引用本文:
梁珊, 宗敏华, 娄文勇. 酶法催化二氧化碳制备高附加值化学品研究进展[J]. 化学学报,
2019, 77(11): 1099-1114.
doi:
10.6023/A19060240

Citation: Liang Shan, Zong Minhua, Lou Wenyong. Recent Advances in Enzymatic Catalysis for Preparation of High Value-Added Chemicals from Carbon Dioxide[J]. Acta Chimica Sinica, 2019, 77(11): 1099-1114. doi: 10.6023/A19060240
Citation: Liang Shan, Zong Minhua, Lou Wenyong. Recent Advances in Enzymatic Catalysis for Preparation of High Value-Added Chemicals from Carbon Dioxide[J]. Acta Chimica Sinica, 2019, 77(11): 1099-1114. doi: 10.6023/A19060240
酶法催化二氧化碳制备高附加值化学品研究进展
摘要:
现代工业的发展不断消耗煤、石油、天然气等碳化石燃料,并产生了大量的温室气体CO2,使人类面临能源和环境的双重挑战,开发绿色能源、控制CO2对环境的影响迫在眉睫.CO2是一种廉价的碳源,可通过化学法、光化学法、电化学法或酶法等转化为高附加值含碳化学品,实现CO2的资源化循环利用,是解决全球碳排放所带来的能源和环境危机的双赢策略.受CO2胞内天然生物转化的启发,酶法为CO2的循环利用带来了新的机遇,相比于传统的化学及光、电化学法,可表现出绿色、高效、温和、高选择性等优点,有望为CO2高值化利用带来新的契机.有鉴于此,本文从胞内CO2生物转化机理出发,综述了国内外近年来多种单酶及多酶级联催化CO2高值化利用的最新研究进展,并交叉论述了固定化酶催化体系的构建、酶定向进化和改造、酶催化过程调控等内容,总结了酶法转化目前存在的缺陷和不足,并提出了展望,以期为酶法催化CO2高值化利用提供一定的参考和借鉴.
English
Recent Advances in Enzymatic Catalysis for Preparation of High Value-Added Chemicals from Carbon Dioxide
Abstract:
With the rapid development of modern industry, coal, petroleum, natural gas and other fossil fuels have been excessively consumed, along with an increasing large quantities of greenhouse gases (e.g. carbon dioxide, CO2) are produced. It is urgent to develop sustainable green energy and abate the detriment of carbon dioxide on global environment. CO2 is a cheap carbon source that can be converted into high value-added chemicals by chemical, photochemical, electrochemical or enzymatic methods to realize the recycling of CO2. It is a win-win strategy to solve the energy and environmental crisis caused by global carbon emissions. Inspired by natural CO2 metabolic process, enzymatic transformation provides an alternative strategy for efficient recycling of CO2. Compared with traditional chemical, photochemical or electrochemical methods, the enzymatic route holds advantages of green, high efficiency, mild and excellent selectivity, which is expected to bring new revolutionary opportunities for efficient utilization of CO2. Thus, in this present review, we firstly introduce the brief background about enzymatic conversion for CO2 capture, sequestration and utilization. Next, we depict six major routes of the CO2 metabolic process in cells, which are taken as the inspiration source for the construction of enzymatic systems in vitro. Subsequently, recent advances in enzymatic conversion of CO2 that catalyzed by various single enzymes and multi-enzyme cascade systems are systematically reviewed. Some emerging approaches for construction of immobilized single-or multi-enzyme systems, directed evolution and artificial modification of enzymes, and cofactor regulation during the enzymatic processes are also discussed. Finally, the defects and shortcomings of enzymatic approaches are summarized, and the future perspectives are finally put forward. Based on this present review, we aim to provide theoretical reference and practical basis for more efficient enzymatic utilization of CO2 to produce high value-added chemicals.
-
Key words:
- CO2
- / enzymatic conversion
- / high value conversion
- / reduction
- / cascade catalysis
- / energy
- / environment
-
1. 引言
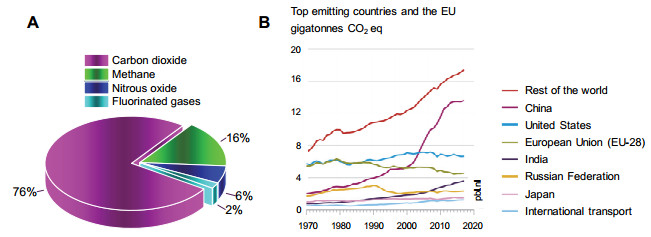
二氧化碳(Carbon Dioxide, CO2)是空气中主要成分之一, 也是一种常见的温室气体, 常温常压下是一种无色无味、不可燃的气态化合物[1].近200年来, 随着现代工业的发展, 煤、石油、天然气等碳化石燃料被不断消耗, 产生了大量的CO2、甲烷、氮氧化物等温室气体, 其中CO2约占76%(图 1A), 能源消耗的同时还带来了全球变暖、气候变化、冰川消融、海洋酸化、环境污染等一系列问题[2~5].据统计, 2018年全球CO2排放量达到509亿吨, 且持续增长, 我国排放量超过120亿吨(图 1B)[6], 由此造成的能源和环境问题将严重影响人类的生存和发展.近年来, 人类在CO2减排方面做了大量的努力, 但受到人口增长、经济发展、社会进步等因素的影响, 人类不得不消耗大量能源, CO2排放量仍在持续增长[4], 由CO2带来的全球性问题是人类必须面对的巨大危机和挑战.
图 1
随着科学技术的发展, 人类对CO2问题有了更多的思考. CO2含有丰富的碳元素, 可进行生物矿化或转化为能源、燃料、大分子聚合物等高附加值含碳化学品[7].近年来, CO2捕获、封存、还原和转化技术不断发展, 特别是利用CO2生产CO、甲烷、甲醇、甲醛和甲酸等能源和燃料类化合物, 既能满足日益增长的化工和能源需求, 又可实现CO2的有效减排, 是应对能源和环境问题的一种双赢策略, 具有极高的科研和应用价值, 已成为全球科学界持续性研究热点之一[4, 7, 8].目前, CO2固定、还原和转化技术主要包括化学催化、光化学催化、电化学催化、光电协同催化、酶法催化等[9~12].然而, 前四种催化方法存在很大的局限性:一方面, 化学及光、电化学法普遍需要较高的工作温度和压力, 或额外的电能或光能, 操作困难, 成本高昂[13]; 另一方面, 由于CO2化学性质稳定、激活困难、产物复杂等因素, 高性能催化剂的选择和设计仍然存在较大的技术瓶颈, 造成反应效率、产物的选择性及产率普遍较低[7].
受卡尔文循环、三羧酸循环等胞内CO2天然生物转化的启发[14], 酶法为CO2的还原和转化带来了新的思路, 酶法胞外转化CO2引起了人们的广泛关注.相比于传统光化学及电化学催化等方法, 酶法具有优越的选择性(立体特异性、区域和化学选择性)、高反应效率、条件温和、环境友好性等优点[7, 13, 15, 16], 有望为CO2高值化利用带来革命性契机.近年来, 酶法捕获及催化转化CO2方面的研究大量涌现, 但酶法也存在易失活、难回收、高成本等问题, 因此, 与CO2生物转化相关的酶的设计和改造技术也随之发展, 为解决酶法转化CO2相关技术瓶颈提供了新的策略[17].针对以上所述关键问题, 本文从胞内CO2生物转化机理出发, 对体(胞)外单酶及多酶级联催化CO2高值化利用近年来国内外最新研究进展进行了综述, 交叉论述了固定化酶催化体系的构建、酶定向进化和改造、酶催化过程调控等内容, 并提出了总结和展望, 以期为CO2高值化利用提供一定的参考.
2. CO2胞内酶法转化机理
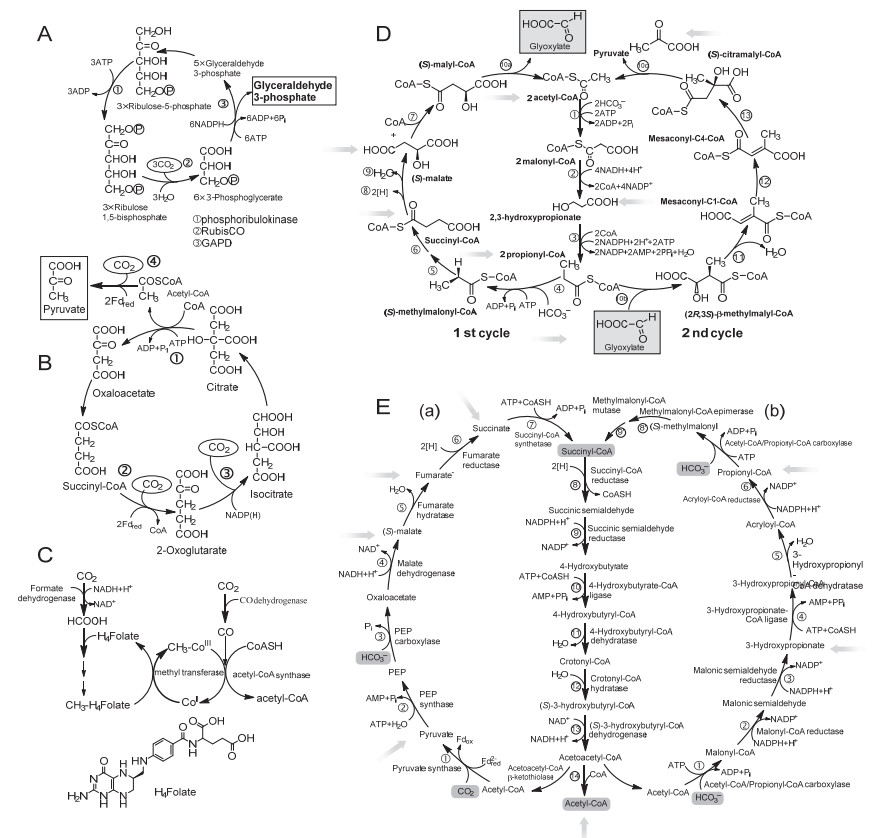
在自然界中, 固定或转化CO2产生有机物是生命产生的先决条件, 代表了自然界生物进化的开端[18].作为控制大气中CO2浓度的重要方式之一, CO2胞内代谢和转化主要有六种途径[19]:卡尔文循环、还原性柠檬酸循环、还原性乙酰辅酶A途径、3-羟基丙酸循环、3-羟基丙酸/4-羟基丁酸循环和二羧酸/4-羟基丁酸循环, 这些途径对全球碳循环有重大影响, 其中酶在催化CO2固定和转化过程中起着决定性作用, 这些代谢途径为CO2体外酶法转化及反应体系的构建提供了重要参考[20, 21].
卡尔文循环(图 2A)是地球上最重要的生物合成途径之一, 大多数光合生物(如植物、藻类、蓝细菌和大多数需氧或兼性需氧菌)通过该循环途径将CO2在胞内进行固定和转化, 是自然界中CO2转化的最主要方法[22].卡尔文循环是光合作用中碳反应的一部分, 可分为三个阶段并涉及到三种酶, 三个阶段包括CO2固定、还原和受体(二磷酸核酮糖)的再生, 对应的三种的酶分别为核酮糖-1, 5-二磷酸羧化酶/加氧酶(固定CO2形成3C化合物)、磷酸甘油醛脱氢酶(催化产生3-磷酸甘油醛)以及磷酸核酮糖激酶(催化二磷酸核酮糖再生), 循环过程需消耗三磷酸腺苷(ATP)及还原型辅酶Ⅱ(NADPH), CO2以3-磷酸甘油醛的形式离开卡尔文循环, 最终实现CO2的固定和转化[22].
图 2
还原性柠檬酸循环也称逆向三羧酸循环(图 2B), 最初是在厌氧泥生绿菌(Chlorobium limicola, 一种光养绿色硫细菌)中发现的, 也存在于一些以氢或硫酸盐为电子供体的细菌中, 在微需氧菌甚至严格厌氧菌中也发现了这种循环[7, 18].该循环途径是柠檬酸循环的逆反应, 包括四个羧化反应步骤:首先, 酮戊二酸合成酶催化琥珀酰辅酶A与CO2还原羧化产生酮戊二酸; 然后, 异柠檬酸脱氢酶催化酮戊二酸产生异柠檬酸, 异柠檬酸随后发生异构化并由ATP柠檬酸裂解酶催化产生草酰乙酸酯和乙酰辅酶A; 接着, 乙酰辅酶A与CO2在丙酮酸合成酶作用下生成丙酮酸; 最后, 丙酮酸激酶激化丙酮酸产生磷酸烯醇式丙酮酸. CO2经还原性柠檬酸循环产生了草酰乙酸, 并通过一系列酶促反应最终转化成琥珀酰辅酶A[7, 18, 20], CO2和水经此循环路径转化成含碳有机化合物.
还原性乙酰辅酶A途径由Wood和Ljungdahl等阐明(图 2C), 因此又称Wood-Ljungdahl途径, 是产乙酸细菌利用甲酸脱氢酶(FDH)还原CO2产甲酸或利用一氧化碳脱氢酶(CODH)将CO2还原为CO的物质和能量代谢过程[23~25].与卡尔文循环和还原性柠檬酸循环不同, 还原性乙酰辅酶A途径是一条非循环路径: CO2最初通过NADH依赖型甲酸脱氢酶转化为甲酸, 随后被四氢叶酸捕获、还原生成甲基四氢叶酸, 甲基化四氢叶酸在钴铁硫蛋白甲基转移酶的作用下形成甲基化钴蛋白(CH3—CoⅢ); 一氧化碳脱氢酶催化CO2还原为CO, 乙酰辅酶A合成酶捕获CH3—CoⅢ甲基, 并催化CO、CoASH和甲基产生乙酰辅酶A[7, 25, 26].
3-羟基丙酸循环(图 2D)是一种存在于嗜热光合绿丝菌属中的新型CO2固定途径.此循环的特征代谢中间物为3-羟基丙酸, 该化合物可用于合成许多重要的化工产品, 如丙烯酸、丙二酸等, 具有很高的工业价值[27].在此循环过程中CO2被乙酰辅酶A和丙酰辅酶A羧化固定, 最终形成β-甲基苹果酰辅酶A.作为一种新型自养固碳途径, 3-羟基丙酸循环具有与其他固碳途径不同的鲜明特点: (1)它是一个双循环偶联的代谢过程, 在第一个循环中, 两分子以碳酸氢根形式存在的CO2被固定, 形成初级产物乙醛酸; 在第二个循环中, 乙醛酸和丙酰辅酶A缩合形成β-甲基苹果酰辅酶A, 最后形成丙酮酸, 并且再生出乙酰辅酶A. (2)循环途径中涉及19步反应, 但只需13种酶, 其中包括几个多功能酶, 这在其他的自养途径中并不常见. (3)该途径中的关键中间产物3-羟基丙酸在生物代谢过程中的功能尚待阐明.
3-羟基丙酸/4-羟基丁酸循环是发现于金属球菌属中的一种新的CO2循环途径(图 2E, b), 这种循环也存在于微需氧菌或严格厌氧菌和古生菌中.这种循环途径涉及到的酶属于氧耐受型, 其中4-羟基丁酰辅酶A脱水酶是其中一种重要的酶, 它参与梭菌属中γ-氨基丁酸的发酵过程[28], 另外还涉及到乙酰辅酶A羧化酶、丙酰辅酶A羧化酶和丙酮酸合成酶三种重要的酶[7].该循环可被分为两个部分:第一部分通过3-羟基丙酸将乙酰辅酶A和两个碳酸氢根离子转化成琥珀酰辅酶A[29, 30]; 第二部分通过4-羟基丁酸酯将琥珀酰辅酶A转化为两分子乙酰辅酶A, 这是自养型泉古生菌属的共同之处[29, 31].该循环中琥珀酰辅酶A由乙酸酯和两分子碳酸氢盐形成, 这一点与3-羟基丙酸循环类似.
二羧酸/4-羟基丁酸循环(图 2E, a)是在低氧压力下进行兼性有氧能量代谢的循环过程, 主要在自养厌氧型热变形菌属和除硫菌属中发挥作用[18], 该循环与3-羟基丙酸/4-羟基丁酸循环类似, 不同的是羧化过程涉及到铁氧化还原酶(丙酮酸合成酶)和磷酸烯醇丙酮酸盐(PEP)羧化酶[19].二羧酸/4-羟基丁酸循环也可分为两部分[18]:第一部分, 一分子CO2和一分子碳酸氢盐通过C4-二羧酸转化成琥珀酰辅酶A; 第二部分, 琥珀酰辅酶A经4-羟基丁酸酯转化为两个乙酰辅酶A, 一个可用于生物合成, 另外一个用作下一轮循环的CO2受体.该循环开始于丙酮酸合成酶催化乙酰辅酶A还原羧化为丙酮酸, 随后将丙酮酸转化为磷酸烯醇丙酮酸(PEP), PEP羧化为草酰乙酸, 最终转化为琥珀酰辅酶A[32].
3. 单酶催化转化CO2研究进展
3.1 固氮酶还原CO2
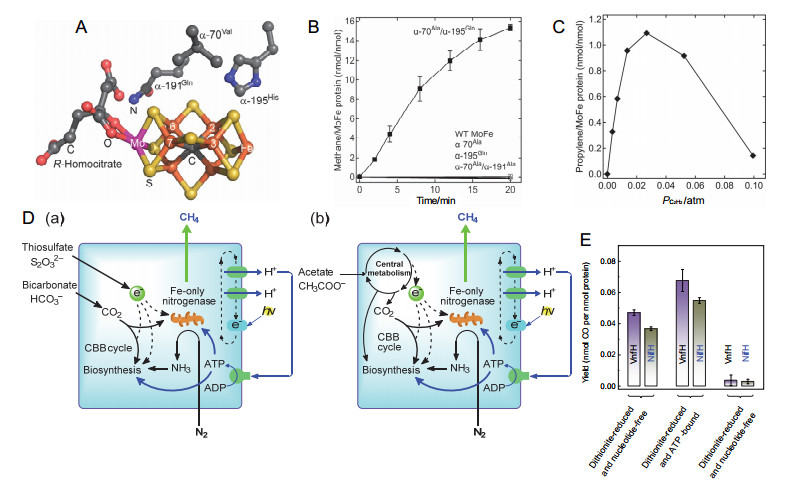
固氮酶(Nitrogenase)对全球氮循环极为重要, 在ATP的参与下可催化氮气(N2)发生多电子/质子转移(还原), 生成两个氨(NH3)分子(式1), 催化中心发生在固氮酶钼铁辅因子(7Fe-9S-1Mo1C-R-homocitrate, 图 3A)上[33~35], 一些固氮酶还具有还原乙炔等惰性小分子化合物双键或三键的能力[33, 36].通常情况下N2的多电子转移反应是较难发生的, 但一些固氮酶活性中心钼原子被钒取代后表现出了优异的CO还原能力, 产生了短链烯烃(例如乙烯、丙烯)或烷烃(例如乙烷)等物质.受此启发, 自20世纪80年代以来, 研究者对该酶是否也可催化CO2发生多电子转移还原为CO或CH4产生了浓厚的兴趣[37~39].近年来的研究结果表明, 固氮酶钼铁蛋白辅因子附近氨基酸被部分取代后, 可催化CO2发生双电子或八电子转移, 以低速率催化还原为CO或CH4[39, 40](式2, 3), 为人工转化CO2提供了新的途径.
图 3
 图 3. (A) 固氮酶钼铁蛋白辅因子及其关键氨基酸残基; (B)天然和重组钼铁蛋白催化CH4形成与时间的关系; (C) α-70Ala/α-195Gln钼铁蛋白催化CO2和C2H2产丙烯与C2H2分压的关系[39]; (D) R. palustris表达的天然唯铁固氮酶催化CO2产CH4代谢途径: (a)硫代硫酸盐(S2O32–)氧化供电子, CO2来源于重碳酸盐; (b) CH3COO–氧化供电子, CO2来源于乙酸[41]; (E)几种状态下的铁蛋白固氮酶VnfH和NifH体外还原CO2生成CO[42]Figure 3. Nitrogenase MoFe cofactor with some key amino acid residues; (B) CH4 formation as a function of time with natural or remodeled MoFe proteins; (C) The relationship between propylene formation and C2H2 partial pressure that catalyzed by α-70Ala/α-195Gln MoFe protein[39]; (D) Metabolic routes of CH4 production by R. palustris expressing wild-type Fe-only nitrogenase: electrons are originated from oxidation of thiosulfate (a) or acetate (b), and CO2 are generated from bicarbonate (a) or the oxidation of acetate (b)[41]; (E) In vitro reduction of CO2 to CO by vnfH or NifH in different forms[42]
图 3. (A) 固氮酶钼铁蛋白辅因子及其关键氨基酸残基; (B)天然和重组钼铁蛋白催化CH4形成与时间的关系; (C) α-70Ala/α-195Gln钼铁蛋白催化CO2和C2H2产丙烯与C2H2分压的关系[39]; (D) R. palustris表达的天然唯铁固氮酶催化CO2产CH4代谢途径: (a)硫代硫酸盐(S2O32–)氧化供电子, CO2来源于重碳酸盐; (b) CH3COO–氧化供电子, CO2来源于乙酸[41]; (E)几种状态下的铁蛋白固氮酶VnfH和NifH体外还原CO2生成CO[42]Figure 3. Nitrogenase MoFe cofactor with some key amino acid residues; (B) CH4 formation as a function of time with natural or remodeled MoFe proteins; (C) The relationship between propylene formation and C2H2 partial pressure that catalyzed by α-70Ala/α-195Gln MoFe protein[39]; (D) Metabolic routes of CH4 production by R. palustris expressing wild-type Fe-only nitrogenase: electrons are originated from oxidation of thiosulfate (a) or acetate (b), and CO2 are generated from bicarbonate (a) or the oxidation of acetate (b)[41]; (E) In vitro reduction of CO2 to CO by vnfH or NifH in different forms[42]$ {{\rm{N}}_2} + 8{{\rm{H}}^ + } + 16{\rm{ATP}} + 8{{\rm{e}}^ - } \to 2{\rm{N}}{{\rm{H}}_3} + {{\rm{H}}_2} + 16{\rm{ADP}} + 16{{\rm{P}}_{\rm{i}}}{\rm{}} $
(1) $ {\rm{C}}{{\rm{O}}_{\rm{2}}} + {\rm{8}}{{\rm{H}}^ + } + {\rm{8}}{{\rm{e}}^{\rm{ - }}} \to {\rm{C}}{{\rm{H}}_{\rm{4}}} + {\rm{2}}{{\rm{H}}_{\rm{2}}}{\rm{O}} $
(2) $ {\rm{C}}{{\rm{O}}_{\rm{2}}} + {\rm{2}}{{\rm{H}}^ + }{\rm{2}}{{\rm{e}}^{\rm{ - }}} \to {\rm{CO}} + {{\rm{H}}_{\rm{2}}}{\rm{O}} $
(3) 重组固氮酶催化CO2甲烷化的研究较多. 2012年, 犹他州立大学Seefeldt等[39]研究发现固氮酶钼铁蛋白辅因子附近的氨基酸序列(α-195, α-70, α-191等, 图 3A)在控制底物结合和还原中起关键作用, 这些氨基酸序列被某些分子取代后可以改变固氮酶的催化行为, 双取代钼铁蛋白固氮酶(α-70Val→Ala, α-195His→Gln)可有效催化CO2还原产生CH4, 1 nmol钼铁蛋白经20 min催化反应后可产生16 nmol甲醇(图 3B), 催化速率取决于CO2的局部压(或
$ HCO_3^ - $ 浓度)和通过固氮酶的电子通量, 当CO2局部压力达到0.45 atm时, 最高反应速率可达到21 nmol CH4/nmol钼铁蛋白.这种双取代钼铁蛋白还可催化CO2与乙烯(HC≡CH)发生还原耦合产生丙烯(H2C=CH—CH3), 在最佳电子通量条件下, 丙烯的生成量随着乙炔分压的增加而增加, 当乙炔分压达到0.027 atm时丙烯的产量随乙炔浓度的升高而迅速减少(图 3C), 这可能是由于乙炔对CO2还原的抑制所致. 2018年, 华盛顿大学Harwood研究团队[41]报道了一种唯铁固氮酶(iron-only nitrogenase)生物还原CO2产CH4的新途径(图 3D), 这种分离自沼泽红假单胞菌(Rhodopseudomonas palustris, R. palustris)的固氮酶可分别催化CO2、质子以及N2产生CH4、H2和NH3. R. palustris与甲基单胞菌(一种利用甲烷为碳源的细菌)共培养时可使其生长良好, 表明CH4生产成功.在其他固氮细菌中表达这种铁固氮酶时也可催化CO2还原产生CH4, 表明这种酶催化CO2甲烷化是其一般属性, 环境中的固氮微生物只有9%含有这种固氮酶的编码基因.这项研究不仅有助于理解CO2微生物酶法代谢产甲烷的过程和机理, 对控制环境中的CO2浓度、清洁能源的生产等均有重要意义.除甲烷之外, 固氮酶催化CO2还原为CO和甲酸也有见于报道[42, 43].例如2016年加州大学Hu等[42]报道了两种具有类似一氧化碳脱氢酶作用的天然含铁固氮酶NifH和VnfH, 来源于棕色固氮菌Azotobacter vinelandi, 能够将CO2转化为CO.这两种固氮酶能够通过蛋白铁硫簇的氧化还原在体内外两种环境中实现CO2的还原.在体外反应体系中, 在还原剂连二亚硫酸盐存在的情况下, VnfH和NifH都能够在无核苷酸或ATP结合状态下将CO2还原成CO(图 3E); 在氧化剂二磺酸盐存在的情况下, VnfH和NifH在CO2还原过程中都表现的不活跃(图 3E), 表明这些Fe蛋白的活性起源于它们+1价还原态铁硫簇.同年, Seefeldt等[43]报道了固氮酶还原CO2为甲酸、CO和CH4的反应路径, 发现CO2甲酸化的速率是甲烷化或CO化的十余倍, 并用量子力学的方法阐明了一条“氢化物直接转移”的反应途径, 为CO2的人工酶法转化提供了新的参考.值得注意的是, 固氮酶转化CO2所产生的中间产物往往性质活跃, 为此途径带来了许多障碍, 在未来的研究中, 如何稳定关键中间产物、理性设计反应路径、提高酶催化效率将是人们重点关注的方向.
3.2 碳酸酐酶催化CO2生物矿化
近年来, 碳酸酐酶(Carbonic Anhydrase, CA)在CO2捕获和转化领域的应用引起人们极大的关注.碳酸酐酶是一种含锌金属酶, 于1940年在奶牛的红细胞中被首次发现, 是一种典型的裂解酶类, 广泛存在于动物、植物和微生物组织中, 可催化CO2发生水合反应生成
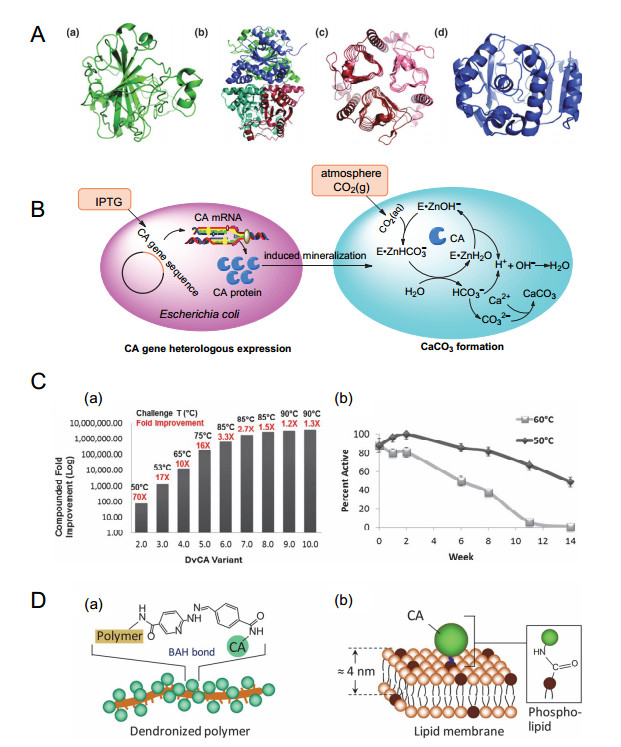
${\rm{HCO}}_{\rm{3}}^{\rm{ - }} $ 和质子$\left( {{\rm{C}}{{\rm{O}}_{\rm{2}}} + {{\rm{H}}_{\rm{2}}}{\rm{O}} \to {{\rm{H}}^ + }{\rm{HCO}}_{\rm{3}}^{\rm{ - }}} \right)$ , 转化速率可达到1×106 mol/s[7, 44].一般情况下, CO2在溶剂中的吸收较慢, 但CA可加速吸收并显示出较低的解吸热, 捕获和封存CO2所需的能量较低[45].根据酶分子氨基酸序列不同, 碳酸酐酶可分为α, β, γ, δ, ζ和η六种类型(图 4A), 这些CA并没有明显的氨基酸序列相似性[46, 47].其中α和β两种构型的CA研究较多(图 4A: a, b), 二者的氨基酸序列具有60%的同源性[48]. α-CA最初发现于人体, 现代研究发现所有动物来源的CA都属于α类型, 哺乳动物几乎所有组织中都含有参与机体多种生命活动的α-CA, 至少存在14种不同的同工酶, 其中催化CO2水合效率最高、研究也最为透彻的是CA-Ⅱ(图 4A, a)[49, 50], 它是一种催化特性良好的单体蛋白, 已广泛应用于CO2捕获及仿生矿化.图 4
 图 4. (A) 几种CA晶体结构示意图(a. α型; b. β型; c. γ型; d. ζ型)[45]; (B)异源表达CA及其催化CO2形成CaCO3示意图[54]; (C)定向进化提高CA稳定性(a.每轮定性进化后DvCA酶综合特性改善倍数; b. 8次定向进化后DvCA在4.2 mol/L MDEA中的储藏稳定性)[56]; (D) CA固定化示意图(a.枝状聚合物共轭固定CA; b.脂质体生物膜共轭固定CA)[57, 59, 60]Figure 4. (A) Depiction of the crystal structures of CA from different families, a~d represent the types of α, β, γ and ζ, respectively[45]; (B) Schematic illustration of heterologously expressed CA for promoting CaCO3 formation[54]; (C) CA stability enhancement by direct evolution: (a) Compounded fold improvement after each round of evolution; (b) The storage stability of DvCA8.0 (8 rounds evolution) when exposed to 4.2 mol/L MDEA[56]; (D) Diagrams showing the immobilization of carbonic anhydrase: (a) Dendronized polymer-CA conjugates; (b) Lipid bilayer membranes-conjugated CA[57, 59, 60]
图 4. (A) 几种CA晶体结构示意图(a. α型; b. β型; c. γ型; d. ζ型)[45]; (B)异源表达CA及其催化CO2形成CaCO3示意图[54]; (C)定向进化提高CA稳定性(a.每轮定性进化后DvCA酶综合特性改善倍数; b. 8次定向进化后DvCA在4.2 mol/L MDEA中的储藏稳定性)[56]; (D) CA固定化示意图(a.枝状聚合物共轭固定CA; b.脂质体生物膜共轭固定CA)[57, 59, 60]Figure 4. (A) Depiction of the crystal structures of CA from different families, a~d represent the types of α, β, γ and ζ, respectively[45]; (B) Schematic illustration of heterologously expressed CA for promoting CaCO3 formation[54]; (C) CA stability enhancement by direct evolution: (a) Compounded fold improvement after each round of evolution; (b) The storage stability of DvCA8.0 (8 rounds evolution) when exposed to 4.2 mol/L MDEA[56]; (D) Diagrams showing the immobilization of carbonic anhydrase: (a) Dendronized polymer-CA conjugates; (b) Lipid bilayer membranes-conjugated CA[57, 59, 60]2001年, Bond等[51]第一次报道了牛碳酸酐酶BCA对CO2的捕获和转化效果, 评价了在不同离子(盐碱卤水、浓盐水、海水)存在下市售BCA的性能, 结果表明在一定浓度的NOx(氮氧化物)和SOx(硫氧化物)存在条件下, BCA在类海水溶液中表现出良好的CO2捕获能力.在接下来的十几年中, 从生物系统中提取、纯化和表征CA, 并将其用于CO2高效捕获和生物矿化取得了重大进展[52].作为CA催化CO2水合及生物矿化的经典案例之一, 2007年, Mahinpey等[53]研究了碳酸酐酶在CO2捕获中的应用, 系统探究了牛碳酸酐酶(bovine CA, α-CA-Ⅲ)对CO2水合及碳酸钙沉淀的影响, 催化反应过程如式(4)~(8)所示.结果表明α-CA-Ⅲ可有效促进CO2水合并最终产生CaCO3沉淀, 水合反应速率随酶浓度和温度的升高而增加, 在酶的存在下, 可促进碳酸钙的沉淀, 但酶浓度对沉淀的影响并不明显, 温度对碳酸钙沉淀影响较大, 温度较高时, 沉淀较少.另外, α-CA-Ⅲ酶活性并不受pH影响, 但pH对沉淀影响较大.动力学分析结果表明, α-CA-Ⅲ催化CO2转化活化能和催化速率常数分别为700.91 cal/mol和0.65 s-1. 2016年, Lian等[54]研究了胶质芽孢杆菌(Bacillus mucilaginosus)CA基因在大肠杆菌中的异源克隆表达及其捕获CO2形成CaCO3作用机制(图 4B), 克隆了5种CA基因并成功表达了CA4, 可在短时间内(40 min)催化CaCO3晶体形成, 为进一步探究CA介导的CO2生物矿化提供了参考.
$ {{\rm{C}}{{\rm{O}}_{\rm{2}}}\left( {{\rm{gaseous}}} \right) \to {\rm{C}}{{\rm{O}}_{\rm{2}}}\left( {{\rm{aqueous}}} \right)} $
(4) $ {{\rm{C}}{{\rm{O}}_{\rm{2}}}\left( {{\rm{aqueous}}} \right) + {{\rm{H}}_{\rm{2}}}{\rm{O}} \to {{\rm{H}}_{\rm{2}}}{\rm{C}}{{\rm{O}}_{\rm{3}}}{\rm{}}} $
(5) $ {{\rm{H}}_{\rm{2}}}{\rm{C}}{{\rm{O}}_{\rm{3}}} \to {\rm{HCO}}_{\rm{3}}^{\rm{ - }} + {{\rm{H}}^ + } $
(6) $ {\rm{HCO}}_{\rm{3}}^{\rm{ - }} \to {\rm{CO}}_{\rm{3}}^{{\rm{2 - }}} + {{\rm{H}}^ + } $
(7) $ {\rm{C}}{{\rm{a}}^{\rm{2}}}^ + + {\rm{CO}}_{\rm{3}}^{{\rm{2 - }}} \to {\rm{CaC}}{{\rm{O}}_{\rm{3}}} \downarrow $
(8) CA在催化CO2生物矿化方面有大量的文献记载, 有望作为CO2封存及高值化利用的有效途径, 但作为一种天然酶类, 存在稳定性差、效率低、易失活等缺点, 定向进化和固定化技术是改善CA催化性能的有效方法, 近年来有大量的研究聚焦.定向进化(directed evolution), 是一种蛋白质的非合理设计, 可以人为创造特殊的进化条件, 模拟自然进化机制, 在体外改造基因, 并定向选择出所需性质的突变CA酶.例如, 2005年, Tawfik等[55]通过双位点突变对CA-Ⅱ进行定向进化, 将其乙酸萘激活活性提高了40倍; 2014年, Alvizo等[56]对来源于脱硫弧菌(Desulfovibrio vulgaris)的β-CA进行定向进化, 大大提高了催化稳定性, 可耐受107 ℃、pH>10的4.2 mol/L碱性胺(MDEA)溶液, 热稳定性及耐碱性是天然游离酶的400万倍; 储藏稳定性也大大提高, 在MDEA中50和60 ℃下半衰期分别为14和16周(图 4C), 相比之下, 未突变野生型CA在50 ℃半衰期仅为15 min, 60 ℃下甚至无活性.固定化技术已研究的比较成熟, 已有文献对CA固定化进行综述[57, 58], 但CA固定化新材料、新方法、新理念仍在不断创新和发展, 前景广阔.例如, 2018年, Walde等[59]研究了BCA在硅酸盐表面通过与树枝状聚合物进行固定化(图 4D), 共轭固定化BCA吸附于玻璃微吸管和玻璃纤维过滤器内, 最终设计出了用于CO2捕获和转化的流过式反应器, 固定化酶体系表现出了良好的催化活性(例如: de-PG21000- BAH179-BCA100, Γapp=6.5×10−4 U/cm2, pH=7.2)和催化稳定性(例如: de-PG21000-BAHy-BCAz, 41天内保留97%原始酶活); 2017年, Yoshimoto等[60]研究了一种负电荷脂质体共轭固定化CA, 这种脂质体同时可吸附钙离子, 因此固定化CA表现出了优异的CO2水合及矿化特性, 对Ca2+的消耗率可达94%.崔建东等[61]利用同轴静电纺丝技术制备出了一种中空纳米纤维结构材料, 实现了CA的原位包埋, 这种固定化CA在75 ℃水浴处理2.0 h后仍可保留74.4%的原始酶活(游离酶仅为9.1%), 重复使用11次剩余活性仍有81.9%, 大大提高了酶稳定性和重复利用率.
3.3 甲酸脱氢酶催化CO2产甲酸
甲酸是酶法还原CO2的重要产物或中间产物之一, 是一种重要的化工原料及有机合成中间体, 可用于青贮保存、动物饲料添加剂、纺织品整理和制浆造纸等, 可进一步转化为甲烷和氢气, 是一种潜在的能源物质, 研究表明甲酸与H2可在相近的电位下被氧化, 在制备低温燃料电池方面有巨大潜力[9, 62, 63].甲酸脱氢酶(formate dehydrogenase, FDH)在辅因子NADH作用下可催化CO2还原并转化为甲酸(式9), 反应机理可解释如下:氢化物从NADH吡啶环的C4原子直接转移到CO2的C原子上, CO2和NADH的位置很接近, 以便于氢化物转移, 在甲酸/甲酸盐生成之后, 留下具有双极构象的NAD+[64].同时, FDH也可催化CO2甲酸化逆反应, 将甲酸氧化为CO2, 伴随着NAD+还原为NADH(图 5A), 因此控制逆反应的发生对FDH催化CO2甲酸化十分重要[65]. FDH通常有两种类型, 一种是金属非依赖-NAD+依赖型FDH (Metal independent NAD+-dependent FDH), 另一种是金属依赖型FDH (Metal dependent FDH), 前者包括2-含氧酸家族的NAD+依赖型D型特异性脱氢酶, 广泛存在于细菌、酵母、真菌和植物中; 后者包括一些原核酶类, 这些原核酶的活性位点上通常结合一分子钨(W)或钼(Mo)离子[4, 66], 其中Mo-FDH研究较多, 其催化活性位点如图 5B所示, 包含两分子钼蝶呤鸟嘌呤二核苷酸(氧化态Mo, MoⅥ)辅因子, 一个蛋白衍生配体(半胱氨酸或硒代半胱氨酸)和一个硫代基团, 它们与钼离子在畸变六配位三棱镜几何结构中发生配位[66~68].
图 5
 图 5. (A) FDH催化CO2甲酸化及其逆反应[64]; (B) Mo-FDH催化活性位点结构示意图(a.硫桥聚焦视角; b. Cys196基团聚焦视角)[66]; (C) CCGCMAQSP人工光催化还原CO2产甲酸及其NADH再生过程示意图; (D)几种催化剂在NADH再生系统中还原CO2产甲酸化的光催化活性(a. NADH再生速率; b. HCOOH产量)[73]; (E) BODIPY, CCG-BODIPY, NH2-TPP以及CCG-NH-TPP的光催化活性(a. NADH再生速率; b. HCOOH产量)[75]Figure 5. (A) Sketch of FDH for catalysis of CO2 to produce HCOOH and the reverse reaction[64]; (B) The active site of the Mo-FDH in two views: focusing on the sulfido (a) and Cys196 (b) groups, respectively[66]; (C) CCGCMAQSP catalyzed artificial photosynthesis of HCOOH from CO2 and the photoregeneration of NADH under visible light; (D) Photocatalytic activities of various catalysts for synthesis of HCOOH from CO2 in the NADH photoregeneration system (a. Yield of NADH; b. HCOOH production)[73]; (E) Photocatalytic activities of BODIPY, CCG-BODIPY, NH2-TPP and CCG-NH-TPP (a. Yield of NADH; b. HCOOH production)[75]
图 5. (A) FDH催化CO2甲酸化及其逆反应[64]; (B) Mo-FDH催化活性位点结构示意图(a.硫桥聚焦视角; b. Cys196基团聚焦视角)[66]; (C) CCGCMAQSP人工光催化还原CO2产甲酸及其NADH再生过程示意图; (D)几种催化剂在NADH再生系统中还原CO2产甲酸化的光催化活性(a. NADH再生速率; b. HCOOH产量)[73]; (E) BODIPY, CCG-BODIPY, NH2-TPP以及CCG-NH-TPP的光催化活性(a. NADH再生速率; b. HCOOH产量)[75]Figure 5. (A) Sketch of FDH for catalysis of CO2 to produce HCOOH and the reverse reaction[64]; (B) The active site of the Mo-FDH in two views: focusing on the sulfido (a) and Cys196 (b) groups, respectively[66]; (C) CCGCMAQSP catalyzed artificial photosynthesis of HCOOH from CO2 and the photoregeneration of NADH under visible light; (D) Photocatalytic activities of various catalysts for synthesis of HCOOH from CO2 in the NADH photoregeneration system (a. Yield of NADH; b. HCOOH production)[73]; (E) Photocatalytic activities of BODIPY, CCG-BODIPY, NH2-TPP and CCG-NH-TPP (a. Yield of NADH; b. HCOOH production)[75]$ {\rm{C}}{{\rm{O}}_{\rm{2}}} + {\rm{NADPH}} \to {\rm{HCO}}{{\rm{O}}^{\rm{ - }}} + {\rm{NA}}{{\rm{D}}^ + } $
(9) 2008年, Moliner等[64]详细研究了金属非依赖-NAD+依赖型FDH的催化机制, 他们提出在催化过程中NADH烟酰胺部分被FDH活性中心氨基酸残基Thr 282, Asp 308, Ser 334, His 332及Gln 332固定, CO2导入催化系统后定向靠近NADH, 从而被FDH氨基酸残基Asn 146, Ile 122和Arg 284固定, 氢化物从烟酰胺的C4向CO2的C原子转移, 通过氢键与蛋白质骨架的氨基酸残基进行特异性相互作用, 酶催化环境适合氢化物转移.在氢化物离子转变之后, 甲酸根离子从系统中释放出来, 留下双极构象的NAD+.金属依赖型FDH还原CO2机制与非依赖-NAD+依赖型FDH完全不同, 在催化过程中, 氧化态酶金属中心接受两个电子, 从Mo(Ⅵ)或W(Ⅵ)变为Mo(Ⅳ)或W(Ⅳ), 结构从扭曲三角棱柱变为四角锥形, 基底平面被4个S原子包围(每两个来自于一个不同的吡喃蛋白配体), 顶端固定第五个S原子, 在这种良好催化环境中, CO2与FDH活性中心结合, C=O双键断裂, 与组氨酸H原子结合, 形成C—H键, 精氨酸残基使甲酸根离子发生定向转移, 质子被组氨酸残基清除[69, 70].
FDH胞外催化CO2甲酸化系统研究始于20世纪70年代, 至今已有近50年发展历史. 1984年, Parkinson等[71]较早研究了CO2胞外甲酸化, 采用p型磷化铟作为光电反应阴极还原甲基紫精, 从而成功介导FDH与CO2的连接还原生成甲酸, 产率为80%~93%;受此启发, 2006年, 天津大学Jiang等[72]以氧化甲基紫原为电子受体, 利用草酸假单胞菌(Pseudomonas oxalaticus) FDH将CO2还原成甲酸, 还利用凝胶包埋固定化技术提高了FDH的稳定性; 2012年, Baeg等[73]研究了一种来源于富马酸合成互营杆菌的含钨FDH还原CO2产甲酸, 表现出了当时已知的最高的催化活性; 类似地, 2014年Choe等[74]研究发现一种产硫杆菌FDH (Thiobacillus FDH, TBsFDH)在天然pH条件下催化CO2还原活性明显高于一些其他来源的FDH以及常见的商业化博伊丁假丝酵母FDH (Candida boidinii FDH, CbFDH), 比CbFDH高出5.8倍, 其主要原因可解释为TbsFDH蛋白骨架上有一个延长的N端和C端环状结构.
CO2酶法甲酸化所需的辅因子NADH价格昂贵, 是制约酶法催化CO2甲酸化的一个重要因素, 为此, 近年来有大量关于FDH催化CO2甲酸化辅因子再生及调控的研究报道.例如, 2012年, Baeg等[73]报道了一种基于石墨烯的新型可见光活性光催化剂CCGCMAQSP的合成, 通过石墨烯与生色团(例如多蒽醌取代卟啉)的共价结合, 并在催化体系中引入FDH, 构建了一种人工光-酶协同催化剂及反应体系(图 5C), 可高效催化CO2产甲酸(产量为110.55 μmol, 图 5D, a), 石墨的巨大表面积和极高的载流子迁移率使电子转移到铑配合物中的可能性增加, 最终加速了NADH再生, 再生率为45.54%(图 5D, b). 2014年, 该团队又报道了一种基于石墨烯的光-生物耦合催化体系(CCG-BODIPY)[75], 该体系NADH再生率进一步提高, 达到54.02%±0.61%(图 5E, a), 甲酸产量达到了144.2±1.8 μmol(图 5E, b).近年来, 研究者在辅因子升级调控方面虽然做了大量的研究, 但仍存在一定的瓶颈, 具有较大的研究空间.
3.4 一氧化碳脱氢酶催化CO2产CO
CO是CO2还原化利用的又一种重要产物, 是许多化学合成反应的重要原料, 例如费托合成制备烃类(Fischer-Tropsch Process)、孟山都反应(Monsanto Process)及卡迪瓦反应(Cativa Process)制乙酸等, 同时, 一氧化碳也具有很高的燃料价值, 可制备水煤气, 并且容易转化为液体燃料甲醇[76].许多研究者利用电催化剂或光催化剂将CO2转化为CO[77], 但该反应是一个双电子跃迁的过程, 并不容易进行, 大多数情况下都需要大量的光能或电能驱动反应, 造成了很大的能源浪费[4].作为一种绿色、高效、专一的催化手段, 酶法为CO2转化为CO提供了新的思路, 一氧化碳脱氢酶(carbon monodioxide dehydrogenase, CODH)可高效还原CO2产生CO[78], 自20世纪80年代被发现以来, 近年来备受关注. CODH有两种类型, 一种是来自专性厌氧菌(例如Moorella thermoacetica, Carboxydothermus hydrogeno- formans和Methanosarcina barkerii)的氧敏感性CODH, 具有[Fe4S4Ni]活性位点, 因此也被称为[NiFe]CODH, 这些酶对CO氧化的周转频率高达40000 s–1 (70 ℃下使用甲基紫精阳离子作为氧化剂), CO2还原周转频率为45 s–1 [79]; 另一种是来源于Oligotropha carboxidovorans的空气稳定型CODH, 包含[MoSCu], 这种CODH的CO氧化周转率较低, 由于Cu中心还原不足, 也无法催化CO2还原为CO[79, 80]. [NiFe]CODH和[MoSCu]CODH均含有双金属活性位点.
[NiFe]CODH是催化CO2还原为CO最重要的一种CODH, 如图 6A所示[79], 其活性中心由Ni和Fe通过临近的铁硫簇(Fe3S4)刚性桥连组装形成, 在这种状态下, 配位不饱和NiⅡ在一种平面T形环境中结合三个S配体; Fe1内配位层包括一个组氨酸配体(H261)、半胱氨酸配体(C295)、μ3-硫化物配体和一个氢原子.这些配位原子也接近Ni原子, 使Ni和Fe紧密桥连, Ni表面上的空位配位表明在催化过程中两个金属中心之间可能存在协同作用[81, 82]. [NiFe]CODH催化还原CO2机理可总结为双电子转移ECE机制, 如图 6B所示, 最先发生的是一个电子转移过程(E), 形成还原态Ni中心域(NiⅠ); 随后发生化学反应步骤(C), CO2与还原的Ni中心域结合; 最后发生第二次电子转移(E)[79].在化学反应步骤中, CO2通过C原子与Ni原子结合, 形成Ni—C键, 同时, 一个羧酸氧原子(O2)与质子化的组氨酸残基(H93)形成氢键, Fe1失水形成CO2复合物, CO2分子中的另一个氧原子O1与Fe1结合并与质子化的赖氨酸残基(K563)形成氢键, C—O1键的断裂和水的损失产生了NiⅡCO中间体, 这种NiⅡCO中间体容易释放CO, 加水后可再生初始的NiⅡ络合物, 从而完成催化循环. CO2在酶中的结合、催化甚至释放过程涉及双金属中心的激活, 以及第二配位球中某些定位残基的额外稳定作用[7].
图 6
 图 6. (A) [NiFe]CODH活性中心球棍模型; (B) [NiFe]CODH催化CO2生成CO机理[79]; (C) [NiFe]CODH吸附固定于热解石墨边缘电极快速催化CO2产CO示意图[80]; (D)钌基金半导体光敏催化剂耦合[NiFe]CODH人工光催化还原CO2示意图[76]Figure 6. (A) Ball-and-stick drawing of the active site of [NiFe]CODH; (B) Mechanism for reduction of CO2 to CO by [NiFe]CODH[79]; (C) [NiFe]CODH anchored on a pyrolytic graphite "edge" electrode for catalysis of rapid conversion of CO2 to CO[80]; (D) Schematic illustration of the CO2 photoreduction system using [NiFe]CODH attached to a Ru-based semiconductor photocatalyst[76]
图 6. (A) [NiFe]CODH活性中心球棍模型; (B) [NiFe]CODH催化CO2生成CO机理[79]; (C) [NiFe]CODH吸附固定于热解石墨边缘电极快速催化CO2产CO示意图[80]; (D)钌基金半导体光敏催化剂耦合[NiFe]CODH人工光催化还原CO2示意图[76]Figure 6. (A) Ball-and-stick drawing of the active site of [NiFe]CODH; (B) Mechanism for reduction of CO2 to CO by [NiFe]CODH[79]; (C) [NiFe]CODH anchored on a pyrolytic graphite "edge" electrode for catalysis of rapid conversion of CO2 to CO[80]; (D) Schematic illustration of the CO2 photoreduction system using [NiFe]CODH attached to a Ru-based semiconductor photocatalyst[76]CODH体外催化CO2还原为CO常结合光催化或电催化进行[83]. 2003年, Shin等[84]较早研究了CODH体外转化CO2, 结果表明CODH是电催化CO2还原的一种良好的酶, 几乎没有过电位, 并且表现出了高选择性及高转化率, 这是由于CODH中具有独特的C-簇结构, 每分子C-簇转换数(turnover number)可达700 h–1, 最适pH为6.3 (0.1 mol/L PBS).然而, 受pH等因素影响, 电催化后酶活性降低, CO产率下降.为了解决这一局限, 2007年, Armstrong等[80]将一种来源于Carboxydothermus hydrogenoformans的[NiFe]CODH吸附固定于热解石墨边缘(pyrolytic graphite edge, PGE)电极(图 5C), 实现了CO2/CO的快速、高效相互转换.在此过程中, 固定化CODH催化CO2与CO之间转化的标准还原电位升高, 导致所需过电位降低, 并提高了产物选择性.这些研究表明CODH作为电催化协同催化剂具有很大的应用潜力.光催化方面, Armstrong团队也做了大量研究, 2010年, 他们把光催化结合到CO2的酶法转化中, 构建了人工光合作用系统.如图 6D所示, 他们将酶催化剂[NiFe]CODH和钌基光敏催化剂[RuII(bipy)2-(4, 4′-(PO3H2)2-bipy)]Br2 (RuP; bipy=2, 2′-联吡啶)杂化吸附固定在n型MOx半导体纳米粒子(例如P25 TiO2、锐钛矿型TiO2、金红石型TiO2、ZnO、SrTiO3等)上, 构建了光-酶偶联催化体系.通过可见光激发, RuP将电子注入到MOx半导体导电带中, 这些电子随后转移至[NiFe]CODH中, 并通过铁硫簇(Fe3S4)转移至活性位点, 将CO2还原为CO, 表现出了较高的活性[76].鉴于上述提到的CO的重要性及酶法转化的优势, 酶法人工转化CO2产CO仍具有一定的研究空间, 但需要不断改进和创新, 以弥补天然酶催化法的不足, 在后续的研究中, 光、电、酶、新型纳米催化剂及其协同催化将是新的研究热点[77].
3.5 脱羧酶催化CO2羧化反应
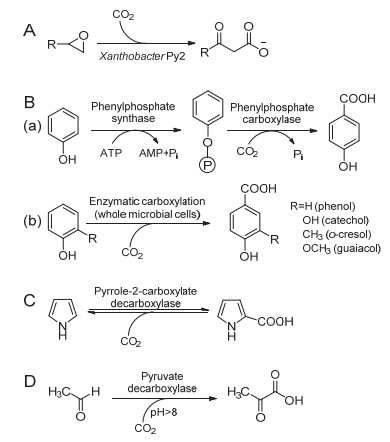
羧基化是一种在底物中引入羧酸基的化学反应, 在胞内由脱羧酶催化进行, 主要目的是提高胞内有毒化合物的亲水性, 从而降低它们对亲脂性生物成分(如细胞膜、蛋白质等)的亲和力, 以降低毒性[7].脱羧酶可催化底物与CO2发生羧基化反应, 产生相应的羧酸类化合物, 反应几乎不消耗能量或耗能较低, 是CO2高值化利用的一种重要途径, 自20世纪90年代以来引起了人们的广泛关注[9].酶催化CO2羧基化反应根据底物的不同可分为4种类型[85]: (1)环氧化物羧基化, (2)芳香族化合物羧基化, (3)杂环芳烃羧基化, (4)脂肪族化合物羧基化.
Ensign等[86]在1996年报道了脂肪族环氧化物在Xanthobacter Py2细胞提取物中通过羧化反应进行分解代谢产生β-酮酸.在本研究中, Xanthobacter Py2细胞提取物催化环氧丙烷与CO2发生羧基化形成乙酰乙酸和少量的β-羟基丁酸(图 7A).在没有CO2的情况下, 环氧丙烷和1, 2-环氧丁烷被异构化, 分别形成丙酮和丁酮.然而, 由于很难纯化出起相应催化作用的脱羧酶, 因此该方法在体/胞外和实际应用中的潜力有限.
图 7
 图 7. (A) Xanthobacter Py2细胞提取物或全细胞催化环氧化物与CO2羧基化; (B) (a) Thauera aromatica细胞提取物或部分纯化的磷酸苯酯酶催化苯酚与CO2羧化反应; (b) Clostridium hydroxybenzoicum区域选择全细胞催化非活化酚对位羧基化; (C)吡咯-2-羧酸脱羧酶催化吡咯与CO2的羧基化反应; (D)丙酮酸脱羧酶催化乙醛与CO2的羧基化反应[85~87, 89~91]Figure 7. (A) Biocatalytic carboxylation of epoxides with CO2 by the cell extracts or whole-cell of Xanthobacter Py2; (B) (a) Carboxylation of phenol by cell extracts of Thauera aromatica or partially purified phenylesterase; (b) Regioselective para-carboxylation of non-activated phenolic compounds catalyzed by whole-cell of Clostridium hydroxybenzoicum; (C) Carboxylation of pyrrole with CO2 by pyrrole-2-carboxylate decarboxylase; (D) Carboxylation of acetaldehyde with CO2 by pyruvate decarboxylase[85~87, 89~91]
图 7. (A) Xanthobacter Py2细胞提取物或全细胞催化环氧化物与CO2羧基化; (B) (a) Thauera aromatica细胞提取物或部分纯化的磷酸苯酯酶催化苯酚与CO2羧化反应; (b) Clostridium hydroxybenzoicum区域选择全细胞催化非活化酚对位羧基化; (C)吡咯-2-羧酸脱羧酶催化吡咯与CO2的羧基化反应; (D)丙酮酸脱羧酶催化乙醛与CO2的羧基化反应[85~87, 89~91]Figure 7. (A) Biocatalytic carboxylation of epoxides with CO2 by the cell extracts or whole-cell of Xanthobacter Py2; (B) (a) Carboxylation of phenol by cell extracts of Thauera aromatica or partially purified phenylesterase; (b) Regioselective para-carboxylation of non-activated phenolic compounds catalyzed by whole-cell of Clostridium hydroxybenzoicum; (C) Carboxylation of pyrrole with CO2 by pyrrole-2-carboxylate decarboxylase; (D) Carboxylation of acetaldehyde with CO2 by pyruvate decarboxylase[85~87, 89~91]酶催化芳香族化合物羧基化是厌氧菌的一种重要的代谢途径, 其中典型的代表是酶催化酚类羧基化生成极性更强的苯甲酸衍生物.研究者最初采用Thauera aromatica细胞提取物或部分纯化的磷酸苯酯酶催化苯酚与CO2发生羧化反应产生对羟基苯甲酸, 羧基化涉及两步酶催化反应: (1)在ATP的激活作用下, 磷酸苯酯合成酶催化苯酚产生磷酸苯酯; (2)金属(Mg2+或Mn2+和K+)依赖型磷酸苯酯羧化酶区域选择性催化中间产物形成对羟基苯甲酸(图 7B, a)[87].类似于苯酚, 磷酸苯酯羧化酶也能使邻苯二酚羧基化, 并且羧化的过程与苯酚相同, 例如, 在碳酸氢盐存在条件下, 3, 4-二羟基苯甲酸脱羧酶(来自于Clostridium hydroxybenzoicum)可催化儿茶酚逆向羧基化为3, 4-二羟基苯甲酸盐(图 7B, b)[85].除了对位羧基化之外, 一些γ-间苯二酚酸脱羧酶(或2, 6-二羟基苯甲酸脱羧酶, 来源于Agrobacterium tumefaciens, Rhizobium radiobacter或Rhizobium sp.)还可催化1, 3-二羟基苯酚与CO2的区域选择性邻位羧基化反应产生间苯二酚酸[88].
杂环芳烃与CO2发生羧基化反应最典型的代表是吡咯, 作为一种多电子杂环芳烃, 可在吡咯-2-羧酸脱羧酶(来源于Bacillus megaterium和Serratia sp.)及乙酸、丙酸、丁酸等辅因子催化作用下与CO2发生羧基化反应(图 7C), 产生重要的有机合成中间体——吡咯-2-羧酸[85, 89].
对于脂肪族化合物羧基化, 2001年, Maeda等[90]报道了以乙醛和CO2为原料, 经啤酒酵母丙酮酸脱羧酶催化羧基化反应合成丙酮酸的研究(图 7D), 焦磷酸硫胺素为反应辅因子, 优化了pH、底物浓度等反应条件, 结果表明, 以pH 11的碳酸氢钠为缓冲溶液及CO2供体, 丙酮酸产量可达到81%.在此之后, Wang等[91]又报道了丙酮酸与CO2在乳酸脱羧酶的作用下产生L-乳酸, 这也是CO2与脂肪族化合物羧基化反应的典型实例.脱羧酶催化CO2羧基化为CO2的高值化利用提供了一条有效途径, 然而, 由于CO2的热力学稳定性高、反应活性低、亲电能力弱, 因此羧化反应速率及转化率往往不高, 在以后的研究中, 酶催化协同光、电及纳米化学催化以提高脱羧酶催化效率将是人们关注的焦点.
4. 多酶复合级联催化CO2研究进展
如本文第二部分所述, CO2胞内生物转化往往涉及到多种酶的协同级联催化作用, 细胞的层级结构和适宜的微环境保证了一系列催化反应的高效有序进行.受此启发, 研究者开发了多种多酶级联途径转化CO2生产目标高附加值化学品, 取得了不错的进展, 其中研究较多也相对较为热门的是三种脱氢酶体系转化CO2产甲醇的研究, 也涉及到一些糖、醛、酸类等其他化合物[92, 93], 综述如下.
4.1 脱氢酶级联催化CO2还原产甲醇
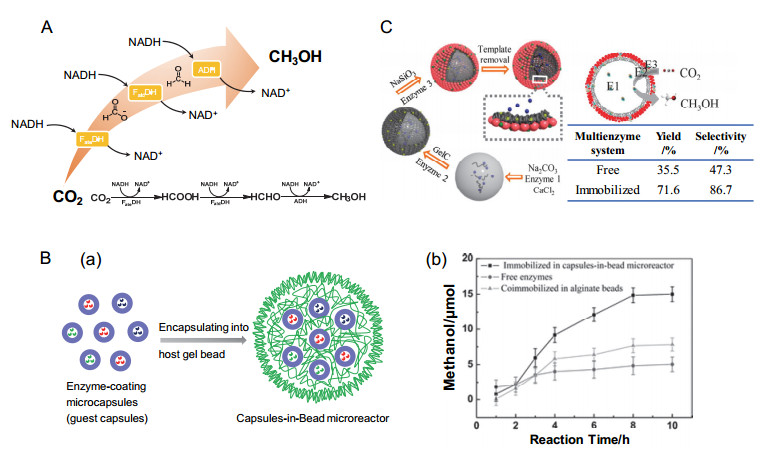
甲醇是一种重要的化工原料及能源物质, 其十六烷值高、燃烧效率高, 世界各地均有广泛分布, 是作为清洁燃料的合适选择[94].甲醇作为燃料主要有四种方式:第一是直接作为燃料; 第二是部分取代生物柴油; 第三是与汽油混合; 第四是转化成二甲醚(二甲醚可在中等压力下液化, 进一步用作柴油的替代品)[4].甲醇通常是由合成天然气制备得到, 但这种方式往往程序复杂、成本昂贵, 经济性不好.近年来, 研究者尝试了包括光催化、电催化、光电协同催化、生物催化等多种方法研究了CO2甲醇化, 取得了一定的研究进展[95~97].作为一种生物催化手段, 多酶级联催化CO2甲醇化近年来引起了较多关注, 其中甲酸脱氢酶(FDH)、甲醛脱氢酶(FaldDH)和乙醇脱氢酶(ADH)在辅因子NADH的作用下级联催化CO2产甲醇是最经典的一种反应模式.与单酶法生产的燃料(CO、甲烷等)相比, 多酶法生产的液态甲醇具有更高的能量容量, 并且易于储运.
1994年, Yoneyama等[98]首次报道了多酶级联催化CO2, 以甲酸脱氢酶(FDH)和甲醇脱氢酶(MDH)为催化剂, 吡咯喹啉醌(PQQ)等为辅因子, 采用电化学的方法, 成功将CO2还原成甲醇, 在温和条件下为CO2甲醇化提供了一条简便的途径.该反应系统中辅因子种类对CO2的还原影响很大, 合适的辅因子可大大提高该多酶级联体系的反应速率.基于脱氢酶可以在合适的辅因子存在下有效地催化CO2的还原这一事实, 1999年, Dave等[16]报道了一种以三种脱氢酶(FDH、FaldDH和ADH)为催化剂, NADH为辅因子, 级联催化CO2还原为甲醇的方法.整个反应过程如图 8A所示, 首先, CO2在FDH的催化作用下产生甲酸, 随后甲酸在FaldDH的作用下产生甲醛, 最后, ADH催化甲醛还原为甲醇, 辅因子NADH参与每一步催化. 2010年, Wang等[99]研究了该多酶级联反应体系的热力学可行性, 发现该反应体系对pH十分敏感, 反应必须在较低的pH值、适宜的离子强度和较高的温度条件下进行, 这种反应条件往往会造成酶的变性或催化活性的降低, 因此需要结合一些固定化手段来提高级联酶催化体系的稳定性.例如, Dave等在多孔硅溶胶-凝胶基质(porous silica sol-gel matrix)中共封装三种脱氢酶, 发现甲醇的产量和产率与游离酶相比都显著增加(产量:游离酶11.2 μmol, 封装酶系29.2 μmol; 产率:游离酶21.0%, 封装酶系91.2%, 均以最大值计), 他们把甲醇产率和产量的提高归因于硅胶纳米孔对多酶体系的保护作用, 这有利于改变反应热力学和最终平衡[16].自此之后, 有大量研究聚焦于多酶体系固定化, 以提高级联催化性能.
图 8
 图 8. (A) NADH介导的脱氢酶(FDH、FaldDH、ADH)级联催化CO2产甲醇示意图[16, 104]; (B) (a)一种基于珠内胶囊支架的多酶级联固定化体系的构建; (b)几种酶催化体系甲醇产量与时间的关系[102]; (C)基于超薄复合杂化微胶囊的多酶固定化体系级联催化CO2产甲醇[103]Figure 8. (A) Schematic diagram of NADH-mediated multi-dehydrogenases (FDH, FaldDH and ADH) for cascade catalysis of CO2 to produce methanol[16, 104]; (B) (a) Capsules-in-bead scaffold for construction of multienzyme cascade system; (b) Plot of methanol formation as a function of reaction time in various enzymatic catalytic systems [102]; (C) Multienzyme cascade system based on ultrathin hybrid microcapsule for conversion of CO2 to methanol[103]
图 8. (A) NADH介导的脱氢酶(FDH、FaldDH、ADH)级联催化CO2产甲醇示意图[16, 104]; (B) (a)一种基于珠内胶囊支架的多酶级联固定化体系的构建; (b)几种酶催化体系甲醇产量与时间的关系[102]; (C)基于超薄复合杂化微胶囊的多酶固定化体系级联催化CO2产甲醇[103]Figure 8. (A) Schematic diagram of NADH-mediated multi-dehydrogenases (FDH, FaldDH and ADH) for cascade catalysis of CO2 to produce methanol[16, 104]; (B) (a) Capsules-in-bead scaffold for construction of multienzyme cascade system; (b) Plot of methanol formation as a function of reaction time in various enzymatic catalytic systems [102]; (C) Multienzyme cascade system based on ultrathin hybrid microcapsule for conversion of CO2 to methanol[103]近20年来, 天津大学姜忠义教授团队[100~103]围绕固定化开展了一系列CO2多酶级联转化的研究.例如, 2006年, 采用海藻酸盐-二氧化硅复合材料将三种脱氢酶封装固定, 与游离酶相比, 酶的活性、贮存稳定性和重用性显著提高, 甲醇产率可达98.1%, 贮存60天及重复使用10次以上均可保留超过76%的甲醇产率.他们推测这种现象是由于海藻酸盐-二氧化硅复合材料为酶催化提供更适宜的微环境, 包括高亲水性、适宜的刚性和柔性、笼约束效应和较短的扩散距离[100]. 2009年, 他们又采用了生物固定化的方法, 在温和条件下, 以鱼精蛋白为诱导剂, 将三种脱氢酶共固定化在TiO2纳米颗粒中, 在这种固定化体系中, 三种脱氢酶可以构建一条将CO2转化成甲醇的“组装线路”, 从而获得高产率的甲醇[101].然而, 三种酶在同一载体中固定化有以下几点弊端: (1)酶的活性可能受到不同酶之间相互作用的不良影响; (2)很难控制固定化过程中单个酶的催化行为.为了使级联反应的不同步骤在特定区域内发生, 同年, 他们构建了珠内胶囊支架(capsules-in-bead scaffold)用于多酶系统的空间分离(图 8B, a), 以含酶碳酸钙微粒为供体模板, 采用仿生矿化和层层自组装相结合的方法制备了客体微囊, 随后, 三种客体微囊共同封装到较大的“宿主”海藻酸盐珠球中, 形成珠内胶囊支架.三种脱氢分区域固定于珠内胶囊支架中, 并最终用于将CO2转化成甲醇.与一锅法包埋的三种脱氢酶及游离酶相比, 包封在珠内胶囊支架中的脱氢酶具有较高的初始活性和更高的甲醇收率(图 8B, b).这是因为不同酶之间的干扰减少、中间产物的局部浓度升高以及中间体从一种酶的活性位点到另一种酶的扩散距离缩短[102].为了进一步实现对单个酶的刚柔性调控, 降低传质阻力, 2014年, 该团队利用仿生矿化和仿生粘附耦合, 构建了超薄复合杂化微胶囊, 用于固定多酶体系转化CO2产甲醇.如图 8C所示, FDH、FaldDH和ADH三种脱氢酶分别固定在囊腔、囊壁有机层和囊壁二氧化硅层中, 该多酶体系甲醇收率和选择性分别为71.6%和86.7%, 显著高于游离多酶体系(35.5%和47.3%)[103].
如图 8A所示, FDH、FaldDH和ADH三种脱氢酶级联催化还原CO2产一分子甲醇需要消耗3分子的辅因子NADH.然而, NADH价格昂贵, 限制了CO2级联酶催化转化在大规模操作中的进一步应用, NADH再生和调控是解决上述问题的重要策略, 已成为近年来的研究热点[104]. 2008年, Wang等[105]较早进行了辅因子NADH再生的研究, 构建了辅因子原位再生系统级联催化CO2产甲醇, 如图 9A所示.将FDH、FaldDH、ADH和GDH(谷氨酸脱氢酶)共同固定在聚苯乙烯微粒上, 在另外的聚苯乙烯微粒上固定NADH以启动级联催化反应, 反应过程中GDH转化为酮戊二酸, 实现了辅因子NADH的原位再生, 与游离酶体系相比, 这种固定化多酶体系显著提高了辅因子的利用效率, 当NADH浓度同为500 μmol/L时, 固定化酶体系甲醇产量是游离酶的10余倍.但是, 这种固定化体系酶活性低于游离酶反应体系, 甲醇得率也较低, 有待于进一步改进. 2015年, Zhang等[106]继续了辅因子再生的研究(图 9B, a), 通过同轴静电纺丝技术制备的阳离子聚电解质掺杂中空纳米纤维(cationic polyelectrolyte-doped hollow nanofibers)可为多酶体系和辅因子提供良好的支撑载体, 这种级联催化和辅因子再生体系可还原CO2产甲醇.将辅因子NADH和四种酶(FDH、FaldDH、ADH和GDH)加入同轴静电纺丝的芯相(core-phase)溶液中, 原位包埋在中空纳米纤维腔内(阳离子聚电解质被预先溶解), 通过中空纳米纤维外壳聚电解质和带相反电荷的辅因子之间的离子交换相互作用, 使得辅因子能够有效地在腔内固定和保留.本研究中还使用了CA来加速CO2水合, 这种中空纳米纤维固定的五种酶级联催化体系表现出了良好的甲醇生产能力, 收率高达103.2%, 是当时有文献记录的最高值, 而使用游离酶和辅因子甲醇收率仅为36.17%(图 9B, b).最近, Lee等研究了胞外多酶级联反应实现了CO2甲醇化, 并采用亚磷酸脱氢酶(PTDH)实现了辅因子NADH的再生(图 9C, a), 该级联反应体系甲醇产量可达3.8 mmol/L(图 9C, b), 引入1-乙基-3-甲基咪唑乙酸酯(EMIM-Ac)后可使甲醇产量进一步达到7.86 mmol/L(图 9C, b), 转换数比传统的博伊丁假丝酵母FDH-恶臭假单胞菌FaldDH-YADH级联酶催化体系提高了500余倍(图 9C, c).本研究所表现出的关键控制因素包括一种新的甲醛脱氢酶BmFaldDH、PTDH再生NADH以及EMIM-Ac的引入[107].总结来看, 辅因子再生的方法包括底物耦合再生、酶耦合再生、闭环再生、膜表面再生及固定化再生等, 近年来虽取得了显著的成效, 但尚未能大规模应用, 仍具有较大的研究空间[17].
图 9
 图 9. (A) NADH原位再生驱动三种脱氢酶级联催化CO2产甲醇示意图(GDH:谷氨酸脱氢酶)[105]; (B) (a)同轴静电纺丝技术制备的阳离子聚电解质掺杂中空纳米纤维系留NADH及其级联催化CO2产甲醇示意图; (b)三种中空纳米纤维系留NADH介导的多酶级联催化系统甲醇产率随反应时间的变化曲线(空心点为对应的游离酶系统)[106]; (C) (a)基于PTDH再生辅因子介导的无细胞ClFDH-BmFaldDH-YADH多酶级联体系催化CO2产甲醇示意图; 不同的多酶级联体系催化CO2产甲醇产量(b)及总转换数(c)与时间关系; (蓝: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); 红: ClFDH+BmFaldDH+YADH with PTDH; 紫: CbFDH+BmFaldDH+YADH with PTDH; 绿: ClFDH+PpFaldDH+YADH with PTDH; 黑: CbFDH+PpFaldDH+YADH with PTDH)[107]Figure 9. (A) Schematic representation of enzymatic synthesis of methanol from CO2 with in situ regeneration of NADH (GDH: glutamate dehydrogenase)[105]; (B) (a) Schematic illustration of coaxial electrospinning for construction of cationic polyelectrolyte-doped hollow nanofiber tethering of NADH for methanol synthesis from CO2; (b) Plots of methanol yield as a function of reaction time for methanol synthesis from CO2 by using different multienzyme systems (Open legends represent results from free multienzyme systems, and the filled legends represent results from the hollow nanofiber-supported multienzymes systems, respectively)[106]; (C) Schematic illustration of reduction of CO2 into methanol using multi-enzymatic cascade system with NADH regeneration by PTDH (a); Time profile of methanol production (b) and total turnover number (c) by multi-enzymatic cascade reactions (blue: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); red: ClFDH+BmFaldDH+YADH with PTDH; purple: CbFDH+BmFaldDH+YADH with PTDH; green: ClFDH+PpFaldDH+YADH with PTDH; black: CbFDH+PpFaldDH+YADH with PTDH)[107]
图 9. (A) NADH原位再生驱动三种脱氢酶级联催化CO2产甲醇示意图(GDH:谷氨酸脱氢酶)[105]; (B) (a)同轴静电纺丝技术制备的阳离子聚电解质掺杂中空纳米纤维系留NADH及其级联催化CO2产甲醇示意图; (b)三种中空纳米纤维系留NADH介导的多酶级联催化系统甲醇产率随反应时间的变化曲线(空心点为对应的游离酶系统)[106]; (C) (a)基于PTDH再生辅因子介导的无细胞ClFDH-BmFaldDH-YADH多酶级联体系催化CO2产甲醇示意图; 不同的多酶级联体系催化CO2产甲醇产量(b)及总转换数(c)与时间关系; (蓝: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); 红: ClFDH+BmFaldDH+YADH with PTDH; 紫: CbFDH+BmFaldDH+YADH with PTDH; 绿: ClFDH+PpFaldDH+YADH with PTDH; 黑: CbFDH+PpFaldDH+YADH with PTDH)[107]Figure 9. (A) Schematic representation of enzymatic synthesis of methanol from CO2 with in situ regeneration of NADH (GDH: glutamate dehydrogenase)[105]; (B) (a) Schematic illustration of coaxial electrospinning for construction of cationic polyelectrolyte-doped hollow nanofiber tethering of NADH for methanol synthesis from CO2; (b) Plots of methanol yield as a function of reaction time for methanol synthesis from CO2 by using different multienzyme systems (Open legends represent results from free multienzyme systems, and the filled legends represent results from the hollow nanofiber-supported multienzymes systems, respectively)[106]; (C) Schematic illustration of reduction of CO2 into methanol using multi-enzymatic cascade system with NADH regeneration by PTDH (a); Time profile of methanol production (b) and total turnover number (c) by multi-enzymatic cascade reactions (blue: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); red: ClFDH+BmFaldDH+YADH with PTDH; purple: CbFDH+BmFaldDH+YADH with PTDH; green: ClFDH+PpFaldDH+YADH with PTDH; black: CbFDH+PpFaldDH+YADH with PTDH)[107]4.2 其它多酶体系催化转化CO2
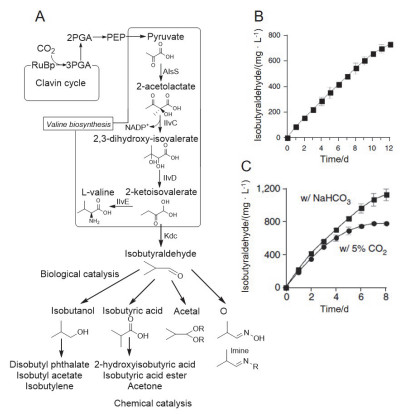
脱氢酶级联催化CO2产甲醇为CO2高值化利用提供了一条有效途径, 但只能生成C1产物, 开发新的多酶级联催化路径生产含多个碳原子的化学品引发了人们更多的思考.早在1952年, Calvin等[108]在光照条件下利用核酮糖-1, 5-二磷酸羧化酶/加氧酶((RuBisCO)产生ATP, 通过光合碳循环形成了多种储能碳化合物.受此启发, 2010年, Montemagno等[109]将卡尔文循环所需的多种酶与纳米级的光磷酸化系统通过Túngara frog表面蛋白Ranaspumin-2偶联, 构建了了一种无细胞的人工光合作用系统, 用于将二氧化碳转化为糖.这种独特的蛋白质表面活性剂使脂质囊泡与耦合的活性酶被集中到泡沫微孔, 可以产生116 nmol•mL-1•h-1的葡萄糖, 化学转化效率接近96%. 2009年, Liao等[110]采用基因手段构建了Synechococcus elongatus PCC7942蓝细菌工程菌, 可过表达核酮糖-1, 5-二磷酸羧化酶/加氧酶(RuBisCO), 通过与乙酰乳酸合成酶(AlsS), 乙酰羟基酸异构还原酶(IlvC), 二羟基酸脱水酶(IlvD)和2-酮酸脱羧酶(Kdc)等协同作用, 五种酶协同催化CO2直接合成异丁醛和异丁醇(图 10A).由于RuBisCO是蓝细菌固碳的关键因素, 这种酶的过度表达可增加工程菌固定CO2的反应速率, 使异丁醛的产率大大提高(图 10B, C), 在8 d内可保持良好活性. 2011年, Wang等[91]报道了一种将CO2和乙醇通过相对简单的多酶级联催化途径转化为L-乳酸, 为生物可降解聚合物的合成提供了一种新的、可持续的替代方法, 反应过程主要包括3个简单酶催化路径: (1)在辅因子NAD+的协同作用下, ADH将乙醇氧化为乙醛; (2)丙酮酸脱羧酶催化CO2和乙醛产生丙酮酸; (3)乳酸脱氢酶(LDH)在NADH存在下将丙酮酸还原为L-乳酸.通过以上三个步骤, 在间歇式反应体系中可将41%乙醇转化为L-乳酸, 第一步和第三步反应可实现NADH/NAD+辅因子再生和循环, 在该多酶级联催化体系中, 每个反应的动力学模型及参数与单一酶反应步骤相同, 因此可由单个反应确定的动力学参数预测混合反应效果.值得注意的是, 生物体系胞内多酶级联转化CO2的途径多种多样, 目前被阐明的还十分有限, 因此在以后的研究中, 从天然多酶级联转化CO2入手, 详细阐明酶催化路径和机制, 对指导体/胞外多酶级联催化CO2高值化利用有重要意义.
图 10
 图 10. Synechococcus elongatus PCC7942蓝藻工程菌胞内多酶催化CO2合成异丁醛(A.反应过程示意图; B.蓝细菌SA590生产异丙醛时间-产量曲线; C. RuBisCO过表达蓝细菌SA665生产异丙醛时间-产量曲线)[110]Figure 10. Intracellular multi-enzymatic synthesis of isobutyraldehyde from CO2 in an engineering cyanobacterium Synechococcus elongatus PCC7942. (A) The pathway for isobutyraldehyde production. Isobutyraldehyde production as a function of time by cyanobacteria SA590 (B), and SA665 with enhanced Rubisco (C), respectively[110]
图 10. Synechococcus elongatus PCC7942蓝藻工程菌胞内多酶催化CO2合成异丁醛(A.反应过程示意图; B.蓝细菌SA590生产异丙醛时间-产量曲线; C. RuBisCO过表达蓝细菌SA665生产异丙醛时间-产量曲线)[110]Figure 10. Intracellular multi-enzymatic synthesis of isobutyraldehyde from CO2 in an engineering cyanobacterium Synechococcus elongatus PCC7942. (A) The pathway for isobutyraldehyde production. Isobutyraldehyde production as a function of time by cyanobacteria SA590 (B), and SA665 with enhanced Rubisco (C), respectively[110]5. 结论和展望
作为一种含碳丰富的温室气体, CO2可通过酶法经一步催化或级联催化转化为甲烷、甲酸、甲醇、CO、
$ {\rm{HCO}}_{\rm{3}}^{\rm{ - }} $ 等高附加值化学品, 尤其是产甲醇、甲烷和CO的研究, 是解决能源和环境危机的有效手段. CO2的酶法转化是一个持续性研究热点, 相关学者在过去的几十年里做了大量的工作, 已经报道了多种关于酶的制备、反应条件优化、反应机理分析、酶分子改造及固定等工作, 取得了一定的成效, 但是仍存在相当多的技术瓶颈, 因此仍将是当下和未来的持续性研究热点.在酶法捕获和转化CO2的研究中, 急需解决的技术瓶颈可总结为以下几个方面: (1)用于转化CO2的酶分离纯化困难, 价格昂贵, 并且非常敏感脆弱, 很容易变性, 温度、pH、强光等苛刻条件均可使其活性降低, 这些不利因素阻碍了酶法转化CO2的大规模使用; (2)对于大规模工业化应用, 适宜的生物酶反应器非常重要, 例如用于太阳能收集的有效系统、易于电子转移的CO2高效还原系统、电荷分离系统、氧化和还原过程的空间分离系统、烃类的选择性合成系统等; (3)昂贵的辅因子限制了CO2级联酶催化转化在大规模操作中的进一步应用; (4)酶法转化虽然有较强的专一性, 但不可避免地会发生某些副反应, 产生一些复杂的中间产物, 这为目标高附加值产物的分离带来了挑战.针对上述问题, 笔者认为酶法转化CO2的研究应当聚焦于以下几个方面: (1)采用传统生物技术及现代合成生物学的方法人工设计开发高产量、高催化活性、高稳定性、低价格的酶是解决酶法催化CO2高值化利用的关键手段, 最终提高经济可行性; (2)辅因子是限制酶法转化CO2的一个重要成本和技术因素, 因此, 低成本、低能耗辅因子的再生和循环利用技术将刺激更多的研究工作; 或可探索新的无辅因子酶催化途径; (3)体外多酶级联催化转化CO2只有少数实例, 有必要设计更多的多酶路线或构建新的多酶催化系统, 从而进一步实现从CO2可持续生产燃料、化学品和高分子材料等高附加值产品; (4)酶法结合化学催化、光化学催化、电化学催化及新型纳米催化剂(例如多孔有机骨架[111]、MOF材料[112]等)协同作用将为CO2高值化转化和利用带来新的机遇.总之, 酶法催化转化CO2取得了一定的成效, 但还存在各种困难, 然而随着科学的发展和技术的进步, 各种技术瓶颈有望被个个突破, 从而开创酶法催化CO2制备高附加值化学品的新局面.
-
-
[1]
Air Products and Chemicals, Inc., Carbon Dioxide Product Stewardship Summary, www.airproducts.com, 2018.
-
[2]
Lei, Z.; Xue, Y.; Chen, W.; Qiu, W.; Zhang, Y.; Horike, S.; Tang, L. Adv. Energy Mater. 2018, 8, 1801587. doi: 10.1002/aenm.201801587
-
[3]
Zhang, Z.; Muschiol, J.; Huang, Y.; Sigurdardóttir, S. B.; von Solms, N.; Daugaard, A. E.; Wei, J.; Luo, J.; Xu, B.-H.; Zhang, S.; Pinelo, M. Green Chem. 2018, 20, 4339. doi: 10.1039/C8GC02230E
-
[4]
Sultana, S.; Chandra Sahoo, P.; Martha, S.; Parida, K. RSC Adv. 2016, 6, 44170. doi: 10.1039/C6RA05472B
-
[5]
常世磊, 梁风, 姚耀春, 马文会, 杨斌, 戴永年, 化学学报, 2018, 76, 515. doi: 10.11862/CJIC.2018.038Chang, S.-L.; Liang, F.; Yao, Y.-C.; Ma, W.-H.; Yang, B.; Dai, Y.-N. Acta Chim. Sinica 2018, 76, 515. doi: 10.11862/CJIC.2018.038
-
[6]
Olivier, J. G. J.; Peters, J. A. H. W. Trends in global CO2 and total greenhouse gas emissions:2018 Report, PBL Netherlands Environmental Assessment Agency, The Hague, 2018, 3125, pp. 4~6.
-
[7]
Shi, J.; Jiang, Y.; Jiang, Z.; Wang, X.; Wang, X.; Zhang, S.; Han, P.; Yang, C. Chem. Soc. Rev. 2015, 44, 5981. doi: 10.1039/C5CS00182J
-
[8]
张帅, 李雪冬, 何良年, 化学学报, 2016, 74, 17. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345299.shtmlZhang, S.; Li, X.-D.; He, L.-N. Acta Chim. Sinica 2016, 74, 17. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345299.shtml
-
[9]
Long, N.; Lee, J.; Koo, K.-K.; Luis, P.; Lee, M. Energies 2017, 10, 473. doi: 10.3390/en10040473
-
[10]
Chang, X.; Wang, T.; Yang, P.; Zhang, G.; Gong, J. Adv. Mater. 2018, 31, 1804710.
-
[11]
Chen, Z.; Wang, X.; Liu, L. Chem. Rec. 2019, 19, 1272. doi: 10.1002/tcr.201800100
-
[12]
Kuramochi, Y.; Ishitani, O.; Ishida, H. Coord. Chem. Rev. 2018, 373, 333. doi: 10.1016/j.ccr.2017.11.023
-
[13]
Aresta, M.; Dibenedetto, A.; Quaranta, E. In Reaction Mechanisms in Carbon Dioxide Conversion, Vol. 9, Eds.: Aresta, M.; Dibenedetto, A.; Quaranta, E., Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 347~371.
-
[14]
Mondal, B.; Song, J.; Neese, F.; Ye, S. Curr. Opin. Chem. Biol. 2015, 25, 103. doi: 10.1016/j.cbpa.2014.12.022
-
[15]
Aresta, M.; Quaranta, E.; Liberio, R.; Dileo, C.; Tommasi, I. Tetrahedron 1998, 54, 8841. doi: 10.1016/S0040-4020(98)00475-X
-
[16]
Obert, R.; Dave, B. C. J. Am. Chem. Soc. 1999, 121, 12192. doi: 10.1021/ja991899r
-
[17]
Marpani, F.; Pinelo, M.; Meyer, A. S. Biochem. Eng. J. 2017, 127, 217. doi: 10.1016/j.bej.2017.08.011
-
[18]
Fuchs, G. Annu. Rev. Microbiol. 2011, 65, 631. doi: 10.1146/annurev-micro-090110-102801
-
[19]
Berg, I. A.; Kockelkorn, D.; Ramos-Vera, W. H.; Say, R.; Zarzycki, J.; Fuchs, G. In Carbon Dioxide as Chemical Feedstock, Vol. 3, Ed.: Aresta, M., WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 33~53.
-
[20]
Alissandratos, A.; Easton, C. J. Beilstein J. Org. Chem. 2015, 11, 2370. doi: 10.3762/bjoc.11.259
-
[21]
Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi: 10.1128/AEM.05702-11
-
[22]
Tabita, F. R.; Hanson, T. E.; Li, H.; Satagopan, S.; Singh, J.; Chan, S. Microbiol. Mol. Biol. Rev. 2007, 71, 576. doi: 10.1128/MMBR.00015-07
-
[23]
Ljungdahl, L. G.; Wood, H. G. Annu. Rev. Microbiol. 1969, 23, 515. doi: 10.1146/annurev.mi.23.100169.002503
-
[24]
Ragsdale, S. W.; Kumar, M.; Seravalli, J.; Qiu, D.; Spiro, T. D. In Microbial Growth on C1 Compounds, Eds.: Lidstrom, M. E.; Tabita, F. R., Springer, Dordrecht, 1996, pp. 191~196.
-
[25]
Ragsdale, S. W.; Pierce, E. Biochim. Biophys. Acta 2008, 1784, 1873. doi: 10.1016/j.bbapap.2008.08.012
-
[26]
Gencic, S.; Duin, E. C.; Grahame, D. A. J. Biol. Chem. 2010, 285, 15450. doi: 10.1074/jbc.M109.080994
-
[27]
王洪杰, 倪俊, 张怡, 张玲, 辛越勇, 微生物学报, 2013, 40, 304.Wang, H.-J.; Ni, J.; Zhang, Y.; Zhang, L.; Xin, Y.-Y. Microbiol. China 2013, 40, 304.
-
[28]
Scherf, U.; Buckel, W. Eur. J. Biochem. 1993, 215, 421. doi: 10.1111/j.1432-1033.1993.tb18049.x
-
[29]
Berg, I. A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. Science 2007, 318, 1782. doi: 10.1126/science.1149976
-
[30]
Ishii, M.; Miyake, T.; Satoh, T.; Sugiyama, H.; Oshima, Y.; Kodama, T.; Igarashi, Y. Arch. Microbiol. 1996, 166, 368. doi: 10.1007/BF01682981
-
[31]
Zarzycki, J.; Brecht, V.; Müller, M.; Fuchs, G. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21317. doi: 10.1073/pnas.0908356106
-
[32]
Patel, H. M.; Kraszewski, J. L.; Mukhopadhyay, B. J. Bacteriol. 2004, 186, 5129. doi: 10.1128/JB.186.15.5129-5137.2004
-
[33]
Burgess, B. K.; Lowe, D. J. Chem. Rev. 1996, 96, 2983. doi: 10.1021/cr950055x
-
[34]
Shah, V. K.; Brill, W. J. Proc. Natl. Acad. Sci., U. S. A. 1977, 74, 3249. doi: 10.1073/pnas.74.8.3249
-
[35]
Yang, Z.-Y.; Danyal, K.; Seefeldt, L. C. In Nitrogen Fixation. Methods in Molecular Biology (Methods and Protocols), Vol. 766, Ed.: Ribbe, M. W., Humana Press, Heidelberg, 2011, pp. 9~29.
-
[36]
Rivera-Ortiz, J. M.; Burris, R. H. J. Bacteriol. 1975, 123, 537. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM1150625
-
[37]
Lee, C. C.; Hu, Y.; Ribbe, M. W. Science 2010, 329, 642. doi: 10.1126/science.1191455
-
[38]
Hu, Y.; Lee, C. C.; Ribbe, M. W. Science 2011, 333, 753. doi: 10.1126/science.1206883
-
[39]
Yang, Z.-Y.; Moure, V. R.; Dean, D. R.; Seefeldt, L. C. Proc. Natl. Acad. Sci., U. S. A. 2012, 109, 19644. doi: 10.1073/pnas.1213159109
-
[40]
Seefeldt, L. C.; Rasche, M. E.; Ensign, S. A. Biochemistry 1995, 34, 5382. doi: 10.1021/bi00016a009
-
[41]
Zheng, Y.; Harris, D. F.; Yu, Z.; Fu, Y.; Poudel, S.; Ledbetter, R. N.; Fixen, K. R.; Yang, Z.-Y.; Boyd, E. S.; Lidstrom, M. E.; Seefeldt, L. C.; Harwood, C. S. Nat. Microbiol. 2018, 3, 281. doi: 10.1038/s41564-017-0091-5
-
[42]
Rebelein, J. G.; Stiebritz, M. T.; Lee, C. C.; Hu, Y. Nat. Chem. Biol. 2016, 13, 147.
-
[43]
Khadka, N.; Dean, D. R.; Smith, D.; Hoffman, B. M.; Raugei, S.; Seefeldt, L. C. Inorg. Chem. 2016, 55, 8321. doi: 10.1021/acs.inorgchem.6b00388
-
[44]
Lindskog, S. Inorg. Chim. Acta 1983, 79, 36.
-
[45]
Savile, C. K.; Lalonde, J. J. Curr. Opin. Biotechnol. 2011, 22, 818. doi: 10.1016/j.copbio.2011.06.006
-
[46]
Smith, K. S.; Jakubzick, C.; Whittam, T. S.; Ferry, J. G. Proc. Natl. Acad. Sci., U. S. A. 1999, 96, 15184. doi: 10.1073/pnas.96.26.15184
-
[47]
蔡丽希, 楚云猛, 张光亚, 生物工程学报, 2019, 31, 1. http://www.cnki.com.cn/Article/CJFDTotal-SHWU201901003.htmCai, L.-X.; Chu, Y.-M.; Zhang, G.-Y. Chin. J. Biotech. 2019, 31, 1. http://www.cnki.com.cn/Article/CJFDTotal-SHWU201901003.htm
-
[48]
Lehtonen, J.; Shen, B.; Vihinen, M.; Casini, A.; Scozzafava, A.; Supuran, C.; Parkkila, A.-K.; Saarnio, J.; Kivel, A. J.; Waheed, A.; Sly, W.; Parkkila, S. J. Biol. Chem. 2004, 279, 2719. doi: 10.1074/jbc.M308984200
-
[49]
Loferer, M. J.; Tautermann, C. S.; Loeffler, H. H.; Liedl, K. R. J. Am. Chem. Soc. 2003, 125, 8921. doi: 10.1021/ja035072f
-
[50]
Shekh, A.; Kannan, K.; Mudliar, N. S.; Yadav, R.; Fulke, A.; Sivanesan, S. d.; Chakrabarti, T. Crit. Rev. Environ. Sci. Technol. 2011, 42, 1419.
-
[51]
Bond, G. M.; Stringer, J.; Brandvold, D. K.; Simsek, F. A.; Medina, M.-G.; Egeland, G. Energy Fuels 2001, 15, 309. doi: 10.1021/ef000246p
-
[52]
Yadav, R. R.; Krishnamurthi, K.; Mudliar, S. N.; Devi, S. S.; Naoghare, P. K.; Bafana, A.; Chakrabarti, T. J. Basic Microbiol. 2014, 54, 472. doi: 10.1002/jobm.201300849
-
[53]
Mirjafari, P.; Asghari, K.; Mahinpey, N. Ind. Eng. Chem. Res. 2007, 46, 921. doi: 10.1021/ie060287u
-
[54]
Xiao, L.; Lian, B. Carbonates Evaporites 2016, 31, 39. doi: 10.1007/s13146-015-0239-4
-
[55]
McQ Gould, S.; Tawfik, D. Biochemistry 2005, 44, 5444. doi: 10.1021/bi0475471
-
[56]
Alvizo, O.; Nguyen, L. J.; Savile, C. K.; Bresson, J. A.; Lakhapatri, S. L.; Solis, E. O. P.; Fox, R. J.; Broering, J. M.; Benoit, M. R.; Zimmerman, S. A.; Novick, S. J.; Liang, J.; Lalonde, J. J. Proc. Natl. Acad. Sci., U. S. A. 2014, 111, 16436. doi: 10.1073/pnas.1411461111
-
[57]
Yoshimoto, M.; Walde, P. World J. Microbiol. Biotechnol. 2018, 34, 151. doi: 10.1007/s11274-018-2536-2
-
[58]
刘文芳, 魏利娜, 分子催化, 2016, 30, 182.Liu, W.-F.; Wei, L.-N. J. Mol. Catal. 2016, 30, 182.
-
[59]
Yoshimoto, M.; Schweizer, T.; Rathlef, M.; Pleij, T.; Walde, P. ACS Omega 2018, 3, 10391. doi: 10.1021/acsomega.8b01517
-
[60]
Maeshima, K.; Yoshimoto, M. Enzyme Microb. Technol. 2017, 105, 9. doi: 10.1016/j.enzmictec.2017.06.002
-
[61]
崔建东, 李莹, 姬晓元, 边红杰, 张羽飞, 苏志国, 马光辉, 张松平, 高等学校化学学报, 2014, 35, 1999. doi: 10.7503/cjcu20140059Cui, J.-D.; Li, Y.; Ji, X.-Y.; Bian, H.-J.; Zhang, Y.-F.; Su, Z.-G.; Ma, G.-H.; Zhang, S.-P. Chem. J. Chin. Univ. 2014, 35, 1999. doi: 10.7503/cjcu20140059
-
[62]
Bulushev, D. A.; Ross, J. R. H. ChemSusChem 2018, 11, 821. doi: 10.1002/cssc.201702075
-
[63]
Kawanami, H.; Himeda, Y.; Laurenczy, G. Adv. Inorg. Chem. 2017, 70, 395. doi: 10.1016/bs.adioch.2017.04.002
-
[64]
Castillo, R.; Oliva, M.; Martí, S.; Moliner, V. J. Phys. Chem. B 2008, 112, 10012. doi: 10.1021/jp8025896
-
[65]
Neuhauser, W.; Steininger, M.; Haltrich, D.; Kulbe, K. D.; Nidetzky, B. Biotechnol. Bioeng. 1998, 60, 277. doi: 10.1002/(SICI)1097-0290(19981105)60:3<277::AID-BIT2>3.0.CO;2-E
-
[66]
Dong, G.; Ryde, U. J. Biol. Inorg. Chem. 2018, 23, 1243. doi: 10.1007/s00775-018-1608-y
-
[67]
Boyington, J. C.; Gladyshev, V. N.; Khangulov, S. V.; Stadtman, T. C.; Sun, P. D. Science 1997, 275, 1305. doi: 10.1126/science.275.5304.1305
-
[68]
Schrapers, P.; Hartmann, T.; Kositzki, R.; Dau, H.; Reschke, S.; Schulzke, C.; Leimkühler, S.; Haumann, M. Inorg. Chem. 2015, 54, 3260. doi: 10.1021/ic502880y
-
[69]
Mota, C. S.; Rivas, M. G.; Brondino, C. D.; Moura, I.; Moura, J. J. G.; González, P. J.; Cerqueira, N. M. F. S. A. J. Biol. Inorg. Chem. 2011, 16, 1255. doi: 10.1007/s00775-011-0813-8
-
[70]
Dobbek, H. Coord. Chem. Rev. 2011, 255, 1104. doi: 10.1016/j.ccr.2010.11.017
-
[71]
Parkinson, B. A.; Weaver, P. F. Nature 1984, 309, 148. doi: 10.1038/309148a0
-
[72]
Lu, Y.; Jiang, Z.-Y.; Xu, S.-W.; Wu, H. Catal. Today 2006, 115, 263. doi: 10.1016/j.cattod.2006.02.056
-
[73]
Yadav, R. K.; Baeg, J.-O.; Oh, G. H.; Park, N.-J.; Kong, K.-j.; Kim, J.; Hwang, D. W.; Biswas, S. K. J. Am. Chem. Soc. 2012, 134, 11455. doi: 10.1021/ja3009902
-
[74]
Choe, H.; Joo, J. C.; Cho, D. H.; Kim, M. H.; Lee, S. H.; Jung, K. D.; Kim, Y. H. PLoS One 2014, 9, e103111. doi: 10.1371/journal.pone.0103111
-
[75]
Yadav, R. K.; Baeg, J.-O.; Kumar, A.; Kong, K.-j.; Oh, G. H.; Park, N.-J. J. Mater. Chem. A 2014, 2, 5068. doi: 10.1039/c3ta14442a
-
[76]
Woolerton, T. W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A. J. Am. Chem. Soc. 2010, 132, 2132. doi: 10.1021/ja910091z
-
[77]
许辰宇, 林伽毅, 潘富强, 邓博文, 王智化, 周俊虎, 陈云, 马京程, 顾志恩, 张彦威, 化学学报, 2017, 75, 699. doi: 10.11862/CJIC.2017.051Xu, C.-Y.; Lin, J.-Y.; Pan, F.-Q.; Den, B.-W.; Wang, Z.-H.; Zhou, J.-H.; Chen, Y.; Ma, J.-C.; Gu, Z.-E.; Zhang, Y.-W. Acta Chim. Sinica 2017, 75, 699. doi: 10.11862/CJIC.2017.051
-
[78]
Jeoung, J.-H.; Martins, B. M.; Dobbek, H. In Metalloproteins: Methods and Protocols, Vol. 3, Ed.: Hu, Y., Springer, New York, 2019, pp. 37~54.
-
[79]
Appel, A. M.; Bercaw, J. E.; Bocarsly, A. B.; Dobbek, H.; DuBois, D. L.; Dupuis, M.; Ferry, J. G.; Fujita, E.; Hille, R.; Kenis, P. J. A.; Kerfeld, C. A.; Morris, R. H.; Peden, C. H. F.; Portis, A. R.; Ragsdale, S. W.; Rauchfuss, T. B.; Reek, J. N. H.; Seefeldt, L. C.; Thauer, R. K.; Waldrop, G. L. Chem. Rev. 2013, 113, 6621. doi: 10.1021/cr300463y
-
[80]
Parkin, A.; Seravalli, J.; Vincent, K. A.; Ragsdale, S. W.; Armstrong, F. A. J. Am. Chem. Soc. 2007, 129, 10328. doi: 10.1021/ja073643o
-
[81]
Jeoung, J.-H.; Dobbek, H. Science 2007, 318, 1461. doi: 10.1126/science.1148481
-
[82]
Miyazaki, S.; Koga, Y.; Matsumoto, T.; Matsubara, K. Chem. Commun. 2010, 46, 1932. doi: 10.1039/b924716e
-
[83]
伍一洲, 石家福, 丁菲, 赵晶晶, 邹晓燕, 王曼茹, 张少华, 佟振伟, 张松平, 姜忠义, 中国科学:化学, 2017, 47, 315.Wu, Y.-Z.; Shi, J.-F.; Ding, F.; Zhao, J.-J.; Zou, X.-Y.; Wang, M.-R.; Zhang, S.-H.; Tong, Z.-W.; Zhang, S.-P.; Jiang, Z.-Y. Sci. Sin. Chim. 2017, 47, 315.
-
[84]
Shin, W.; Lee, S. H.; Shin, J. W.; Lee, S. P.; Kim, Y. J. Am. Chem. Soc. 2003, 125, 14688. doi: 10.1021/ja037370i
-
[85]
Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi: 10.1039/B807875K
-
[86]
Allen, J. R.; Ensign, S. A. J. Bacteriol. 1996, 178, 1469. doi: 10.1128/jb.178.5.1469-1472.1996
-
[87]
Boll, M.; Fuchs, G. Biol. Chem. 2005, 386, 989. doi: 10.1515/BC.2005.115
-
[88]
Huang, J.; He, Z.; Wiegel, J. J. Bacteriol. 1999, 181, 5119.
-
[89]
Wieser, M.; Yoshida, T.; Nagasawa, T. J. Mol. Catal. B:Enzym. 2001, 11, 179. doi: 10.1016/S1381-1177(00)00038-2
-
[90]
Miyazaki, M.; Shibue, M.; Ogino, K.; Nakamura, H.; Maeda, H. Chem. Commun. 2001, 18, 1800.
-
[91]
Tong, X.; El-Zahab, B.; Zhao, X.; Liu, Y.; Wang, P. Biotechnol. Bioeng. 2011, 108, 465. doi: 10.1002/bit.22938
-
[92]
刘文芳, 侯延慧, 侯本象, 赵之平, 中国化学工程学报, 2014, 22, 1328.Liu, W.-F.; Hou, Y.-H.; Hou, B.-X.; Zhao, Z.-P. Chinese J. Chem. Eng. 2014, 22, 1328.
-
[93]
刘文芳, 侯本象, 侯延慧, 赵之平, 催化学报, 2012, 33, 730.Liu, W.-F.; Hou, B.-X.; Hou, Y.-H.; Zhao, Z.-P. Chinese J. Catal. 2012, 33, 730.
-
[94]
Li, R.; Wang, Z.; Ni, P.; Zhao, Y.; Li, M.; Li, L. Fuel 2014, 128, 180. doi: 10.1016/j.fuel.2014.03.011
-
[95]
Jadhav, S. G.; Vaidya, P. D.; Bhanage, B. M.; Joshi, J. B. Chem. Eng. Res. Des. 2014, 92, 2557. doi: 10.1016/j.cherd.2014.03.005
-
[96]
Goeppert, A.; Czaun, M.; Jones, J.-P.; Surya Prakash, G. K.; Olah, G. A. Chem. Soc. Rev. 2014, 43, 7995. doi: 10.1039/C4CS00122B
-
[97]
Wang, W.-H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; Fujita, E. Chem. Rev. 2015, 115, 12936. doi: 10.1021/acs.chemrev.5b00197
-
[98]
Kuwabata, S.; Tsuda, R.; Yoneyama, H. J. Am. Chem. Soc. 1994, 116, 5437. doi: 10.1021/ja00091a056
-
[99]
Baskaya, F. S.; Zhao, X.; Flickinger, M. C.; Wang, P. Appl. Biochem. Biotechnol. 2010, 162, 391. doi: 10.1007/s12010-009-8758-x
-
[100]
Xu, S.-W.; Lu, Y.; Li, J.; Jiang, Z.-Y.; Wu, H. Ind. Eng. Chem. Res. 2006, 45, 4567. doi: 10.1021/ie051407l
-
[101]
Sun, Q.; Jiang, Y.; Jiang, Z.; Zhang, L.; Sun, X.; Li, J. Ind. Eng. Chem. Res. 2009, 48, 4210. doi: 10.1021/ie801931j
-
[102]
Jiang, Y.; Sun, Q.; Zhang, L.; Jiang, Z. J. Mater. Chem. 2009, 19, 9068. doi: 10.1039/b914268a
-
[103]
Wang, X.; Li, Z.; Shi, J.; Wu, H.; Jiang, Z.; Zhang, W.; Song, X.; Ai, Q. ACS Catal. 2014, 4, 962. doi: 10.1021/cs401096c
-
[104]
Aresta, M.; Dibenedetto, A.; Baran, T.; Angelini, A.; Labuz, P.; Macyk, W. Beilstein J. Org. Chem. 2014, 10, 2556. doi: 10.3762/bjoc.10.267
-
[105]
El-Zahab, B.; Donnelly, D.; Wang, P. Biotechnol. Bioeng. 2008, 99, 508. doi: 10.1002/bit.21584
-
[106]
Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. ACS Nano 2015, 9, 4600. doi: 10.1021/acsnano.5b01278
-
[107]
Singh, R. K.; Singh, R.; Sivakumar, D.; Kondaveeti, S.; Kim, T.; Li, J.; Sung, B. H.; Cho, B.-K.; Kim, D. R.; Kim, S. C.; Kalia, V. C.; Zhang, Y.-H. P. J.; Zhao, H.; Kang, Y. C.; Lee, J.-K. ACS Catal. 2018, 8, 11085. doi: 10.1021/acscatal.8b02646
-
[108]
Calvin, M.; Massini, P. Experientia 1952, 8, 445. doi: 10.1007/BF02139287
-
[109]
Wendell, D.; Todd, J.; Montemagno, C. Nano Lett. 2010, 10, 3231. doi: 10.1021/nl100550k
-
[110]
Atsumi, S.; Higashide, W.; Liao, J. C. Nat. Biotechnol. 2009, 27, 1177. doi: 10.1038/nbt.1586
-
[111]
王维, 闫卓君, 元野, 孙福兴, 赵明, 任浩, 朱广山, 化学学报, 2014, 72, 557. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344408.shtmlWang, W.; Yan, Z.-J.; Yuan, Y.; Sun, F.-X.; Zhao, M.; Ren, H.; Zhu, G.-S. Acta Chim. Sinica 2014, 72, 557. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344408.shtml
-
[112]
贾江涛, 王蕾, 赵晴, 孙福兴, 朱广山, 化学学报, 2013, 71, 1492. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract343577.shtmlJia, J.-T.; Wang, L.; Zhao, Q.; Sun, F.-X.; Zhu, G.-S. Acta Chim. Sinica 2013, 71, 1492. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract343577.shtml
-
[1]
-


图 2 CO2胞内转化机理示意图. (A)卡尔文循环; (B)还原性柠檬酸循环; (C)还原性乙酰辅酶A途径; (D) 3-羟基丙酸二循环; (E)二羧酸/4-羟基丁酸循环(a)和3-羟基丙酸/4-羟基丁酸循环(b)[7, 18~20]
Figure 2 Schematics of CO2 metabolic process in cells. (A) Calvin cycle; (B) Reductive citric acid cycle; (C) Reductive acetyl-CoA route; (D) 3-Hydroxypropionate bi-cycle; (E) Dicarboxylate/4-hydroxybutyrate cycle (a) and 3-hydroxypropionate/4-hydroxybutyrate cycle (b) [7, 18~20]
图 3 (A) 固氮酶钼铁蛋白辅因子及其关键氨基酸残基; (B)天然和重组钼铁蛋白催化CH4形成与时间的关系; (C) α-70Ala/α-195Gln钼铁蛋白催化CO2和C2H2产丙烯与C2H2分压的关系[39]; (D) R. palustris表达的天然唯铁固氮酶催化CO2产CH4代谢途径: (a)硫代硫酸盐(S2O32–)氧化供电子, CO2来源于重碳酸盐; (b) CH3COO–氧化供电子, CO2来源于乙酸[41]; (E)几种状态下的铁蛋白固氮酶VnfH和NifH体外还原CO2生成CO[42]
Figure 3 Nitrogenase MoFe cofactor with some key amino acid residues; (B) CH4 formation as a function of time with natural or remodeled MoFe proteins; (C) The relationship between propylene formation and C2H2 partial pressure that catalyzed by α-70Ala/α-195Gln MoFe protein[39]; (D) Metabolic routes of CH4 production by R. palustris expressing wild-type Fe-only nitrogenase: electrons are originated from oxidation of thiosulfate (a) or acetate (b), and CO2 are generated from bicarbonate (a) or the oxidation of acetate (b)[41]; (E) In vitro reduction of CO2 to CO by vnfH or NifH in different forms[42]
图 4 (A) 几种CA晶体结构示意图(a. α型; b. β型; c. γ型; d. ζ型)[45]; (B)异源表达CA及其催化CO2形成CaCO3示意图[54]; (C)定向进化提高CA稳定性(a.每轮定性进化后DvCA酶综合特性改善倍数; b. 8次定向进化后DvCA在4.2 mol/L MDEA中的储藏稳定性)[56]; (D) CA固定化示意图(a.枝状聚合物共轭固定CA; b.脂质体生物膜共轭固定CA)[57, 59, 60]
Figure 4 (A) Depiction of the crystal structures of CA from different families, a~d represent the types of α, β, γ and ζ, respectively[45]; (B) Schematic illustration of heterologously expressed CA for promoting CaCO3 formation[54]; (C) CA stability enhancement by direct evolution: (a) Compounded fold improvement after each round of evolution; (b) The storage stability of DvCA8.0 (8 rounds evolution) when exposed to 4.2 mol/L MDEA[56]; (D) Diagrams showing the immobilization of carbonic anhydrase: (a) Dendronized polymer-CA conjugates; (b) Lipid bilayer membranes-conjugated CA[57, 59, 60]
图 5 (A) FDH催化CO2甲酸化及其逆反应[64]; (B) Mo-FDH催化活性位点结构示意图(a.硫桥聚焦视角; b. Cys196基团聚焦视角)[66]; (C) CCGCMAQSP人工光催化还原CO2产甲酸及其NADH再生过程示意图; (D)几种催化剂在NADH再生系统中还原CO2产甲酸化的光催化活性(a. NADH再生速率; b. HCOOH产量)[73]; (E) BODIPY, CCG-BODIPY, NH2-TPP以及CCG-NH-TPP的光催化活性(a. NADH再生速率; b. HCOOH产量)[75]
Figure 5 (A) Sketch of FDH for catalysis of CO2 to produce HCOOH and the reverse reaction[64]; (B) The active site of the Mo-FDH in two views: focusing on the sulfido (a) and Cys196 (b) groups, respectively[66]; (C) CCGCMAQSP catalyzed artificial photosynthesis of HCOOH from CO2 and the photoregeneration of NADH under visible light; (D) Photocatalytic activities of various catalysts for synthesis of HCOOH from CO2 in the NADH photoregeneration system (a. Yield of NADH; b. HCOOH production)[73]; (E) Photocatalytic activities of BODIPY, CCG-BODIPY, NH2-TPP and CCG-NH-TPP (a. Yield of NADH; b. HCOOH production)[75]
图 6 (A) [NiFe]CODH活性中心球棍模型; (B) [NiFe]CODH催化CO2生成CO机理[79]; (C) [NiFe]CODH吸附固定于热解石墨边缘电极快速催化CO2产CO示意图[80]; (D)钌基金半导体光敏催化剂耦合[NiFe]CODH人工光催化还原CO2示意图[76]
Figure 6 (A) Ball-and-stick drawing of the active site of [NiFe]CODH; (B) Mechanism for reduction of CO2 to CO by [NiFe]CODH[79]; (C) [NiFe]CODH anchored on a pyrolytic graphite "edge" electrode for catalysis of rapid conversion of CO2 to CO[80]; (D) Schematic illustration of the CO2 photoreduction system using [NiFe]CODH attached to a Ru-based semiconductor photocatalyst[76]
图 7 (A) Xanthobacter Py2细胞提取物或全细胞催化环氧化物与CO2羧基化; (B) (a) Thauera aromatica细胞提取物或部分纯化的磷酸苯酯酶催化苯酚与CO2羧化反应; (b) Clostridium hydroxybenzoicum区域选择全细胞催化非活化酚对位羧基化; (C)吡咯-2-羧酸脱羧酶催化吡咯与CO2的羧基化反应; (D)丙酮酸脱羧酶催化乙醛与CO2的羧基化反应[85~87, 89~91]
Figure 7 (A) Biocatalytic carboxylation of epoxides with CO2 by the cell extracts or whole-cell of Xanthobacter Py2; (B) (a) Carboxylation of phenol by cell extracts of Thauera aromatica or partially purified phenylesterase; (b) Regioselective para-carboxylation of non-activated phenolic compounds catalyzed by whole-cell of Clostridium hydroxybenzoicum; (C) Carboxylation of pyrrole with CO2 by pyrrole-2-carboxylate decarboxylase; (D) Carboxylation of acetaldehyde with CO2 by pyruvate decarboxylase[85~87, 89~91]
图 8 (A) NADH介导的脱氢酶(FDH、FaldDH、ADH)级联催化CO2产甲醇示意图[16, 104]; (B) (a)一种基于珠内胶囊支架的多酶级联固定化体系的构建; (b)几种酶催化体系甲醇产量与时间的关系[102]; (C)基于超薄复合杂化微胶囊的多酶固定化体系级联催化CO2产甲醇[103]
Figure 8 (A) Schematic diagram of NADH-mediated multi-dehydrogenases (FDH, FaldDH and ADH) for cascade catalysis of CO2 to produce methanol[16, 104]; (B) (a) Capsules-in-bead scaffold for construction of multienzyme cascade system; (b) Plot of methanol formation as a function of reaction time in various enzymatic catalytic systems [102]; (C) Multienzyme cascade system based on ultrathin hybrid microcapsule for conversion of CO2 to methanol[103]
图 9 (A) NADH原位再生驱动三种脱氢酶级联催化CO2产甲醇示意图(GDH:谷氨酸脱氢酶)[105]; (B) (a)同轴静电纺丝技术制备的阳离子聚电解质掺杂中空纳米纤维系留NADH及其级联催化CO2产甲醇示意图; (b)三种中空纳米纤维系留NADH介导的多酶级联催化系统甲醇产率随反应时间的变化曲线(空心点为对应的游离酶系统)[106]; (C) (a)基于PTDH再生辅因子介导的无细胞ClFDH-BmFaldDH-YADH多酶级联体系催化CO2产甲醇示意图; 不同的多酶级联体系催化CO2产甲醇产量(b)及总转换数(c)与时间关系; (蓝: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); 红: ClFDH+BmFaldDH+YADH with PTDH; 紫: CbFDH+BmFaldDH+YADH with PTDH; 绿: ClFDH+PpFaldDH+YADH with PTDH; 黑: CbFDH+PpFaldDH+YADH with PTDH)[107]
Figure 9 (A) Schematic representation of enzymatic synthesis of methanol from CO2 with in situ regeneration of NADH (GDH: glutamate dehydrogenase)[105]; (B) (a) Schematic illustration of coaxial electrospinning for construction of cationic polyelectrolyte-doped hollow nanofiber tethering of NADH for methanol synthesis from CO2; (b) Plots of methanol yield as a function of reaction time for methanol synthesis from CO2 by using different multienzyme systems (Open legends represent results from free multienzyme systems, and the filled legends represent results from the hollow nanofiber-supported multienzymes systems, respectively)[106]; (C) Schematic illustration of reduction of CO2 into methanol using multi-enzymatic cascade system with NADH regeneration by PTDH (a); Time profile of methanol production (b) and total turnover number (c) by multi-enzymatic cascade reactions (blue: ClFDH+BmFaldDH+YADH with PTDH and EMIM-Ac (1%); red: ClFDH+BmFaldDH+YADH with PTDH; purple: CbFDH+BmFaldDH+YADH with PTDH; green: ClFDH+PpFaldDH+YADH with PTDH; black: CbFDH+PpFaldDH+YADH with PTDH)[107]
图 10 Synechococcus elongatus PCC7942蓝藻工程菌胞内多酶催化CO2合成异丁醛(A.反应过程示意图; B.蓝细菌SA590生产异丙醛时间-产量曲线; C. RuBisCO过表达蓝细菌SA665生产异丙醛时间-产量曲线)[110]
Figure 10 Intracellular multi-enzymatic synthesis of isobutyraldehyde from CO2 in an engineering cyanobacterium Synechococcus elongatus PCC7942. (A) The pathway for isobutyraldehyde production. Isobutyraldehyde production as a function of time by cyanobacteria SA590 (B), and SA665 with enhanced Rubisco (C), respectively[110]
-

 下载:
下载:




 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 106
- 文章访问数: 5058
- HTML全文浏览量: 1265
 下载:
下载:
