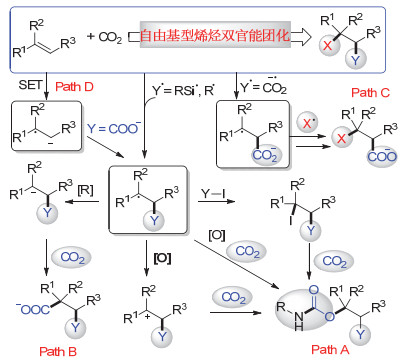
图 1.
二氧化碳参与的自由基型烯烃双官能团化反应
Figure 1.
Radical-type difunctionalization of alkenes with CO2

二氧化碳(CO2)是空气的重要组分, 也是重要的C1资源.由于其无毒、廉价易得、储量丰富且可循环再生等特点, CO2是合成化学中的理想C1合成子[1].利用CO2参与化学转化, 通过形成C—C和C—O等化学键, 可以高效构筑一系列具有良好生物活性的分子骨架和功能材料等具有高附加值的产品, 具有潜在的工业应用前景.因此, 基于CO2这一C1合成子的化学转化一直备受关注[2]并取得了一系列的成果:例如, 通过CO2与过渡金属催化[3]、光催化[4]、电催化[5]、碱促进[6]、离子液体等的结合[7], 实现多种羧基化、羰基化等产物的合成; 通过对二氧化碳的还原, 实现化合物甲基化、甲酰化[8]的转化或直接将二氧化碳转化为甲酸[9]等能源性物质; 更奇特的是, 利用二氧化碳的特殊性质, 将二氧化碳作为化学辅助试剂, 实现化合物的氧化、官能团化[10].
另一方面, 烯烃是有机合成中最重要的合成前体之一, 取代类型多样, 且来源广泛, 易于制备.此外, 烯烃参与的官能团化反应种类繁多, 且选择性可以调控, 易于快速合成具有重要官能团的分子, 是有机合成化学的重要工具[11].其中, CO2作为C1合成子参与的烯烃官能团化反应, 可以直接合成羧酸、噁唑啉酮和醇类等重要有机化合物.近年来人们在该领域进行了大量研究, 开发了多样的催化体系, 如过渡金属催化、光催化、电催化和有机小分子催化等[12].
CO2参与的烯烃官能团化反应主要分为烯烃单官能团化反应和双官能团化反应.其中, CO2参与的烯烃单官能团化反应最为常见, 主要包括发展较为成熟的氢羧基化反应[13]和还原羟甲基化[14]等反应.与之相比, CO2参与的烯烃双官能团化反应, 例如碳羧基化反应、硫羧基化反应和硅羧基化反应等, 可以同时构建多个化学键, 形成结构多样、高度官能团化的有机化合物, 具有更高的价值.
然而, 相比于烯烃单官能团化反应而言, CO2参与的烯烃双官能团化反应报道相对较少, 具有较大的挑战性.首先, 由于其热力学稳定性和动力学惰性, CO2是一种反应活性较低的亲电试剂, 不易直接与烯烃反应, 甚至和一些有机金属试剂都不容易反应(如:需要高温高压促进芳基钯物种和CO2反应), 因此需要开发高活性体系活化烯烃(如:将烯烃转化为高活性的烷基金属物种或者碳负离子[12d, 12e, 13], 进而进攻CO2)或者CO2(如:通过单电子还原CO2形成自由基负离子, 然后直接进攻烯烃[15]); 其次, CO2参与的烯烃双官能团化反应属于多组分反应, 还存在着诸多选择性问题: (1)化学选择性.如前所述, CO2参与的烯烃氢羧基化反应是CO2参与的烯烃官能团化反应中研究最多的形式.该反应主要经由烯烃对金属氢物种的插入形成亲核性的有机金属物种, 进而对CO2进攻得到羧酸衍生物.相比而言, 烯烃对其他金属物种中的碳金属键或者杂原子金属键进行插入, 进而与CO2反应的研究较少, 其原因可能在于:类似于烯烃的碳碳双键, CO2中的碳氧双键也可以与反应体系中的金属物种直接发生背景反应(如迁移插入反应、氧化环金属化反应等); (2)区域选择性.在CO2参与的烯烃双官能团化反应中, 除了使用结构对称的烯烃或者发生双羧基化反应外, 其他反应类型均存在成键的区域选择性问题; (3)对映选择性.相比于化学选择性和区域选择性, CO2参与的烯烃双官能团化反应中的对映选择性控制则更加困难.尽管有很多尝试, CO2参与的不对称碳碳键形成反应还存在巨大挑战, 目前能够实现高对映选择性的例子还很少.
综上所述, CO2参与的烯烃双官能团化反应存在诸多挑战.尽管如此, 人们通过大量尝试, 开发了一些新型策略, 尤其是通过近年来蓬勃发展的自由基化学, 利用高活性的自由基参与反应, 在相对温和的条件下实现了多种CO2参与的烯烃双官能团化反应.根据具体历程, 我们将CO2参与的自由基型烯烃双官能团化反应分为四类: (1)自由基进攻烯烃形成新的碳自由基中间体, 随后被单电子氧化形成碳正离子, 再进一步被碳酸盐或者氨基甲酸盐捕获, 或者直接发生氧化型的碳氧成键反应, 又或者先发生自由基攫碘形成烷基碘, 进而被氨基甲酸盐捕获形成碳氧键(图 1, path A); (2)自由基进攻烯烃形成新的碳自由基中间体后, 被单电子还原为碳负离子, 进而进攻CO2形成碳碳键(图 1, path B); (3) CO2在强还原体系中被还原为二氧化碳自由基负离子, 进攻烯烃形成较为稳定的碳自由基中间体, 进一步形成碳碳键或者碳杂原子键(图 1, path C); (4)烯烃在强还原体系中被单电子还原为烯基自由基负离子, 随后进攻CO2形成较为稳定的碳自由基中间体, 进一步被单电子还原为碳负离子, 最后进攻第二分子的CO2形成丁二酸类衍生物(图 1, path D).本文将重点围绕最近几年CO2参与的这几类自由基化学历程的烯烃双官能团化反应进行总结, 并对该领域的进一步发展进行展望.
噁唑啉酮广泛存在于药物分子当中.例如, 利奈唑酮(Linezolid)、美他沙酮(Skelaxin)和佐米曲普坦(Zolmitriptan)等都含有噁唑啉酮结构.另外, 噁唑啉酮也是有机合成化学中广泛使用的反应中间体和手性辅基[16].对于此类化合物的制备传统上多采用光气、氨基甲酸酯等与β-氨基乙醇、β-卤代乙醇、1, 2-乙二醇等反应.而鉴于该类化合物的重要性, 有必要开拓更为多样有效的制备方式, 其中, 通过烯丙胺与CO2实现烯烃双键的双官能团化反应制备噁唑啉酮不仅能够有效地利用CO2这一优良的C1合成子, 同时烯丙胺合成方法众多, 可以更好的实现噁唑啉酮的多样化制备.但是通过烯丙胺与CO2实现烯烃双键的双官能团化反应制备噁唑啉酮的方法目前还很有限, 取代类型单一, 不能满足药物开发对多样性的需求.针对该问题, 近年来人们通过自由基化学, 利用自由基前体、烯丙胺与CO2参与的三组分反应成功实现噁唑啉酮合成的同时, 可以引入多种类型的取代基团, 具有重要的合成价值.
2016年, 我们课题组[17]以Togni试剂为自由基前体, 首次报道了铜催化的CO2参与的烯丙胺的氧-三氟甲基化反应, 得到了一系列含CF3基团的重要噁唑啉酮类化合物(图 2).在这类多组分反应中, 主要存在着以下挑战: (1)氮-三氟甲基化与氧-三氟甲基化之间的化学选择性. Sodeoka和Cho分别报道了在温和条件下三氟甲基化试剂参与的烯丙胺中碳碳双键的氮三氟甲基化反应.反应中如何有效地生成氨基甲酸中间体并优先于C—N键形成C—O键, 对该反应至关重要; (2) endo和exo环化之间的区域选择性.除化学选择性外, 环化步骤必须具有区域选择性才能以良好的收率得到单一杂环; (3)产物的稳定性问题. CF3的α位上的H具有较强的酸性, 碱性条件下可能会导致2-噁唑啉酮通过脱除一分子CO2而发生分解反应.为克服以上挑战, 我们组经过大量条件筛选后, 成功的在101 kPa的CO2氛围中和室温下高选择性地实现了CO2参与的烯丙胺的氧-三氟甲基化反应.该反应底物范围广、官能团兼容性好、条件温和、整体反应收率高, 且能够实现克级规模反应, 产品易于衍生.
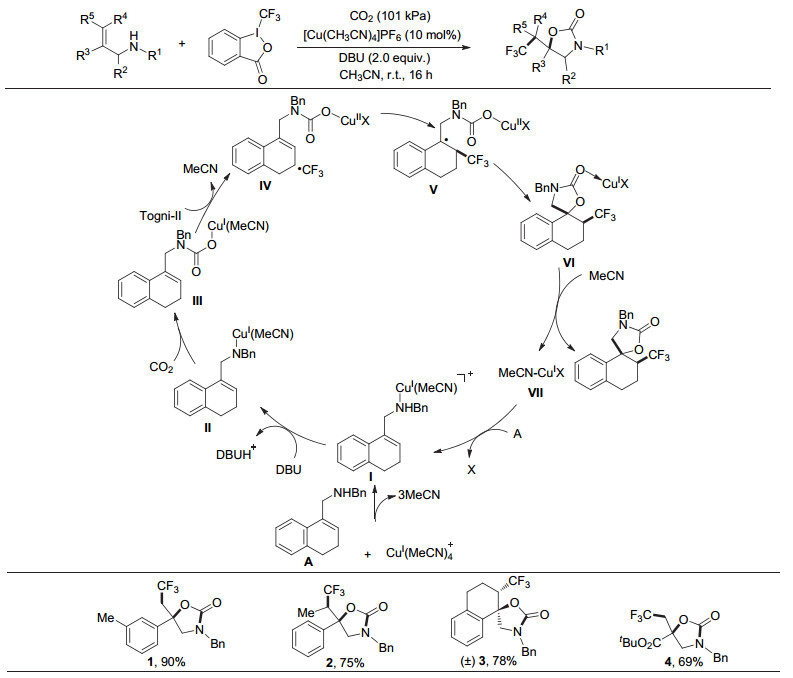
随后, 为了对该反应有更为深入的理解, 我们课题组与蓝宇课题组[18]合作, 通过密度泛函理论(DFT)计算详细地研究了该反应的机理.研究结果表明, 烯丙胺A中的N原子与Cu(Ⅰ)催化剂前体配位形成络合物Ⅰ, 该络合物在碱的作用下失去质子形成络合物Ⅱ.计算证明去质子这一步为整个催化循环的决速步.随后, CO2插入到N—Cu键中形成中间体Ⅲ, 该中间体中的亚铜离子进一步与亲电性的Togni试剂发生单电子转移并形成氨基甲酸铜(Ⅱ)中间体Ⅳ和游离的三氟甲基自由基.该三氟甲基自由基可进一步进攻烯烃并形成稳定的苄基自由基物种Ⅴ, 经过分子内的自由基-自由基偶联得到螺环铜物种Ⅵ, Ⅵ经过配体交换最终得到氧-三氟甲基化产物并释放出活性Cu(Ⅰ)中间体Ⅰ.另外, 计算表明, 缺电子Cu(Ⅱ)中心和亲电的三氟甲基自由基不能反应产生铜(Ⅲ)中间体; 前线分子轨道理论分析表明, 与Togni试剂Ⅱ发生单电子转移的是中性的Cu(Ⅰ)物种, 而不是阳离子Cu(Ⅰ)物种; 此外, 非对映选择性可能源于大位阻的三氟甲基和羰基部分之间的空间位阻排斥.
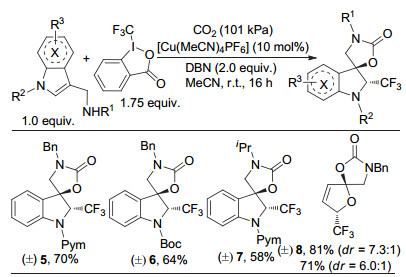
在此基础上, 我们课题组和蓝宇课题组[19]又进一步合作实现了铜催化的CO2参与的自由基型去芳构化反应(图 3).多种吲哚和呋喃可以发生氧-三氟甲基化的去芳构化反应, 并以中等到较高的收率及较好的dr值得到重要C-3螺环二氢吲哚和不饱和螺环缩醛.值得强调的是, 对于吲哚甲胺类底物, 吲哚氮上不同取代基对反应有较大影响.当该取代基为吸电子基团时, 反应才可以较好的进行.其中, 嘧啶基取代的底物反应性最好.该反应官能团兼容性较好, 常见的官能团, 如卤素(氟、氯、溴)、醚、硝基和酯基等, 都能很好的兼容.此外, 糠胺类衍生物的反应不仅可以构建双五元环螺缩醛, 还可以构建五六元环螺缩醛.
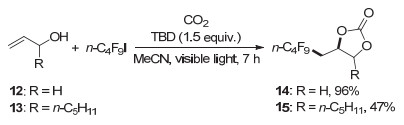
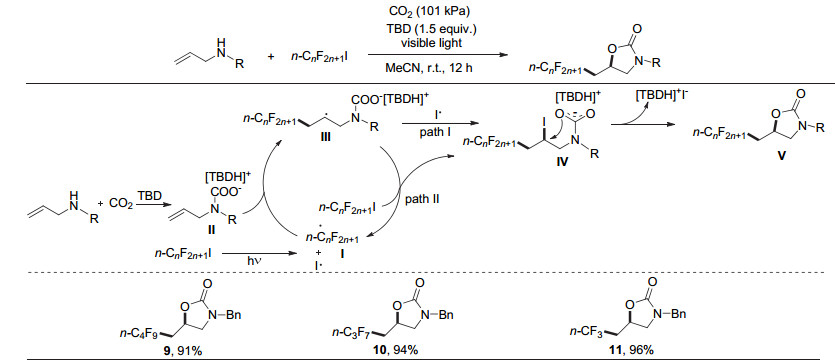
2017年, 何良年课题组[20]使用全氟烷基碘作为自由基前体, 1, 5, 7-三氮杂二环[4.4.0]癸-5-烯(TBD)作碱, 首次实现了可见光驱动CO2参与的烯丙胺的氧-全氟烷基化反应, 高效构建噁唑啉酮的同时引入重要的全氟烷基(图 4).该反应条件温和, 无需过渡金属.全氟烷基碘试剂由于活性较高, 可以直接在光照下均裂, 因此无需加额外的光敏剂.另外, 该类反应也可以使用烯丙醇作为底物(图 5), 得到全氟烷基化的环碳酸酯.
机理方面, 该反应首先通过可见光引发全氟烷基碘(如n-C4F9I)发生C—I键的均裂, 产生碘自由基和全氟烷基自由基, 随后全氟烷基自由基对原位形成的氨基甲酸盐中的碳碳双键加成, 得到全氟烷基取代的仲碳自由基物种Ⅲ, 该物种随后被碘自由基或全氟烷基碘代物捕获, 形成关键的碘代全氟烷基化氨基甲酸盐中间体Ⅳ.最后, 经分子内亲核环化得到全氟烷基化的噁唑啉酮Ⅴ.
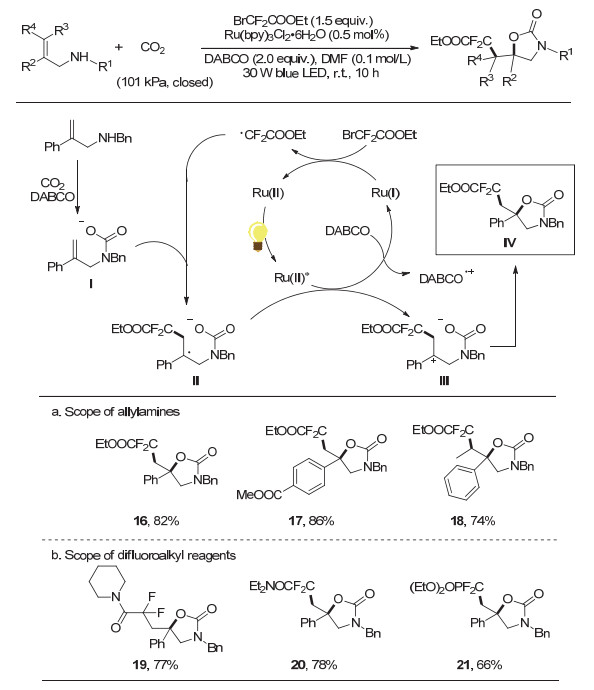
2018年, 在实现了铜催化烯丙胺的氧-三氟甲基化反应的基础上, 我们课题组考虑到可见光催化是实现类似自由基化学的重要方法, 进一步发展了可见光催化CO2参与的烯丙胺的氧-二氟烷基化反应[21](图 6), 为各种重要的二氟烷基取代噁唑啉酮及其衍生物的制备提供了新途径, 具有良好的官能团兼容性、广泛的底物范围、可克级规模化、反应条件温和环境友好等特点.
机理研究表明, 反应体系中的三乙烯二胺(DABCO)对激发态的光催化剂Ru(Ⅱ)*进行还原猝灭产生Ru(Ⅰ).作者认为, 该Ru(Ⅰ)可以单电子还原BrCF2- COOEt形成二氟烷基自由基.该自由基与氨基甲酸盐Ⅰ反应产生苄基自由基Ⅱ, 随后苄基自由基Ⅱ被激发态的Ru(Ⅱ)氧化形成苄基正离子Ⅲ, 然后进行分子内环化得到目标产物Ⅳ.
2017年, 我们课题组[22]报道了可见光驱动钯催化C—H键与非活化的烷基溴代物的自由基型烷基化反应.在该反应中, 我们推测活性钯(0)络合物可与叔丁基溴在光照下发生单电子转移(SET)过程, 产生叔丁基自由基.考虑到如前所述的几例自由基型噁唑啉酮合成方法都需要使用活性较高的亲电试剂, 不能适用于活性较低的普通烷基卤代物, 我们课题组[23]在2018年进一步实现了CO2参与的可见光驱动钯催化的烯丙胺和非活化烷基溴代物的氧-烷基化反应(图 7).该三组分反应以Pd(PPh3)4为催化剂, 反应条件温和, 底物适用范围广且产物易于衍生.反应中各种非活化的烷基溴代物都可以参与反应, 生成重要的2-噁唑啉酮类化合物.总体而言, 一级烷基溴代物的反应性比二、三级烷基溴代物的反应性差, 可能与相应烷基自由基的稳定性有关.
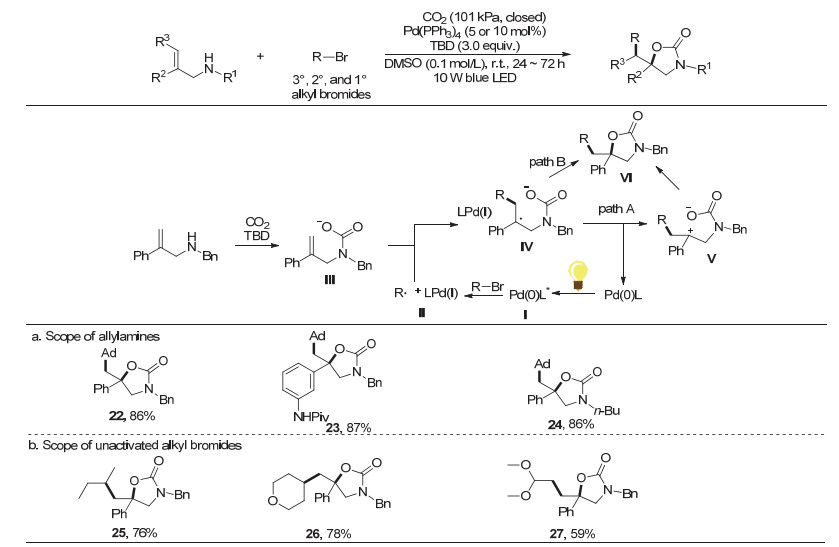
机理研究表明, 该反应历程中有烷基自由基的存在.作者认为, 该反应首先通过光激发Pd(0)催化剂引发催化循环.激发态的Pd(0)络合物Ⅰ和烷基溴代物经过单电子转移产生相应的Pd(Ⅰ)络合物和烷基自由基Ⅱ.同时, 底物烯丙胺在TBD存在下与CO2原位形成氨基甲酸盐Ⅲ.随后, 烷基自由基Ⅱ与氨基甲酸盐Ⅲ中的烯烃加成产生更稳定的苄基自由基物种Ⅳ.接着, Pd(I)物种和中间体Ⅳ经单电子转移生成中间体Ⅴ, 同时再生Pd(0)催化剂(path A), 最后中间体Ⅴ发生分子内亲核反应得到所需的噁唑啉酮Ⅵ.我们认为, 中间体Ⅳ也可能经历类似于图 2所示的自由基-自由基偶联, 直接形成碳氧键并重生Pd(0)催化剂(path B, 图 7).
如前所述, CO2参与的芳基烯烃羧基化反应以氢羧基化为主, 近年来人们通过自由基化学成功实现了双官能团化的α羧基化反应, 主要包括碳羧基化和硅羧基化两类反应.从反应机制上讲, 主要是自由基前体在光催化条件下产生自由基, 随后进攻烯烃, 形成碳自由基, 碳自由基再进一步被还原为碳负离子, 最后进攻CO2生成取代的α羧酸等重要化合物(path B, 图 1).
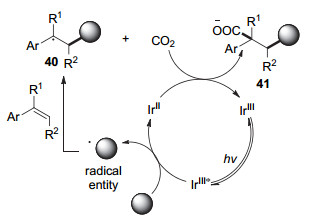
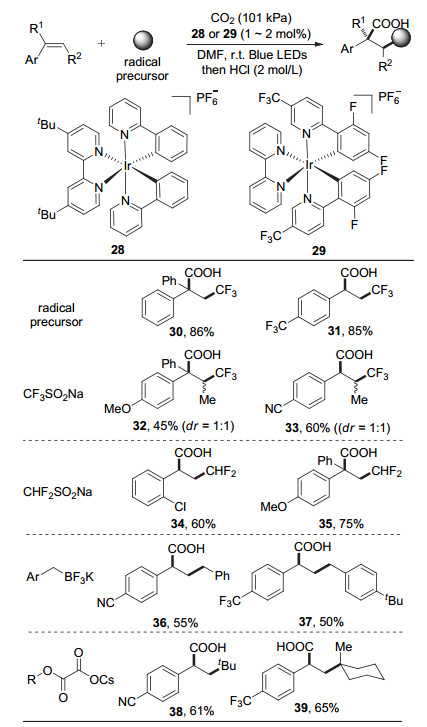
2017年, Martin课题组[24]报道了可见光促进、CO2参与的芳基烯烃的双官能团化羧基化反应(图 8).该反应使用金属Ir络合物作为光敏剂, 以Langlois试剂(三氟甲磺酸钠)、二氟甲磺酸钠、苄基三氟硼酸钾盐或草酸单酯酸盐为自由基前体, 在氧化还原中性条件下实现了CO2参与的芳基烯烃的选择性碳羧基化反应.
根据机理实验, 作者提出了以下机理(图 9).激发态的光敏剂Ir(Ⅲ)*通过单电子氧化自由基前体产生碳自由基物种并生成Ir(Ⅱ), 随后该碳自由基物种对芳基烯烃加成得到苄基自由基物种(40), 此物种被还原态的光敏剂Ir(Ⅱ)单电子还原为苄基碳负离子(41)并与CO2反应得到羧基化产物.整个反应的条件简单温和, 不需要额外加入有机金属还原剂、空气敏感的有机金属试剂及特殊的配体, 为实现碳羧基化反应提供了新的策略.此外, 该反应可引入重要的含氟砌块和多种烷基, 具有广阔的应用前景.但反应底物主要为芳基烯烃等活化烯烃, 体系并不适用一般非活化的烯烃.
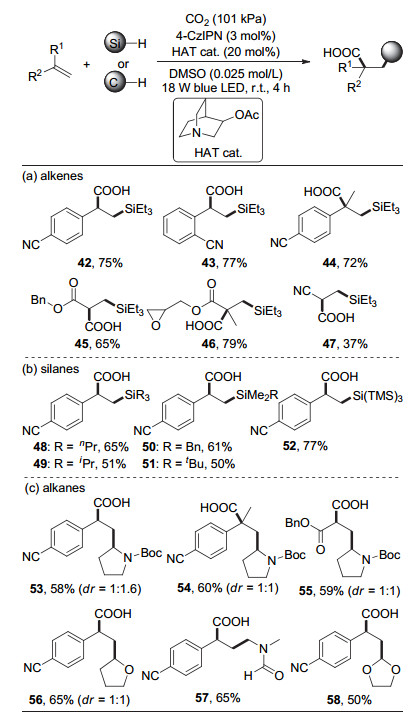
2018年, 吴杰课题组[25](图 10)利用光催化剂和氢原子转移催化剂的协同作用, 首次实现了光催化CO2参与的烯烃硅羧基化反应.此外, 该策略也可以胺类或者醚类为原料, 通过碳氢键直接官能团化实现烯烃的碳羧基化反应.反应以4CzIPN作为光催化剂, 3-乙酰氧基奎宁为氢原子转移催化剂, 在室温和蓝光照射下, 一系列取代苯乙烯与硅烷在101 kPa的CO2氛围下反应, 以较高的收率生成烯烃硅羧基化产物.在用叔丁氧羰基(Boc)保护的四氢吡咯作为自由基前体时, 该策略同样可以实现烯烃的碳羧基化反应, 并得到重要的γ-氨基酸类产物.该反应在氧化还原中性条件下通过硅氢键活化或碳氢键活化实现烯烃的双官能团化, 反应无需外加氧化剂, 具有很好的原子经济性.此外, 该反应条件温和, 可利用连续流技术实现克级反应.该反应除了贫电子的芳基烯烃, 还适用于丙烯酸酯、丙烯酰胺、烯基砜、丙烯腈等贫电子烯烃, 但对于非活化的普通烯烃并不兼容.
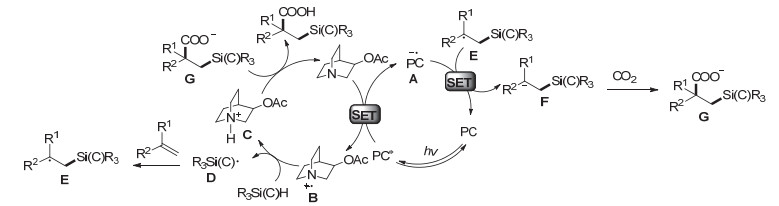
机理研究表明(图 11), 激发态的4CzIPN*首先被3-乙酰氧基奎宁还原淬灭得到4CzIPN自由基负离子A, 同时生成自由基阳离子中间体B; 随后, 自由基阳离子B促进硅氢键或碳氢键的氢原子转移产生硅或碳自由基D, 同时产生奎宁阳离子C, 硅或碳自由基D与烯烃加成形成碳自由基物种E; 接着, 碳自由基物种E被还原态的4CzIPN (A)还原得到碳负离子中间体F, 随后亲核进攻CO2得到β-硅基羧酸或含有γ-杂原子的羧酸产物.
相较于α-羧基化反应, 通过烯烃双官能团化生成β-羧基化产物, 无论从CO2的活化模式还是从反应位点选择性来讲, 都更具有挑战性.目前实现该类反应的例子非常少.
2017年, 我们课题组[26]首次报道了可见光驱动铁促进CO2参与的苯乙烯和丙烯酸酯的硫羧基化反应(图 12).反应以叔丁醇钾作碱, 氯化铁为催化剂, 在室温条件下, 不同种类的苯乙烯和丙烯酸酯都可以很好地反应, 并以非常高的收率得到重要的β-硫代羧酸产物.
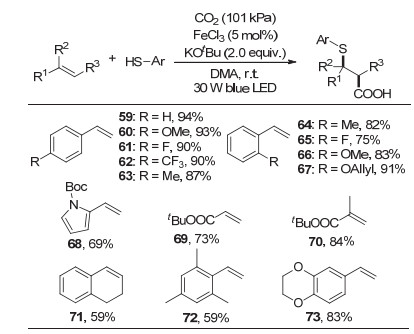
值得一提的是, 与以往报道的烯烃α-选择性羧基化反应不同, 该反应的羧基化发生在苯乙烯的β位, 具有很好的区域选择性.除此之外, 该反应条件温和, 具有高的化学选择性和非对映选择性, 生成的产物易于转化为生物活性物种和重要药物分子.
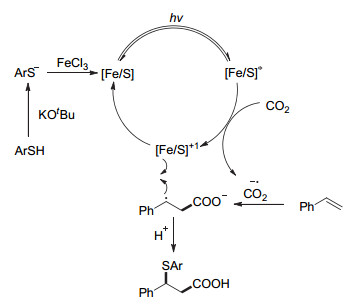
经过自由基捕获实验和自由基钟实验, 我们推测该反应可能经历自由基历程.虽然目前还缺乏证据我们认为该反应的可能机理如图 13所示.首先, 叔丁醇钾作为碱实现硫酚的去质子, 并促进硫铁复合物的形成, 硫铁复合物作为光致氧化还原催化剂或电子转移促进剂.在可见光照射下, 激发态硫铁复合物与CO2发生单电子转移形成CO2自由基负离子和氧化态的硫铁复合物, 而后CO2自由基负离子与苯乙烯加成得到稳定的苄基自由基物种, 最后与氧化态硫铁复合物反应得到β-硫代羧酸产物, 并重生活性硫铁复合物.
该反应提供了新型的二氧化碳活化模式, 为后续二氧化碳的活化和利用提供了很好的借鉴, 但目前实现的硫羧基化反应, 底物范围还具有一定的局限性, 比如底物之一的烯烃, 目前只适用于活化烯烃(包括苯乙烯类化合物、丙烯酸酯类化合物); 硫源中, 供电子性的硫酚化合物反应较好, 贫电子硫酚以及普通硫醇在该反应中不兼容.
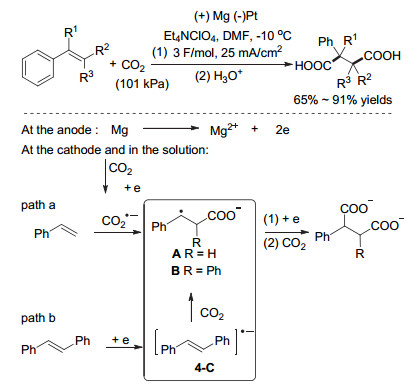
除通过光催化实现烯烃双官能团化反应外, 电催化烯烃双官能团化反应也有相关报道, 较为常见的反应类型为烯烃的双羧基化反应, 可以构建在药物、材料等工业中有着非常重要作用的丁二酸类衍生物.早在2001年, Senboku和Tokuda组[27]就报道了CO2参与的苯基丁二酸的合成(图 14).反应中, 在镁为阳极、铂为阴极的条件下, 苯基取代的烯烃与二氧化碳高效的反应并以中等或较好的收率得到目标产物苯基丁二酸.反应中, 作者提出了两条可能的路径:路径a, 由于苯乙烯及其衍生物相较于二氧化碳有更低的还原电势, 所以CO2在反应体系中被还原为CO2自由基负离子后进攻烯烃, 形成自由基中间体A, 中间体A进一步被还原为碳负离子并直接与另一分子CO2反应形成二酸.路径b, 对于1, 2-二苯基乙烯类化合物, 由于还原电势类似于CO2, 烯烃类化合物可被还原为烯烃自由基负离子, 进而与CO2反应生成类似A的中间体B, 中间体B进一步被还原为碳负离子并进一步与CO2反应生成二酸(图 1, path D).
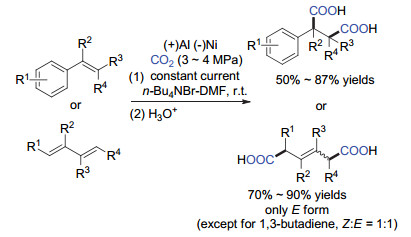
随后江焕峰课题组用非贵金属Ni代替Pt作为阴极, Al作为牺牲阳极, 高效地实现了芳基取代的烯烃[28]和1, 3-丁二烯[29]的双羧基化反应(图 15), 分别构建重要的丁二酸和己二酸类衍生物, 具有很重要合成意义.该类反应收率良好, 对于1, 3-丁二烯类化合物几乎可以单一的E式构型得到目标产物(1, 3-丁二烯除外, 得到的是1:1的混合物), 阴极电极除了Ni电极, Cu电极也有不错的效果.
虽然CO2参与的自由基型烯烃双官能团化反应已经取得一些重要进展, 为相关领域的发展提供了很好的借鉴, 但是该领域目前还处于发展阶段, 仍然面临众多挑战, 需要进一步发展, 主要表现在以下几个方面.
一、CO2参与自由基型反应构建噁唑啉酮类杂环化合物的方法存在以下不足: (a)已合成的噁唑啉酮类化合物的取代类型还局限于引入烷基, 还需进一步利用杂原子自由基参与类似转化, 合成具有杂原子的噁唑啉酮; (b)此类反应一般基于(path A, 图 1)这样的路径, 只实现了分子内关环反应, 目前还未实现更具挑战性的分子间反应; (c)该类反应的立体选择性还不能很好控制, 需要进一步深入研究.此外, 该类方法应该可以拓展至杂原子自由基前体参与的反应以及其他重要含羰基杂环(如环碳酸酯)的高效合成中.
二、通过烯烃自由基型双官能团化反应实现烯烃的碳羧基化、硅羧基化和硫羧基化等羧基化反应, 虽然反应机制不尽相同, 但都能够在温和的反应条件下实现较好的选择性和较高的收率, 具有较好的潜在工业化应用前景.该类转化目前还有以下几个问题需进一步解决: (a)底物普适性有待进一步提高.目前底物范围还比较局限, 主要集中在芳基烯烃、丙烯酸酯等这类活化烯烃, 而对于非活化的普通烯烃还不能实现其双官能团化, 可能原因是反应过程中非活化烯烃产生的自由基中间体稳定性较差, 也难于进一步还原为碳负离子, 即使生成的羧酸盐也容易发生脱羧, 不利于催化反应进行; (b)产物类型存在一定局限.目前只实现了碳羧基化、硫羧基化、硅羧基化、膦羧基化[30]反应等少数几个类型, 而其他类型, 如自由基型氮羧基化等非常重要的反应目前还未见报道; (c)从选择性来看, 目前虽然实现特定区域选择性, 但还不能通过条件简单变化实现两种区域选择性的调控; 此外, 对映选择性的控制目前还非常困难, 如何发展新型不对称催化体系实现手性羧酸的合成具有重大挑战.除此而外, 相关反应机理的研究目前尚不够深入, 实验数据尚不能完全说明提出的机理, 所以相关反应机理研究也将是该领域未来发展的重点.
作者相信, 随着自由基化学的快速发展、二氧化碳活化模式以及烯烃双官能团化催化体系的不断创新, 化学家们一定可以实现更多更有价值的二氧化碳参与的自由基型烯烃双官能团化反应, 变废为宝, 将二氧化碳化学转化变得真正有用, 从而实现工业化, 为相关精细化学品的合成、以及资源和能源问题的解决提供新的思路.
(a) Aresta, M. Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010. (b) He, L.-N. Carbon Dioxide Chemistry, Science Press, Beijing, 2013 (in Chinese). (何良年, 二氧化碳化学, 科学出版社, 北京, 2013). (c) Centi, G.; Perathoner, S. Green Carbon Dioxide: Advances in CO2 Utilization, Wiley-VCH, Weinheim, 2014.
Selected reviews on CO2 utilization to generate the C-O/C-C bonds, see: (a) Huang, K.; Sun, C.-L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435. (b) Martin, R.; Kleij, A. W. ChemSusChem 2011, 4, 1259. (c) Tsuji, Y.; Fujihara, T. Chem. Commun. 2012, 48, 9956. (d) He, M.; Sun, Y.; Han, B. Angew. Chem., Int. Ed. 2013, 52, 9620. (e) Zhang, L.; Hou, Z. Chem. Sci. 2013, 4, 3395. (f) Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Nat. Commun. 2015, 6, 5933. (g) Börjesson, M.; Moragas, T.; Gallego, D.; Martin, R. ACS Catal. 2016, 6, 6739. (h) Zhang, L.; Han, Z.; Zhang, L.; Li, M.; Ding, K. Chin. J. Org. Chem. 2016, 36, 1824(in Chinese). (张琳莉, 韩召斌, 张磊, 李明星, 丁奎岭, 有机化学, 2016, 36, 1824) (i) Zhu, Q.; Wang, L.; Xia, C.; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813(in Chinese). (朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813) (j) Zhang, W.; Guo, C.; Lu, X. Chin. J. Catal. 2016, 37, 215. (k) Zhang, H.; Sun, H.; Li, X. Chin. J. Org. Chem. 2016, 36, 2843(in Chinese). (仉花, 孙宏建, 李晓燕, 有机化学, 2016, 36, 2843.) (l) Zhang, S.; Li, X.; He, L.-N. Acta Chim. Sinica 2016, 74, 17(in Chinese). (张帅, 李雪冬, 何良年, 化学学报, Acta Chim. Sinica 2016, 74, 17. (m) Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Green Chem. 2017, 19, 3707. (n) Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. ChemSusChem 2017, 10, 1337. (o) Luo, J.; Larrosa, I. ChemSusChem 2017, 10, 3317. (p) Zhang, Z.; Ju, T.; Ye, J.-H.; Yu, D.-G. Synlett 2017, 28, 741. (q) Zou, B.; Hu, C. Chin. J. Chem. 2017, 35, 541. (r) Li, Y.; Wang, Z.; Liu, Q. Chin. J. Org. Chem. 2017, 37, 1978(in Chinese). (李勇, 王征, 刘庆彬, 有机化学, 2017, 37, 1978) (s) Zhang, W.; Zhang, N.; Guo, C.; Lü, X. Chin. J. Org. Chem. 2017, 37, 1309(in Chinese). (张文珍, 张宁, 郭春晓, 吕小兵, 有机化学, 2017, 37, 1309) (t) Feng, J.; Zeng, S.; Feng, J.; Dong, H.; Zhang, X. Chin. J. Chem. 2018, 36, 961. (u) Zhao, Y.; Liu, Z. Chin. J. Chem. 2018, 36, 455; (v) Zhang, Y.; Cen, J.; Xiong, W.; Qi, C.; Jiang, H. Prog. Chem. 2018, 30, 547(in Chinese). (张宇, 岑竞鹤, 熊文芳, 戚朝荣, 江焕峰, 化学进展, 2018, 30, 547.) (w) Wang, L.; Sun, W.; Liu, C. Chin. J. Chem. 2018, 36, 353. (x) Chen, Y.-G.; Xu, X.-T.; Zhang, K.; Li, Y.-Q.; Zhang, L.-P.; Fang, P.; Mei, T.-S. Synthesis 2018, 50, 35. (y) Wang, S.; Xi, C. Chem. Soc. Rev. 2019, 48, 382. (z) Chen, Z.; Liu, J.; Cui, H.; Zhang, L.; Su, C. Acta Chim. Sinica 2019, 77, 242(in Chinese). (陈之尧, 刘捷威, 崔浩, 张利, 苏成勇, 化学学报, 2019, 77, 242.)
(a) Sasano, K.; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2013, 135, 1251. (b) Sekine, K.; Sadamitsu, Y.; Yamada, T. Org. Lett. 2015, 17, 5706. (c) Moragas, T.; Gaydou, M.; Martin, R. Angew. Chem., Int. Ed. 2016, 55, 5053. (d) Miao, B.; Li, S.; Li, G.; Ma, S. Org. Lett. 2016, 18, 2556. (e) Nogi, K.; Fujihara, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2016, 138, 5547. (f) Gholap, S. S.; Takimoto, M.; Hou, Z. Chem. Eur. J. 2016, 22, 8547. (g) Yan, S.-S.; Zhu, L.; Ye, J.-H.; Zhang, Z.; Huang, H.; Zeng, H.; Li, C.-J.; Lan, Y.; Yu, D.-G. Chem. Sci. 2018, 9, 4873. (h) Song, L.; Zhu, L.; Zhang, Z.; Ye, J.-H.; Yan, S.-S.; Han, J.-L.; Yin, Z.-B.; Lan, Y.; Yu, D.-G. Org. Lett. 2018, 20, 3776. (i) Fu, L.; Li, S.; Cai, Z.; Ding, Y.; Guo, X.; Zhou, L.; Yuan, D.; Sun, Q.; Li, G. Nat. Catal. 2018, 1, 469. (j) Xiong, W. F.; Yan, D. H.; Qi, C. R.; Jiang, H. F. Org. Lett. 2018, 20, 672. (k) Wang, S.; Xi, C. J. Org. Lett. 2018, 20, 4131. (l) Song, L.; Cao, G.-M.; Zhou, W.; Ye, J.-H.; Zhang, Z.; Tian, X.-Y.; Li, J.; Yu, D.-G. Org. Chem. Front. 2018, 5, 2086. (m) Cai, Z.; Li, S.; Gao, Y.; Li, G. Adv. Synth. Catal. 2018, 360, 4005. (n) Huang, R.; Li, S.; Fu, L.; Li, G. Asian J. Org. Chem. 2018, 7, 1376. (o) Gao, Y.; Cai, Z.; Li, S.; Li, G. Org. Lett. 2019, 21, 3663. (p) Yan, S.-S.; Wu, D.-S.; Ye, J.-H.; Gong, L.; Zeng, X.; Ran, C.-K.; Gui, Y.-Y.; Li, J.; Yu, D.-G. ACS Catal. 2019, 9, 6987.
(a) Seo, H; Katcher, M. H.; Jamison, T. F. Nat. Chem. 2017, 9, 453. (b) Meng, Q.; Wang, S.; König, B. Angew. Chem., Int. Ed. 2017, 56, 13426. (c) Shimomaki, K.; Murata, K.; Martin, R.; Iwasawa, N. J. Am. Chem. Soc. 2017, 139, 9467. (d) Liao, L.-L.; Cao, G.-M.; Ye, J.-H.; Sun, G.-Q.; Zhou, W.-J.; Gui, Y.-Y.; Yan, S.-S.; Shen, G.; Yu, D.-G. J. Am. Chem. Soc. 2018, 140, 17338. (e) Ju, T.; Fu, Q.; Ye, J.-H.; Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Tian, X.-Y.; Luo, S.-P.; Li, J.; Yu, D.-G. Angew. Chem. Int. Ed. 2018, 57, 13897. (f) Fan, X.; Gong, X.; Ma, M.; Wang, R.; Walsh, P. J. Nat. Commun. 2018, 9, 4936.
(a) Wang, H.; Lin, M.-Y.; Fang, H. J.; Chen, T. T.; Lu, J.-X. Chin. J. Chem. 2007, 25, 913. (b) Wang, H.; Du, Y. F.; Lin, M. Y.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1745. (c) Jiao, K.; Li, Z.; Xu, X.; Zhang, L.; Li, Y.; Zhang, K.; Mei, T.-S. Org. Chem. Front. 2018, 5, 2244.
(a) Xin, Z.; Lescot, C.; Friis, S. D.; Daasbjerg, Kim; Skrydstrup, T. Angew. Chem. Int. Ed. 2015, 54, 6862. (b) Zhang, W.; Yang, M. W.; Lv, X. Green Chem. 2016, 18, 4181. (c) Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Wang, L.; He, Y.-Q.; Ye, J.-H.; Li, J.; Zhi, Y.-G.; Yu, D.-G. Angew. Chem., Int. Ed., 2016, 55, 7068. (d) Wang, S.; Shao, P.; Du, G.; Xi, C. J. Org. Chem. 2016, 81, 6672.
(a) Hu, J.; Ma, J.; Zhu, Q.; Zhang, Z.; Wu, C.; Han, B. Angew. Chem. Int. Ed. 2015, 54, 5399. (b) Gao, X.; Yu, B.; Yang, Z.; Zhao, Y.; Zhang, H.; Hao, L.; Han, B.; Liu, Z. ACS Catal. 2015, 5, 6648. (c) Zhao, Y.; Wu, Y.; Yuan, G.; Hao, L.; Gao, X.; Yang, Z.; Yu, B.; Zhang, H.; Liu, Z. Chem. Asian J. 2016, 11, 2735.
(a) Li, Y.; Fang, X.; Junge, K.; Beller, M. Angew. Chem. Int. Ed. 2013, 52, 9568. (b) Zhang, Z.; Sun, Q.; Xia, C.; Sun, W. Org. Lett. 2016, 18, 6316. (c) Zhang, Y.; Wang, H.; Yuan, H.; Shi, F. ACS Sustainable Chem. Eng. 2017, 5, 5758. (d) Ren, X.; Zheng, Z.; Zhang, L.; Wang, Z.; Xia, C.; Ding, K. Angew. Chem., Int. Ed. 2017, 56, 310.
(a) Lehn, J.-M.; Ziessel, R. Proc. Natl. Acad. Sci. USA 1982, 79, 701. (b) Burgess, S. A.; Kendall, A. J.; Tyler, D. R.; Linehan, J. C.; Appel, A. M. ACS Catal. 2017, 7, 3089.
(a) Pupo, G.; Properzi, R.; List, B. Angew. Chem., Int. Ed. 2016, 55, 6099. (b) Riemer, D.; Mandaviya, B.; Schilling, W.; Götz, A. C.; Kühl, T.; Finger, M.; Das, S. ACS Catal. 2018, 8, 3030. (c) Roy, T.; Kim, M. J.; Yang, Y.; Kim, S.; Kang, G.; Ren, X.; Kadziola, A.; Lee, H.-Y.; Baik, M.-H. Lee, J.-W. ACS Catal. 2019, 9, 6006.
For selected reviews, see: (a) Cao, M.-Y.; Ren, X.; Lu, Z. Tetrahedron Lett. 2015, 56, 3732. (b) Chen, J.-R.; Yu, X.-Y.; Xiao, W.-J. Synthesis 2015, 47, 604. (c) Koike, T.; Akita, M. Acc. Chem. Res. 2016, 49, 1937. (d) Koike, T.; Akita, M. Chem 2018, 4, 409. (e) Li, W.; Xu, W.; Xie, J.; Yu, S.; Zhu, C. Chem. Soc. Rev. 2018, 47, 654. (f) Wu, X.; Wu, S.; Zhu, C. Tetrahedron Lett. 2018, 59, 1328.
(a) Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230. (b) Zhang, Z.; Ye, J.-H.; Wu, D.-S.; Zhou, Y.-Q.; Yu, D.-G. Chem. Asian J. 2018, 13, 2292. (c) Peshkov, V. A.; Pereshivko, O. P.; Nechaev, A. A; Peshkov, A. A.; Vander Eycken, E. V. Chem. Soc. Rev. 2018, 47, 3861. (d) Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Angew. Chem., Int. Ed. 2018, 57, 15948. (e) Yan, S.-S.; Fu, Q.; Liao, L.-L.; Sun, G.-Q.; Ye, J.-H.; Gong, L.; Bo-Xue, Y.-Z.; Yu, D.-G. Coord. Chem. Rev. 2018, 374, 439. (f) Cao, Y.; He, X.; Wang, N.; Li, H.-R.; He, L.-N. Chin. J. Chem. 2018, 36, 644. (g) Hou, J.; Li, J.-S.; Wu, J. Asian J. Org. Chem. 2018, 7, 1439. (h) Tan, F.; Yin, G. Chin. J. Chem. 2018, 36, 545. (i) Yeung, C. S. Angew. Chem., Int. Ed. 2019, 58, 5492.
Luan, Y.-X.; Ye, M. Tetrahedron Lett. 2018, 59, 853. doi: 10.1016/j.tetlet.2018.01.035
(a) Tominaga, K.-I.; Sasaki, Y. Catal. Commun. 2000, 1, 1. (b) Tominaga, K.-i.; Sasaki, Y. J. Mol. Catal. A: Chem. 2004, 220, 159. (c) Liu, Q.; Wu, L.; Fleischer, I.; Selent, D.; Franke, R.; Jackstell, R.; Beller, M. Chem.-Eur. J. 2014, 20, 6888. (d) Tani, Y.; Kuga, K.; Fujihara, T.; Terao, J.; Tsuji, Y. Chem. Commun. 2015, 51, 13020. (e) Gui, Y.-Y.; Hu, N.; Chen, X.-W.; Liao, L.-L.; Ju, T.; Ye, J.-H.; Zhang, Z.; Li, J.; Yu, D.-G. J. Am. Chem. Soc. 2017, 139, 17011.
Seo, H.; Liu, A.-F.; Jamison, T. F. J. Am. Chem. Soc. 2017, 139, 13969. doi: 10.1021/jacs.7b05942
(a) Evans, D. A.; Bartroli, J.; Shih, L. T. J. Am. Chem. Soc. 1981, 103, 2127. (b) Pandit, N.; Singla, R. K.; Shrivastava, B. Int. J. Med. Chem. 2012, 2012, 159285. (c) Ed.: Acton, Q. A., Oxazolidinones-Advances in Research and Application, Scholarly Editions, Atlanta, U.S., 2012.
Ye, J.-H.; Song, L.; Zhou, W.-J.; Ju, T.; Yin, Z.-B.; Yan, S.-S.; Zhang, Z.; Li, J.; Yu, D.-G. Angew. Chem. Int. Ed. 2016, 55, 10022. doi: 10.1002/anie.201603352
Zhu, L.; Ye, J.-H.; Duan, M.; Qi, X.; Yu, D.-G.; Bai, R.; Lan, Y. Org. Chem. Front. 2018, 5, 633. doi: 10.1039/C7QO00838D
Ye, J.-H.; Zhu, L.; Yan, S.-S.; Miao, M.; Zhang, X.-C.; Zhou, W.-J.; Li, J.; Lan, Y.; Yu, D.-G. ACS Catal. 2017, 7, 8324. doi: 10.1021/acscatal.7b02533
Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Green Chem. 2017, 19, 1240. doi: 10.1039/C6GC03200A
Yin, Z.-B.; Ye, J.-H.; Zhou, W.-J.; Zhang, Y.-H.; Ding, L.; Gui, Y.-Y.; Yan, S.-S.; Li, J.; Yu, D.-G. Org. Lett. 2017, 20, 190.
Zhou, W.-J.; Cao, G.-M.; Sen, G.; Zhu, X.-Y.; Gui, Y.-Y.; Ye, J.-H.; Sun, L.; Liao, L.-L.; Li, J.; Yu, D.-G. Angew. Chem., Int. Ed. 2017, 56, 15683. doi: 10.1002/anie.201704513
(a) Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Org. Lett. 2018, 20, 3049. (b) For a very recent work, see: Sun, S.; Zhou, C.; Yu, J.-T.; Cheng, J. Org. Lett. 2019, DOI: 10.1021/acs.org-lett.9b02700.
Yatham, V. R.; Shen, Y.; Martin, R. Angew. Chem., Int. Ed. 2017, 56, 10915. doi: 10.1002/anie.201706263
Hou, J.; Ee, A.; Cao, H.; Ong, H.-W.; Xu, J.-H.; Wu J. Angew. Chem., Int. Ed. 2017, 57, 17220.
Ye, J.-H.; Miao, M.; Huang, H.; Yan, S.-S.; Yin, Z.-B.; Zhou, W.-J.; Yu, D.-G. Angew. Chem., Int. Ed. 2017, 56, 15416. doi: 10.1002/anie.201707862
Senboku, H.; Komatsu, H.; Fujimura, Y.; Tokuda, M. Synlett 2001, 2001, 418. doi: 10.1055/s-2001-11417
Yuan, G.-Q.; Jiang, H.-F.; Lin, C.; Liao, S.-J. Electrochim. Acta 2008, 53, 2170. doi: 10.1016/j.electacta.2007.09.023
Li, C.-H.; Yuan, G.-Q.; Ji, X.-C.; Wang, X.-J.; Ye, J.-S.; Jiang, H.-F. Electrochim. Acta 2011, 56, 1529. doi: 10.1016/j.electacta.2010.06.057
For a very recent work on phosphonocarboxylation of alkenes with CO2, see: Fu, Q.; Bo, Z.-Y.; Ye, J.-H.; Ju, T.; Huang, H.; Liao, L.-L.; Yu, D.-G. Nat. Commun. 2019, 10, 3592.

图 1 二氧化碳参与的自由基型烯烃双官能团化反应
Figure 1 Radical-type difunctionalization of alkenes with CO2

图 2 CO2参与的烯丙胺的氧-三氟甲基化
Figure 2 Selective oxytrifluoromethylation of allylamines with CO2

图 3 CO2参与的自由基型去芳构化反应
Figure 3 Radical trifluoromethylative dearomatization of indoles and furans with CO2

图 7 可见光驱动钯催化的烯丙胺的氧-烷基化反应
Figure 7 Oxy-alkylation of allylamines via visible-light-driven palladium catalysis

图 9 CO2参与的芳基烯烃碳羧基化反应机理
Figure 9 A proposed mechanism of dicarbofunctionalization of styrenes with CO2 and radical precursors

图 10 CO2参与的烯烃与硅烷或烷烃的双官能团化反应
Figure 10 Dicarbofunctionalization of alkenes with CO2 and silanes or alkanes

图 11 CO2参与的烯烃与硅烷或烷烃的双官能团化反应机理
Figure 11 A proposed mechanism of dicarbofunctionalization of alkenes with CO2 and silanes or alkanes

图 12 可见光驱动铁促进CO2参与的烯烃与苯硫酚的硫羧基化反应
Figure 12 Visible-light-driven iron-promoted thiocarboxylation of alkenes with CO2

图 13 可见光驱动铁促进CO2参与的烯烃与苯硫酚的硫羧基化反应机理
Figure 13 A proposed mechanism of visible-light-driven iron-promoted thiocarboxylation of alkenes with CO2

图 14 Senboku和Tokuda组电催化CO2参与的烯烃双羧基化反应
Figure 14 Electrosynthesis of 2-phenylsuccinic acids with CO2 by Senboku and Tokuda group

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:





 下载:
下载: