高的金属分散度以及小的微晶尺寸. Marcu等[75]首次报道了基于Co(x)MgAl-LDH前体焙烧得到不同Co含量(原子百分数为1%~20%)的Co(x)MgAlO复合氧化物催化剂用于丙烷的氧化脱氢反应.发现丙烷的ODH反应活性随着Co含量的增加而增加; 然而只有在Co含量较低时, 才能获得最高的乙烯选择性, 其中Co(5)MgAlO和Co(7)MgAlO催化剂在873 K下获得最高的丙烯产率.拉曼(Raman)和光致发光光谱表明, 分散良好的四面体配位钴物种在丙烷氧化脱氢成丙烯反应中起主要作用.除此之外, 利用LDHs层间阴离子可交换的性质, 可以将V元素引入层间, 焙烧之后获得的VMgAlO复合氧化物催化剂也可以作为轻质烷烃氧化脱氢的催化剂[78]. Pt作为过渡金属中最有效的烷烃脱氢催化剂之一, 然而在缺乏改性剂的情况下, 催化剂对烯烃的选择性并不高, 烯烃在Pt表面再吸附产生焦炭而使催化剂失活.通过引入Sn, Ga, In, Zn, Ge等元素与Pt相互作用形成双金属合金可以实现高的烯烃选择性, 产生较少的焦炭[79~82]. Bell等[80~82]以MgGaAl-LDH和MgInAl-LDH为前驱体, 结合浸渍法制备了负载型PtGa和PtIn双金属合金催化剂.将其用于乙烷和丙烷脱氢的反应中发现, 当Ga/Pt和In/Pt比分别为5.4和0.48时, PtGa和PtIn双金属合金催化剂显示出最优活性, 对烯烃的选择性接近100%.研究表明, Ga和In对活性金属Pt的分割效应增强了反应物烷烃的解离吸附, 削弱了产物烯烃的吸附, 由此导致烯烃的高选择性, 最大限度减少焦炭的形成. Belskaya等[83]将Sn引入LDHs层板, 利用焙烧后的MgAl(Sn)Ox载体对氯铂酸进行吸附, 含Sn复合氧化物恢复为LDHs结构的同时将[PtCl6]2-嵌入LDHs层间.透射电子显微镜(TEM)和扩展X射线吸收精细结构(EXAFS)等研究表明载体中Sn可以提高Pt的分散度; XPS数据证明电荷从Sn转移到Pt上.为了揭示氧化物载体中的Sn对Pt催化性能的影响, 对丙烷和正癸烷进行脱氢氧化反应性能评价, 发现引入最佳含量的Sn可以使丙烷转化率增加两倍以上, 丙烯选择性略有提高; 当载体中Sn含量过高时将导致催化剂失活.
图 5

金属位和载体酸碱位之间的协同效应在多相催化中扮演着重要的角色[84, 85], 包括不饱和醛加氢、乙炔选择性加氢以及醇脱氢等反应.本课题组[85]采用原位生长法合成了多级结构NiAl-LDH前体, 通过精确控制还原温度, 构筑了载体酸碱性可控的负载型Ni/NiAl-MMO催化剂(图 6). XRD、Raman和EXAFS等多种原位表征手段证明立方相的类NiO (Al3+掺杂NiO)高度分散在无定型的Al2O3中.借助CO2-TPD和NH3-TPD等表征技术研究发现样品中的中强酸-碱对(Niδ+-Oδ-对)主要来源于类NiO相.将Ni/NiAl-MMO催化剂用于2-辛醇脱氢羰基化反应中, 发现2-辛酮的生成速率高达78.5 mmol•g-1•h-1, 是传统浸渍法制备的Ni/Al2O3催化剂的3.9倍.进一步, 采用时间分辨的原位EXAFS以及动力学同位素(KIE)对催化剂结构-性能相关性进行研究, 发现金属位点Ni0以及载体中类NiO的中强酸-碱对的协同效应加速了动力学关键步骤α-C—H键断裂和O—H键断裂, 从而有效提升了催化剂对2-辛醇脱氢羰基化的催化活性.
图 6

4.3 在选择性加氢反应中的应用
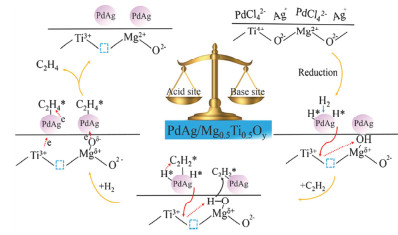
选择性加氢催化剂在石油化工、精细化学品以及生物质利用等领域都扮演着重要的角色[86, 87], 如负载型贵金属催化剂Pd/Al2O3被广泛应用于乙烯装置中C2~C4等组分以及石油裂解中炔烃和二烯烃的脱除.李殿卿课题组[88]以MgGaAl-LDH为前体, 将Pd组分引入, 由于带正电LDH层板与[PdCl4]2-阴离子之间的静电相互作用实现了活性前体的均匀分散.焙烧还原后获得的分散良好的负载型双金属Pd-Ga/MgO-Al2O3催化剂应用到乙炔选择性加氢反应中.在相同的乙炔转化率(90%)下, Pd-Ga (1:5)/MgO-Al2O3, Pd-Ga (2:1)/MgO-Al2O3和Pd/MgO-Al2O3催化剂对乙烯的选择性分别为82%, 75%和55%.相对于单金属Pd/MgO-Al2O3催化剂, 双金属Pd-Ga/MgO-Al2O3催化剂对乙烯表现出更高的选择性. XPS和CO-IR证实了双金属PdGa合金催化剂中Pd与Ga之间的几何效应以及电子效应促进了乙烯在双金属Pd-Ga/MgO-Al2O3催化剂表面的脱附, 进而提高乙烯的选择性.进一步, 基于富缺陷的NiTi-LDH制备了新型负载PdAg纳米合金催化剂[89].该催化剂对乙炔选择性加氢表现优异的催化性能:当乙炔转化率达到90%时, 对乙烯的选择性保持在82%.基于LDHs的结构拓扑转变特性得到的复合金属氧化物具有丰富的Lewis酸碱位点, 其与金属之间的协同效应可以提高反应的活性以及选择性.李殿卿等[90]将PdAg双金属合金负载在不同Mg/Ti比的MgTi-MMO载体上, 用于乙炔选择性加氢反应中(图 7).催化结果表明, 随着Mg/Ti比的增加, 催化剂对于乙炔转化率和乙烯选择性呈现先上升后下降的规律.其中, PdAg/Mg0.5Ti0.5Oy催化剂在70 ℃时转化率接近100%, 选择性为83.8%, 远高于其他Mg/Ti比的催化剂.研究表明, 载体中等强度的酸性位点促进氢溢出效应, PdAg/Mg0.5Ti0.5Oy催化剂具有最明显的氢溢流效应, 促进了氢的活化和解离(氢的活化解离是乙炔加氢的速控步).因此, 有利于提高PdAg/Mg0.5Ti0.5Oy催化剂乙炔加氢的活性.选择性的提高主要归因于几何效应和电子效应.一方面, 载体碱性位点的电子转移和Ti3+缺陷引起Pd电子密度增加, 有利于乙烯的脱附; 另一方面, PdAg/Mg0.5Ti0.5Oy催化剂中PdAg的高合金化程度增加了Pd线性配位位点的数量, 促进了乙烯在催化剂表面的脱附, 从而进一步提高反应选择性.
图 7

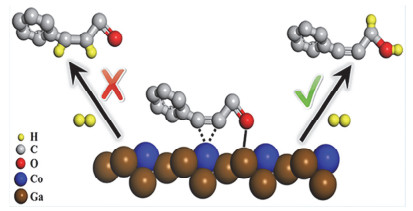
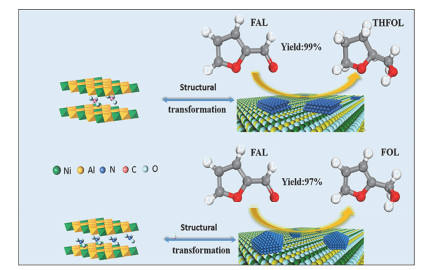
金属间化合物(IMCs)由两种或多种具有特定成分和确定晶体结构的元素组成, 其作为优异的选择性加氢催化剂引起了广泛关注. Pd和Pt等贵金属以及Ni和Co等过渡金属作为第一活性金属通常对氢以及α, β-不饱和醛有吸附和解离的作用. Sn, Ga和In等第二金属作为调节剂可以改变第一金属的电子结构, 并且第二金属的存在也可以分离第一金属的连续金属位点, 从而提高对α, β-不饱和醛的选择性[15, 28, 59, 91].通常, C=O和C=C基团在催化剂表面的竞争吸附对最终产物的选择性起着决定性的作用[92].本课题组[93]将不同物质的量之比的Ni和In通过共沉淀法引入层板制得NixAlyInz-LDHs和NixMgyInz-LDHs前体, 共还原得到不同粒径的高分散负载型Ni-In金属间化合物催化剂.相对于对照样Ni/Al2O3, Ni-In IMCs在不饱和醛酮加氢生成不饱和醇反应中表现出良好的活性以及很高的选择性.其中, 在110 ℃下, Ni2In/Al2O3(粒径5.1 nm)催化剂可获得99%糠醇产率.在五个循环周期内, 催化剂表现出良好的可循环性和稳定性, 这主要归因于氧化物载体对于金属间化合物纳米颗粒的锚定作用增强了催化剂的稳定性. X射线吸收精细结构(XAFS)以及DFT计算结果表明Ni-In之间存在强的电子相互作用.此外, In对于活性组分Ni的分割效应进一步增加了Ni-In IMCs对于不饱和醇的选择性.本课题组[94]进一步基于CoInMgAl-LDH和CoGaMgAl-LDH前驱体经共还原处理获得负载型Co-In和Co-Ga金属间化合物催化剂(图 8). GoGa3催化剂在肉桂醛选择性加氢反应中, 对肉桂醇表现出最高的选择性(96%), 远高于单金属Co催化剂(42%).采用XANES、XPS和CO-IR表征证实了从Ga(或In)到Co的电子转移. FT-IR光谱和DFT计算证实了IMCs中带正电的元素Ga或In充当活性位点并促进极化基团C=O的吸附和活化, 同时抑制C=C的吸附, 由此导致对C=O基团的高选择性.该研究揭示了底物在催化剂表面的吸附构型最终决定了加氢位点以及产物选择性.同时, 基于FT-IR和DFT计算的结果, 提出了在CoGa3催化剂表面上肉桂醛选择性加氢成肉桂醇的反应机理. Kong等[95]首次报道了以LDHs为前体制备的Ni/Al2O3催化剂可以有效将5-羟甲基糠醛(HMF)有选择性地转化为2, 5-二甲基呋喃(DMF).通过调节焙烧温度和反应条件, DMF的产率可以达到91.5%. Yan等[96]通过改进的共沉淀法合成了Cu-Cr催化剂, 该催化剂在糠醛选择性加氢反应中, 实现了对糠醇95%的产率.本课题组[8]利用LDHs层间阴离子可交换的特性, 制备了不同阴离子(NO3-, CO32-)插层的NiAl-LDH前体, 经过拓扑还原后所获得的两种负载型的Ni/MMO-NO3和Ni/MMO-CO3催化剂对糠醛加氢反应表现出完全不同的催化选择性(图 9).其中, Ni/MMO-NO3催化剂对于糠醇具有高选择性(97%), 而Ni/MMO-CO3催化剂仅对四氢糠醇具有选择性(99%). HRTEM, XAFS和CO-IR证实Ni/MMO-CO3催化剂表面主要暴露Ni(111)晶面, 这有利于C=O基团以及呋喃环C=C基团的活化吸附(平躺吸附构型), 从而导致糠醛过加氢成四氢糠醇.相反, Ni/MMO-NO3催化剂表面含有丰富的低配位Ni台阶/边缘位点, 使得糠醛中只有C=O基团在Ni表面发生线式活化吸附(直立吸附构型), 从而实现了糠醇的高选择性.由具有不同层间阴离子的LDHs前体调节Ni纳米颗粒的表面精细结构, 从而对底物糠醛分子吸附构型以及产物的选择性产生极其显著的影响.
图 8

图 9

4.4 在合成气转化中的应用
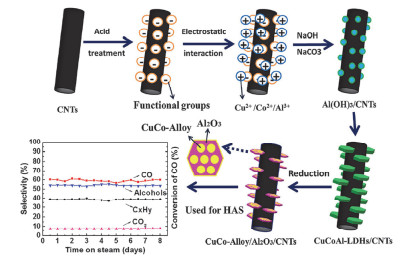
随着石油资源的减少以及能源消耗的增加, 替代燃料的使用和研究引起了研究者们极大的兴趣.由天然气、煤以及生物质资源经合成气(CO+H2)转化为高级醇等液体燃料以及其他高附加值的化学品成为最有前途的方法之一[97, 98].近年来, Cu-Co和Cu-Fe基催化剂被广泛用于合成气转化研究.本课题组[99]以CuFeMg-LDH为前体, 经过焙烧还原过程得到均匀且高度分散的负载型CuFe基催化剂, 其对合成气制醇表现出相当高的CO转化率(56.89%)以及高级醇的选择性(49.07%), 总醇产率达到0.28 g•mLcat-1•h-1, 优于共沉淀法制备的CuFe催化剂.研究表明均匀高分散的CuFe活性位点为H2和CO吸附提供更多的不饱和配位中心; 另外, Cu和Fe之间协同作用有助于提高醇的选择性.进一步, 以CuCoAl-LDH为前体, 通过两步法制备具有核壳结构的负载型Cu@(CuCo-alloy)/Al2O3催化剂[55].将其用于CO加氢制高级醇中发现, Cu:Co为1:2的催化剂获得最优的催化性能, CO转化率为21.5%, C6+伯醇的选择性达到48.9%. Liu等[100]利用共沉淀法制备了不同Cu/Co比的(CuxCoy)2Al-LDHs, 焙烧还原之后得到负载于Al2O3的CuCo合金纳米颗粒.所得到的双金属催化剂在合成气制高级醇中表现出高活性、良好的稳定性和对C2+醇的选择性.在反应温度为250 ℃, 压力为3 MPa, 体积空速(GHSV)为3900 mL•(gcat•h)-1的反应条件下, CO转化率为51.8%, 对醇类的选择性为45.8%, 其中C2+醇占比94.3 wt%.考虑到合成气制高级醇是一个强放热反应, Liu等进一步将导热性良好的碳纤维(CFs)和改性的碳纳米管(CNTs)与CuCoAl-LDH结合形成CuCoAl-LDH/CFs[101]以及CuCoAl-LDH/CNTs复合材料[102], 经拓扑还原后均可得到高度分散的负载型CuCo合金纳米颗粒(图 10), 由于CFs和CNTs的高导热性, 有效抑制了烃类和CO2的产生, 因此CuCo-alloy/Al2O3/ CFs以及CuCo-alloy/Al2O3/ CNTs复合材料比单独的CuCo-alloy/Al2O3具有更好的催化活性以及更高的C2+醇选择性. Sun等[103]成功地基于LDHs制备了高度分散的CuFe基催化剂.在对催化剂物化性能与催化性能的关系进行研究的发现, 产物醇/烃比与四配位的Cu离子含量呈现线性相关, 表明四配位的Cu离子很有可能是CO插入的主要活性位点.此外, Cu/Fe物质的量之为0.5的催化剂, 对高级醇表现出最优的催化性能, 其对乙醇的选择性为20.77%, 对C2+醇选择性高达48.06%.何静等[104]在球形γ-Al2O3表面原位合成了CoGaZnAl-LDH, 经共还原得到的负载型CoGa双金属催化剂可用于合成气转化为乙醇和高级醇, CO转化率在15 h达到43.5%, 醇的选择性为59%, 在醇类产品中, 乙醇和高级醇的比例高达92.8%;且在100 h的稳定性测试中, 负载型CoGa双金属催化剂CO转化率以及醇的选择性基本保持不变, 表现出出色的稳定性, 这主要归因于ZnAl- MMO对于CoGa颗粒的锚定作用.后续的研究表明, Ga原子的存在不仅可以分离Co位点, 而且可以向邻近的Co原子转移电子.其中, 被分割的Co位点起到CO非解离线性吸附作用, 促进CO插入以产生醇, 相邻富电子的Co位点起到CO解离吸附作用, 促进碳碳键偶合.此外, 当Co/Ga为0.6时, CO插入与碳碳键偶合动力学速率的匹配有助于产生更多的乙醇和高级醇. Tan等[19]采用不同方法制备了一系列的双金属CuCo催化剂, 发现通过LDHs前体法得到的双金属CuCo纳米颗粒直径约为6 nm, 尺寸明显小于通过浸渍法得到的双金属CuCo催化剂(20~30 nm).该催化剂在合成气转化中对总醇的选择性为44.0%, 并表现出高的C1~C3醇的选择性.
图 10

5. 总结与展望
LDHs作为一类独特的二维层状材料在催化领域展现出巨大的优势和广阔的应用前景, 引起了国内外研究者越来越多的关注.近年来, 基于LDHs自身的结构优势与结构拓扑转变效应, 可获得高分散、几何和电子结构可调的负载型单金属、双金属催化剂.利用LDHs材料, 可获得对特定目标反应具有高活性和良好选择性的负载型催化剂.此外, 利用LDHs的限域效应和载体的锚定作用, 使得LDHs基负载型催化剂具有优异的稳定性.本文综述了LDHs基负载型催化剂的制备以及其在电化学、氧化脱氢、选择性加氢以及合成气转化等领域应用, 并讨论了催化活性、选择性与催化剂结构的内在关系, 为高性能LDHs基负载型催化剂的设计和合成提供了一定的依据.
尽管调控LDHs基负载型催化剂的组成、形貌以及几何/电子结构可在许多重要的催化反应中获得较为理想的催化效果, 但仍然存在一些亟待解决的问题和挑战. (1)目前人们对LDHs拓扑转变过程的结构演化以及拓扑转变机理仍缺乏清晰的认识, 从而限制了LDHs基负载型催化剂的精确设计和调控. (2)由于多相催化的复杂性以及催化剂的活性位结构在实际反应中可能发生重构, 使得在反应条件下对于催化剂活性位结构的表征和确定变得十分困难. (3)针对同一类反应, 多种LDHs基负载型催化剂体系均具有高活性; 虽然研究者对于催化剂的结构以及反应机理有一定认识, 但是很难达成统一的理论和规律性认识, 严重制约了催化剂活性位结构的精确调控. (4)绝大多数LDHs基负载型催化剂尚处于实验室研究阶段, 距真正工业化应用还有很长一段距离, 基于LDHs催化材料的工业应用仍然是今后研究的重点和难点.
近年来, 材料表征技术的进步以及原位/操作条件下表征技术的发展, 使得对于催化材料活性位结构的鉴定以及反应机理的揭示成为可能.基于这些问题和挑战, 研究的重点可从以下几个方面着手. (1)对于LDHs拓扑转变机理以及结构演变过程中金属原子迁移规律的研究, 可采用原位时间分辨EXAFS和XPS等表征技术监测LDHs焙烧/还原过程中金属原子的配位环境、键长以及电子状态的变化, 有助于对LDHs拓扑转变的本质进行详细而深入的研究, 这将对LDHs基负载型催化剂的结构设计和可控制备具有理论指导意义. (2)通常, 本征活性的差异极大决定了催化剂的性能, 因此, 反应速率或TOF值与金属位、酸碱位或者界面位之间定量关系的建立对于活性位结构的确定尤为重要.为了获得活性位结构的直接证据, 必须采用原位表征技术来跟踪催化反应过程.原位时间分辨漫反射红外光谱和原位/操作条件XAFS等手段可获得实际反应条件下活性中心的结构演化以及电子态变化等信息. (3)采用不同大小和功能的探针可深入了解反应位点的空间以及电子特征, 多种原位/操作条件下原子尺度表征方法可观察反应条件下催化剂的活性位结构与反应物的作用机制.此外, DFT计算可作为研究反应物的吸附能和活化能、过渡态和反应中间体以及模拟整个反应过程的有力工具, 因此先进的原位表征手段与DFT计算的结合对于活性位结构以及反应机理的揭示具有重要意义. (4)为推进LDHs基负载型催化剂的工业化应用, 可将LDHs基负载型催化剂与工业上广泛使用的载体如分子筛或SiO2等结合构筑复合负载型催化剂.为克服上述挑战, 需要开展更多有实用意义和富有创新性的研究, 以促进LDHs基负载型催化材料在催化领域的应用.
-
-
[1]
White, R. J.; Rafael, L.; Budarin, V. L.; Clark, J. H.; Macquarrie, D. J. Chem. Soc. Rev. 2009, 38, 481. doi: 10.1039/B802654H
-
[2]
Wang, Q.; O'Hare, D. Chem. Rev. 2012, 112, 4124. doi: 10.1021/cr200434v
-
[3]
Yu, J.; Wang, Q.; O'Hare, D.; Sun, L. Chem. Soc. Rev. 2017, 46, 5950. doi: 10.1039/C7CS00318H
-
[4]
Xu, M.; Wei, M. Adv. Funct. Mater. 2018, 28, 1802943. doi: 10.1002/adfm.201802943
-
[5]
贾云生, 王火焰, 赵雪松, 刘晓伟, 王一柳, 范群龙, 周健民, 化学学报, 2015, 73, 1207. doi: 10.3866/PKU.WHXB201504142Jia, Y.; Wang, H.; Zhao, X.; Liu, X.; Wang, Y.; Fan, Q.; Zhou, J. Acta Chim. Sinica 2015, 73, 1207. doi: 10.3866/PKU.WHXB201504142
-
[6]
Fan, G.; Li, F.; Evans, D. G.; Duan, X. Chem. Soc. Rev. 2014, 43, 7040. doi: 10.1039/C4CS00160E
-
[7]
李甜甜, 赵继宽, 李尧, 全贞兰, 徐洁, 化学学报, 2017, 75, 485. http://sioc-journal.cn/Jwk_hxxb/CN/Y2017/V75/I5/485Li, T.; Zhao, J.; Li, Y.; Quan, Z.; Xu, J. Acta Chim. Sinica 2017, 75, 485. http://sioc-journal.cn/Jwk_hxxb/CN/Y2017/V75/I5/485
-
[8]
Meng, X.; Yang, Y.; Chen, L.; Xu, M.; Zhang, X.; Wei, M. ACS Catal. 2019, 9, 4226. doi: 10.1021/acscatal.9b00238
-
[9]
Gao, Z.; Liu, F. Q.; Wang, L.; Luo, F. Inorg. Chem. 2019, 58, 3247. doi: 10.1021/acs.inorgchem.8b03327
-
[10]
Xia, C.; Gao, R.; Li, K.; Yang, Y.; Lin, Y.; Yan, D. Chin. J. Chem. 2017, 35, 1701. doi: 10.1002/cjoc.201700136
-
[11]
陈海军, 黄舒怡, 张志宾, 刘云海, 王祥科, 化学学报, 2017, 75, 560. doi: 10.11862/CJIC.2017.075Chen, H.; Huang, S.; Zhang, Z.; Liu, Y.; Wang, X. Acta Chim. Sinica 2017, 75, 560. doi: 10.11862/CJIC.2017.075
-
[12]
王宁, 庞宏伟, 于淑君, 顾鹏程, 宋爽, 王宏青, 王祥科, 化学学报, 2019, 77, 143. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346814.shtmlWang, N.; Pang, H.; Yu, S.; Gu, P.; Song, S.; Wang, H.; Wang, X. Acta Chim. Sinica 2019, 77, 143. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346814.shtml
-
[13]
Bing, W.; Zheng, L.; He, S.; Rao, D.; Xu, M.; Zheng, L.; Wang, B.; Wang, Y.; Wei, M. ACS Catal. 2018, 8, 656. doi: 10.1021/acscatal.7b03022
-
[14]
Yang, Y.; Chen, L.; Chen, Y.; Liu, W.; Feng, H.; Wang, B.; Zhang, X.; Wei, M. Green. Chem. 2019, DOI: 10.1039/C9GC01119F.
-
[15]
Zhou, J.; Yang, Y.; Li, C.; Zhang, S.; Chen, Y.; Shi, S.; Wei, M. J. Mater. Chem. A 2016, 4, 12825. doi: 10.1039/C6TA04542A
-
[16]
Feng, J.; He, Y.; Liu, Y.; Du, Y.; Li, D. Chem. Soc. Rev. 2015, 44, 5291. doi: 10.1039/C5CS00268K
-
[17]
Yan, K.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catal. Sci. Technol. 2017, 7, 1622. doi: 10.1039/C7CY00274B
-
[18]
李小磊, 蒋平平, 卢云, 张伟杰, 董玉明, 化学学报, 2012, 70, 544. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract340751.shtmlLi, X.; Jiang, P.; Lu, Y.; Zhang, W.; Dong, Y. Acta Chim. Sinica 2012, 70, 544. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract340751.shtml
-
[19]
Sun, K.; Gao, X.; Bai, Y.; Tan, M.; Yang, G.; Tan, Y. Catal. Sci. Technol. 2018, 8, 3936. doi: 10.1039/C8CY01074A
-
[20]
王力耕, 余琴, 冯春, 张岩, 胡军, 有机化学, 2019, 39, 1787. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract347143.shtmlWang, L.; Yu, Q.; Feng, C.; Zhang, Y.; Hu, J. Chin. J. Org. Chem. 2019, 39, 1787. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract347143.shtml
-
[21]
Gao, X.; Zhou, Y.; Jing, F.; Luo, J.; Huang, Q.; Chu, W. Chin. J. Chem. 2017, 35, 1009. doi: 10.1002/cjoc.201600865
-
[22]
Li, X.; Zhang, Q.; Wang, H.; Li, Y. Chin. J. Chem. 2017, 35, 196. doi: 10.1002/cjoc.201600667
-
[23]
He, S.; Li, C.; Chen, H.; Su, D.; Zhang, B.; Cao, X.; Wang, B.; Wei, M.; Evans, D. G.; Duan, X. Chem. Mater. 2013, 25, 1040. doi: 10.1021/cm303517z
-
[24]
Chen, H.; He, S.; Cao, X.; Zhang, S.; Xu, M.; Pu, M.; Su, D.; Wei, M.; Evans, D. G.; Duan, X. Chem. Mater. 2016, 28, 4751. doi: 10.1021/acs.chemmater.6b01784
-
[25]
Gao, Z.; Liu, F.; Wang, L.; Luo, F. Appl. Surf. Sci. 2019, 480, 548. doi: 10.1016/j.apsusc.2019.02.219
-
[26]
Wang, Y.; Chao, X.; Zhang, Z.; Liu, D.; Ru, C.; Wang, S. Adv. Funct. Mater. 2018, 28, 1703363. doi: 10.1002/adfm.201703363
-
[27]
Zhao, Y.; Chen, G.; Bian, T.; Zhou, C.; Waterhouse, G. I.; Wu, L. Z.; Tung, C. H.; Smith, L. J.; O'Hare, D.; Zhang, T. Adv. Mater. 2016, 27, 7823.
-
[28]
Chen, Y.; Li, C.; Zhou, J.; Zhang, S.; Rao, D.; He, S.; Wei, M.; Evans, D. G.; Duan, X. ACS Catal. 2015, 5, 5756. doi: 10.1021/acscatal.5b01429
-
[29]
Li, C.; Dou, Y.; Liu, J.; Chen, Y.; He, S.; Wei, M.; Evans, D. G.; Duan, X. Chem. Commun. 2013, 49, 9992. doi: 10.1039/c3cc45697h
-
[30]
Zhang, S.; Fan, G.; Feng, L. Green Chem. 2013, 15, 2389. doi: 10.1039/c3gc40658j
-
[31]
Zhou, L.; Shao, M.; Zhang, C.; Zhao, J.; He, S.; Rao, D.; Wei, M.; Evans, D. G.; Duan, X. Adv. Mater. 2017, 29, 1604080. doi: 10.1002/adma.201604080
-
[32]
Zhang, F.; Zhao, X.; Feng, C.; Bo, L.; Tao, C.; Wei, L.; Lei, X.; Xu, S. ACS Catal. 2011, 1, 232. doi: 10.1021/cs100089v
-
[33]
Liu, Y.; He, Y.; Zhou, D.; Feng, J.; Li, D. Catal. Sci. Technol. 2016, 6, 3027. doi: 10.1039/C5CY01516B
-
[34]
Zhu, Y.; An, Z.; He, J. J. Catal. 2016, 341, 44. doi: 10.1016/j.jcat.2016.06.004
-
[35]
Wang, Z.; Xu, S. M.; Xu, Y.; Tan, L.; Wang, X.; Zhao, Y.; Duan, H.; Song, Y. F. Chem. Sci. 2019, 10, 378. doi: 10.1039/C8SC04480E
-
[36]
Li, C.; Wei, M.; Evans, D. G.; Duan, X. Small 2014, 10, 4469. doi: 10.1002/smll.201401464
-
[37]
Xu, M.; He, S.; Chen, H.; Cui, G.; Zheng, L.; Wang, B.; Wei, M. ACS Catal. 2017, 7, 7600. doi: 10.1021/acscatal.7b01951
-
[38]
Liu, N.; Xu, M.; Yang, Y.; Zhang, S.; Zhang, J.; Wang, W.; Zheng, L.; Hong, S.; Wei, M. ACS Catal. 2019, 9, 2707. doi: 10.1021/acscatal.8b04913
-
[39]
Clarke, J. B.; Hastie, J. W.; Kihlborg, L. H. E.; Metselaar, R.; Thackeray, M. M. Pure Appl. Chem. 1994, 66, 577. doi: 10.1351/pac199466030577
-
[40]
Valente, J. S.; Rodriguez-Gattorno, G.; Valle-Orta, M.; Torres-Garcia, E. Mater. Chem. Phys. 2012, 133, 621. doi: 10.1016/j.matchemphys.2012.01.026
-
[41]
Ferreira, O. P.; Alves, O. L.; Gouveia, D. X.; Souza Filho, A. G.; de Paiva, J. A. C.; Filho, J. M. J. Solid. State. Chem. 2004, 177, 3058. doi: 10.1016/j.jssc.2004.04.030
-
[42]
Zhao, X.; Zhang, F.; Xu, S.; Evans, D. G.; Duan, X. Chem. Mater. 2010, 22, 3933. doi: 10.1021/cm100383d
-
[43]
He, S.; Zhang, S.; Lu, J.; Zhao, Y.; Ma, J.; Wei, M.; Evans, D. G.; Duan, X. Chem. Commun. 2011, 47, 10797. doi: 10.1039/c1cc14360c
-
[44]
Meng, Q.; Yan, H. Mol. Simul. 2017, 43, 1338. doi: 10.1080/08927022.2017.1362107
-
[45]
Costa, D. G.; Rocha, A. B.; Souza, W. F.; Chiaro, S. S. X.; Leit o, A. A. J. Phys. Chem. C 2012, 116, 13679. doi: 10.1021/jp303529y
-
[46]
Zhang, S. T.; Dou, Y.; Zhou, J.; Pu, M.; Yan, H.; Wei, M.; Evans, D. G.; Duan, X. ChemPhysChem 2016, 17, 2754. doi: 10.1002/cphc.201600354
-
[47]
He, S.; An, Z.; Wei, M.; Evans, D. G.; Duan, X. Chem. Commun. 2013, 49, 5912. doi: 10.1039/c3cc42137f
-
[48]
Yan, H.; Lu, J.; Wei, M.; Ma, J.; Li, H.; He, J.; Evans, D. G.; Duan, X. J. Mol. Struct.:Theochem. 2008, 866, 34. doi: 10.1016/j.theochem.2008.06.031
-
[49]
He, Y.; Fan, J.; Feng, J.; Luo, C.; Yang, P.; Li, D. J. Catal. 2015, 331, 118. doi: 10.1016/j.jcat.2015.08.012
-
[50]
Tongsakul, D.; Nishimura, S.; Ebitani, K. ACS Catal. 2013, 3, 2199. doi: 10.1021/cs400458k
-
[51]
Francová, D.; Tanchoux, N.; Gérardin, C.; Trens, P.; Prinetto, F.; Ghiotti, G.; Tichit, D.; Coq, B. Microporous Mesoporous Mater. 2007, 99, 118. doi: 10.1016/j.micromeso.2006.07.038
-
[52]
Wang, L.; Zhang, J.; Zhu, Y.; Xu, S.; Wang, C.; Bian, C.; Meng, X.; Xiao, F.-S. ACS Catal. 2017, 7, 7461. doi: 10.1021/acscatal.7b01947
-
[53]
Sun, T.; Fan, G.; Li, F. Ind. Eng. Chem. Res. 2013, 52, 5538. doi: 10.1021/ie3032795
-
[54]
Zhao, M. Q.; Zhang, Q.; Zhang, W.; Huang, J. Q.; Zhang, Y.; Su, D. S.; Wei, F. J. Am. Chem. Soc. 2010, 132, 14739. doi: 10.1021/ja106421g
-
[55]
Gao, W.; Zhao, Y.; Chen, H.; Chen, H.; Li, Y.; He, S.; Zhang, Y.; Wei, M.; Evans, D. G.; Duan, X. Green Chem. 2015, 17, 1525. doi: 10.1039/C4GC01633E
-
[56]
Wu, J.; Gao, G.; Li, J.; Sun, P.; Long, X.; Li, F. Appl. Catal., B 2017, 203, 227. doi: 10.1016/j.apcatb.2016.10.038
-
[57]
Dung, N. T.; Tichit, D.; Chiche, B. H.; Coq, B. Appl. Catal., A 1998, 169, 179. doi: 10.1016/S0926-860X(98)00006-4
-
[58]
Koike, M.; Li, D.; Nakagawa, Y.; Tomishige, K. ChemSusChem 2012, 5, 2312. doi: 10.1002/cssc.201200507
-
[59]
Li, C.; Chen, Y.; Zhang, S.; Zhou, J.; Wang, F.; He, S.; Wei, M.; Evans, D. G.; Duan, X. ChemCatChem 2014, 6, 824. doi: 10.1002/cctc.201300813
-
[60]
Dresselhaus, M. S.; Thomas, I. L. Nature 2001, 414, 332. doi: 10.1038/35104599
-
[61]
Jingshan, L.; Jeong-Hyeok, I.; Mayer, M. T.; Marcel, S.; Mohammad Khaja, N.; Nam-Gyu, P.; S David, T.; Jin, F. H.; Michael, G. T. Science 2014, 345, 1593. doi: 10.1126/science.1258307
-
[62]
Wang, Q.; Shang, L.; Shi, R.; Zhang, X.; Waterhouse, G. I. N.; Wu, L.-Z.; Tung, C.-H.; Zhang, T. Nano Energy 2017, 40, 382. doi: 10.1016/j.nanoen.2017.08.040
-
[63]
Wang, Q.; Shang, L.; Shi, R.; Zhang, X.; Zhao, Y.; Waterhouse, G. I. N.; Wu, L.-Z.; Tung, C.-H.; Zhang, T. Adv. Energy Mater. 2017, 7, 1700467. doi: 10.1002/aenm.201700467
-
[64]
Qiao, B.; Wang, A.; Yang, X.; Allard, L. F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Nat. Chem. 2011, 3, 634. doi: 10.1038/nchem.1095
-
[65]
Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Angew. Chem., Int. Ed. 2017, 56, 13944. doi: 10.1002/anie.201703864
-
[66]
Li, P.; Wang, M.; Duan, X.; Zheng, L.; Cheng, X.; Zhang, Y.; Kuang, Y.; Li, Y.; Ma, Q.; Feng, Z.; Liu, W.; Sun, X. Nat. Commun. 2019, 10, 1711. doi: 10.1038/s41467-019-09666-0
-
[67]
Zhang, J.; Liu, J.; Xi, L.; Yu, Y.; Chen, N.; Sun, S.; Wang, W.; Lange, K. M.; Zhang, B. J. Am. Chem. Soc. 2018, 140, 3876. doi: 10.1021/jacs.8b00752
-
[68]
Zhao, Y.; Zhang, X.; Jia, X.; Waterhouse, G. I. N.; Shi, R.; Zhang, X.; Zhan, F.; Tao, Y.; Wu, L.-Z.; Tung, C.-H.; O'Hare, D.; Zhang, T. Adv. Energy Mater. 2018, 8, 1703585. doi: 10.1002/aenm.201703585
-
[69]
Jia, X.; Zhang, X.; Zhao, J.; Zhao, Y.; Zhao, Y.; Waterhouse, G. I. N.; Shi, R.; Wu, L.-Z.; Tung, C.-H.; Zhang, T. J. Energy Chem. 2019, 34, 57. doi: 10.1016/j.jechem.2018.09.011
-
[70]
Zhao, Y.; Jia, X.; Chen, G.; Shang, L.; Waterhouse, G. I.; Wu, L. Z.; Tung, C. H.; O'Hare, D.; Zhang, T. J. Am. Chem. Soc. 2016, 138, 6517. doi: 10.1021/jacs.6b01606
-
[71]
He, L.; Huang, Y.; Wang, A.; Wang, X.; Chen, X.; Delgado, J. J.; Zhang, T. Angew. Chem., Int. Ed. 2012, 51, 6191. doi: 10.1002/anie.201201737
-
[72]
Gao, W.; Li, C.; Chen, H.; Wu, M.; He, S.; Wei, M.; Evans, D. G.; Duan, X. Green Chem. 2014, 16, 1560. doi: 10.1039/c3gc41939h
-
[73]
Zhao, J.; Shao, M.; Yan, D.; Zhang, S.; Lu, Z.; Li, Z.; Cao, X.; Wang, B.; Wei, M.; Evans, D. G.; Duan, X. J. Mater. Chem. A 2013, 1, 5840. doi: 10.1039/c3ta10588a
-
[74]
Takato, M.; Yusuke, M.; Hisashi, F.; Tomoo, M.; Koichiro, J.; Kiyotomi, K. Angew. Chem., Int. Ed. 2008, 47, 138. doi: 10.1002/anie.200703161
-
[75]
Mitran, G.; Cacciaguerra, T.; Loridant, S.; Tichit, D.; Marcu, I.-C. Appl. Catal., A 2012, 417~418, 153.
-
[76]
Wang, L.; Zhang, J.; Meng, X.; Zheng, D.; Xiao, F.-S. Catal. Today 2011, 175, 404. doi: 10.1016/j.cattod.2011.03.040
-
[77]
He, Y.; Feng, J.; Brett, G. L.; Liu, Y.; Miedziak, P. J.; Edwards, J. K.; Knight, D. W.; Li, D.; Hutchings, G. J. ChemSusChem 2015, 8, 3314. doi: 10.1002/cssc.201500503
-
[78]
Blanco, S.; Carrazán, S. R. G.; Rives, V. Appl. Catal., A 2008, 342, 93. doi: 10.1016/j.apcata.2008.03.002
-
[79]
Pakhomov, N. A. Kinet. Catal. 2001, 42, 334. doi: 10.1023/A:1010409230898
-
[80]
Sun, P.; Siddiqi, G.; Chi, M.; Bell, A. T. J. Catal. 2010, 274, 192. doi: 10.1016/j.jcat.2010.06.017
-
[81]
Siddiqi, G.; Sun, P.; Galvita, V.; Bell, A. T. J. Catal. 2010, 274, 200. doi: 10.1016/j.jcat.2010.06.016
-
[82]
Sun, P.; Siddiqi, G.; Vining, W. C.; Chi, M.; Bell, A. T. J. Catal. 2011, 282, 165. doi: 10.1016/j.jcat.2011.06.008
-
[83]
Belskaya, O. B.; Stepanova, L. N.; Nizovskii, A. I.; Kalinkin, A. V.; Erenburg, S. B.; Trubina, S. V.; Kvashnina, K. O.; Leont'eva, N. N.; Gulyaeva, T. I.; Trenikhin, M. V.; Bukhtiyarov, V. I.; Likholobov, V. A. Catal. Today 2019, 329, 187. doi: 10.1016/j.cattod.2018.11.081
-
[84]
Shimizu, K. I.; Kon, K.; Shimura, K.; Hakim, S. S. M. A. J. Catal. 2013, 300, 242. doi: 10.1016/j.jcat.2013.01.005
-
[85]
Chen, H.; He, S.; Xu, M.; Wei, M.; Evans, D. G.; Duan, X. ACS Catal. 2017, 7, 2735. doi: 10.1021/acscatal.6b03494
-
[86]
Mckenna, F. M.; Mantarosie, L.; Wells, R. P. K.; Hardacre, C.; Anderson, J. A. Catal. Sci. Technol. 2012, 2, 632. doi: 10.1039/c2cy00479h
-
[87]
Kahsar, K. R.; Schwartz, D. K.; Will, J. M. J. Am. Chem. Soc. 2014, 136, 520. doi: 10.1021/ja411973p
-
[88]
He, Y.; Liang, L.; Liu, Y.; Feng, J.; Ma, C.; Li, D. J. Catal. 2014, 309, 166. doi: 10.1016/j.jcat.2013.09.017
-
[89]
Liu, Y. N.; Feng, J. T.; He, Y. F.; Sun, J. H.; Li, D. Q. Catal. Sci. Technol. 2015, 5, 1231. doi: 10.1039/C4CY01160K
-
[90]
Liu, Y.; Zhao, J.; He, Y.; Feng, J.; Wu, T.; Li, D. J. Catal. 2017, 348, 135. doi: 10.1016/j.jcat.2017.02.020
-
[91]
Stassi, J. P.; Zgolicz, P. D.; Miguel, S. R. D.; Scelza, O. A. J. Catal. 2013, 306, 11. doi: 10.1016/j.jcat.2013.05.029
-
[92]
Ide, M. S.; Bing, H.; Neurock, M.; Davis, R. J. ACS Catal. 2012, 2, 671. doi: 10.1021/cs200567z
-
[93]
Li, C.; Chen, Y.; Zhang, S.; Xu, S.; Zhou, J.; Wang, F.; Wei, M.; Evans, D. G.; Duan, X. Chem. Mater. 2013, 25, 3888. doi: 10.1021/cm4021832
-
[94]
Yang, Y.; Rao, D.; Chen, Y.; Dong, S.; Wang, B.; Zhang, X.; Wei, M. ACS Catal. 2018, 8, 11749. doi: 10.1021/acscatal.8b02755
-
[95]
Kong, X.; Zheng, R.; Zhu, Y.; Ding, G.; Zhu, Y.; Li, Y.-W. Green Chem. 2015, 17, 2504. doi: 10.1039/C5GC00062A
-
[96]
Yan, K.; Chen, A. Energy 2013, 58, 357. doi: 10.1016/j.energy.2013.05.035
-
[97]
Gupta, M.; Smith, M. L.; Spivey, J. J. ACS Catal. 2011, 1, 641. doi: 10.1021/cs2001048
-
[98]
Spivey, J. J.; Egbebi, A. Chem. Soc. Rev. 2007, 36, 1514. doi: 10.1039/b414039g
-
[99]
Gao, W.; Zhao, Y.; Liu, J.; Huang, Q.; He, S.; Li, C.; Zhao, J.; Wei, M. Catal. Sci. Technol. 2013, 3, 1324. doi: 10.1039/c3cy00025g
-
[100]
Cao, A.; Liu, G.; Yue, Y.; Zhang, L.; Liu, Y. RSC Adv. 2015, 5, 58804. doi: 10.1039/C5RA05190H
-
[101]
Wang, L.; Cao, A.; Liu, G.; Zhang, L.; Liu, Y. Appl. Surf. Sci. 2016, 360, 77. doi: 10.1016/j.apsusc.2015.10.234
-
[102]
Cao, A.; Liu, G.; Wang, L.; Liu, J.; Yue, Y.; Zhang, L.; Liu, Y. J. Mater. Sci. 2016, 51, 5216. doi: 10.1007/s10853-016-9823-9
-
[103]
Han, X.; Fang, K.; Zhou, J.; Zhao, L.; Sun, Y. J. Colloid. Interface Sci. 2016, 470, 162. doi: 10.1016/j.jcis.2015.09.062
-
[104]
Ning, X.; An, Z.; He, J. J. Catal. 2016, 340, 236. doi: 10.1016/j.jcat.2016.05.014
-
[1]
-

图 4 (a) 水解沉积制备Ru/CoFe-LDHs的示意图、(b) Ru/CoFe-LDHs纳米片的透射电镜(TEM)图像、(c) Cs校正的Ru/CoFe-LDHs纳米片的STEM图像和(d) Ru/CoFe-LDHs的高角环形暗场扫描透射电镜(HAADF-STEM)图像及相应的Ni, Fe和Ru元素分布图[66]
Figure 4 (a) Schematic illustration for the hydrolysis-deposition to form Ru/CoFe-LDHs, (b) transmission electron microscopy (TEM) images of as-prepared Ru/CoFe-LDHs nanosheets (inset shows the corresponding SAED pattern of Ru/CoFe-LDHs nanosheets marked in orange circle), (c) Cs-corrected STEM image of Ru/CoFe-LDHs nanosheets shows the monoatomic ruthenium dispersed on the surface of LDHs (some of the isolated Ru atoms are marked with red circles) and (d) HAADF-STEM images of the Ru/CoFe-LDHs and corresponding elemental distribution maps of Ni, Fe, and Ru in the Ru/CoFe-LDHs[66]
-

 下载:
下载:



 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 110
- 文章访问数: 5343
- HTML全文浏览量: 1434
 下载:
下载:
