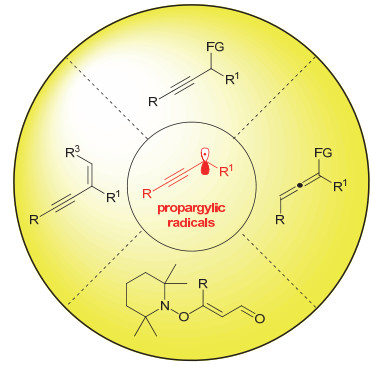
图 1.
炔丙基自由的转化(FG:官能团)
Figure 1.
Transformations of propargylic radicals (FG: functional group)

炔烃作为一类重要的不饱和官能团, 广泛存在于许多天然产物、药物分子和功能材料中.并且, 由于炔烃这一类官能团结构的特殊性, 能够实现向烷烃、烯烃、醇、酮以及芳环等其它许多化合物的转化.因此, 炔烃化合物的合成与转化一直是合成化学学科的一个重要研究领域[1].
炔丙位的官能化反应是实现炔烃合成与转化的一种重要途径, 并已经取得了重要的发展与应用[2].早在1972年, Nicholas等[3]就已经实现了炔丙醇及其衍生物的炔丙位官能化反应.这类反应主要经历了钴稳定的炔丙基正离子中间体, 并且被广泛用于天然产物和药物分子的合成.此外, 钴稳定的炔丙基自由基中间体也有所报道, 主要被用于炔丙基自由基的偶联反应当中[4].美中不足的是, 该类方法需要用到当量的有毒金属试剂、多步的操作且产生当量的金属废弃物. 1994年, Caporusso等[5]首次实现了铜催化的炔丙位胺化反应, 为后期催化不对称的炔丙位官能化反应打下了坚实的基础.在随后的二十多年时间里, 这一领域由于Nishibayashi, 侯雪龙, 胡向平等许多化学家持续不断的努力得到了快速的发展, 并有多篇综述总结了该领域的进展[2, 6].需要指出的是, 这些方法大都经过γ位具有亲电性的亚丙二烯基金属物种中间体, 这就意味着非端炔类化合物难以参与该类反应[7].而炔丙基自由基所参与的炔丙基化反应刚好填补了这一空缺, 能够用于非端炔类化合物的炔丙位官能化反应, 具有十分重要的合成意义.问题在于, 由于炔丙基自由基的炔烃片段与自由基共轭, 使得炔丙基自由基不仅能够直接参与到炔丙位官能化反应中(产生中心手性), 也能够发生自由基迁移异构为联烯自由基之后参与到后续偶联反应, 从而构建烯类化合物(产生轴手性).此外, 由于炔丙基自由基丰富的反应位点和模式, 还可以产生烯炔等其它不同结构的产物.因此, 化学选择性控制和立体选择性控制是自由基途径炔丙位官能化的难点和关键所在.本综述依据炔丙基自由基所参与的反应类型, 对近年来炔丙基自由基参与的反应进行了总结(图 1).
1977年, Kropf等[8a]报道了一例Kharasch类型的炔丙位官能化反应, 在铜盐和过氧酯的存在下能够实现内炔类化合物的炔丙位C—H键氧化反应. 2007年, Christ和Sorokin团队[8b]运用类似的策略实现了烯丙位和炔丙位的选择性C—H键氧化反应.值得注意的是, 作者通过使用手性配体能够以较高的收率和中等的对映选择性得到手性的烯丙位氧化产物, 但炔丙位氧化产物的ee值最高只能达到4%(图 2).
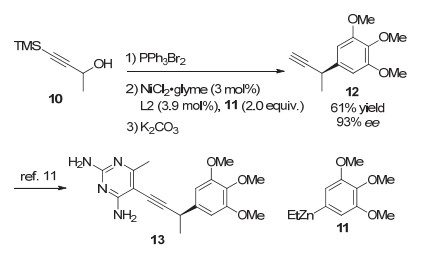
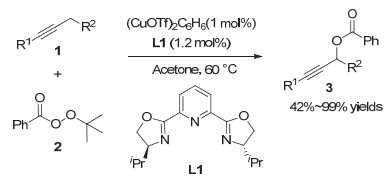
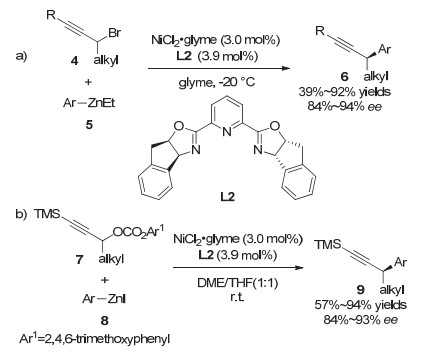
2008年, Fu等[9]实现了镍催化的二级炔丙基溴化物与芳基锌试剂的不对称Negishi交叉偶联反应.该反应在NiCl2·glyme为催化剂、茚取代的手性吡啶双噁唑啉L2为配体的条件下, 能够实现消旋炔丙基溴代物到手性炔丙位芳基化产物的转化.该反应表现出较好的底物普适性, 带有酯基或烯基等官能团的炔丙基溴代物均能很好地适用于该反应, 以较高的收率和对映选择性生成目标化合物(图 3, a). 2012年, Fu等[10]利用类似的策略实现了炔丙酯类化合物的不对称Negishi交叉偶联反应(图 3, b).相比之前的工作, 该反应条件更加温和, 且同样具有较好的底物普适性.这类镍催化的不对称炔丙基偶联反应已成为有机化学中一种有效的合成手段.例如, Fu教授将该策略成功应用于药物中间体的合成中.其通过将商业可得的4-三甲基硅基-3-丁基-2-醇10转化成炔丙基溴, 然后与3, 4, 5-三甲氧基苯基锌11偶联, 最终以93%的ee值合成得到二氢叶酸还原酶抑制剂的关键中间体13(图 4)[11].
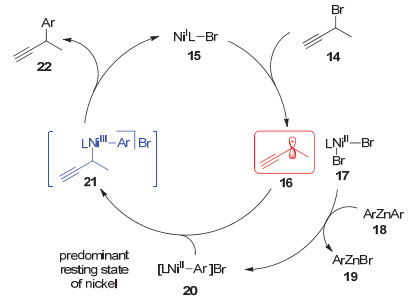
2014年, Fu等[12]对他所发展的镍催化不对称炔丙位芳基化反应进行了详细的机理研究.作者通过EPR(电子顺磁共振波谱)实验、TEMPO(2, 2, 6, 6-四甲基哌啶氧化物)捕获实验等验证了该反应体系中一价镍物种以及炔丙基自由基的存在, 为该反应提出了一个合理的机理.如图 5所示, 一价镍物种首先与炔丙基溴代物发生单电子转移, 生成炔丙基自由基16和二价镍物种17; 接着, 该物种与芳基锌试剂18发生转移金属化并生成二价的芳基镍物种20; 随后, 炔丙基自由基16对芳基镍物种加成生成三价镍物种21, 该三价镍物种还原消除生成目标产物22以及一价镍物种, 最终完成催化循环.
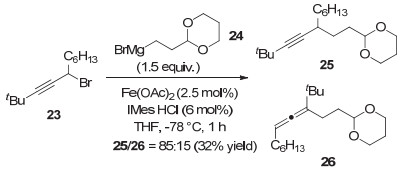
2018年, Cárdenas等[13]运用铁催化的方法实现了炔丙基溴代物与烷基格氏试剂的Kumada-类型偶联反应.该反应仅需1 h就能达到最高83%的收率, 但作者无法完全抑制炔丙位自由基向联烯自由基的异构.因此, 产物中总会混杂一定的联烯产物(图 6).
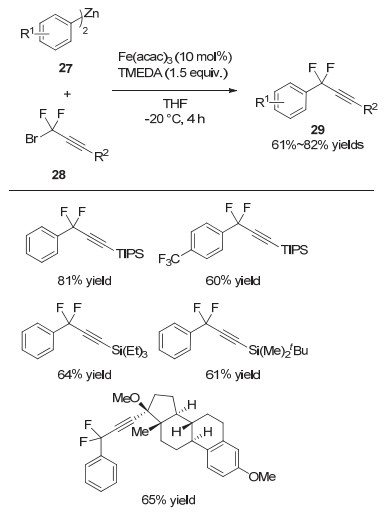
2018年, 张新刚等[14]利用铁催化实现了芳基金属试剂的二氟烷基化反应, 其中偕二氟炔丙基溴类化合物也能够很好地参与到该反应中, 并以较高的收率得到炔丙位官能化的产物.值得注意的是, 当使用雌炔醇衍生的二氟炔丙基溴底物时, 该反应能够以65%的收率得到目标产物, 表明该策略对复杂生物分子的修饰也具有很好的效果(图 7).
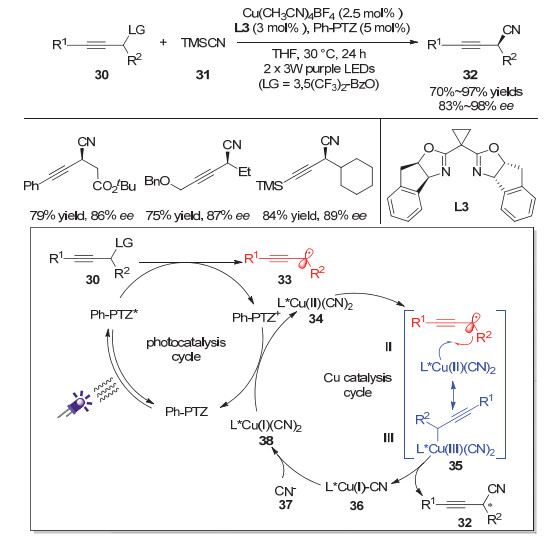
2019年, 肖文精、陆良秋等[15]利用协同的可见光催化和铜催化策略, 实现了不对称的炔丙基自由基氰基化反应.该反应利用副产物羧酸负离子来活化三甲基氰硅烷, 以达到缓慢释放氰基负离子的作用, 从而避免了高浓度氰基负离子与铜配位导致金属催化剂失活的问题(图 8).该反应不仅能够实现苯乙炔类化合物的炔丙位氰基化, 也能实现烷基炔烃、三甲基硅乙炔类化合物的炔丙位氰基化反应, 具有较好的底物普适性和官能团耐受性.此外, 作者通过与重庆大学的蓝宇教授合作, 通过实验和理论计算研究了该反应的自由基途径并且对立体选择控制做出了合理的解释.如图所示, 作者认为激发态的有机光催化剂Ph-PTZ在光照条件下到达激发态, 随后与炔丙酯底物30发生单电子转移生成炔丙基自由基33和氧化态的光催化剂Ph-PTZ+; 该氧化态的光催化剂将一价的氰基铜38氧化成二价的氰基铜物种34, 炔丙基自由基33随后对34加成, 最后通过还原消除得到手性的炔丙腈类化合物32.
除了通过炔丙位碳杂键的断裂生成自由基外, 通过自由基对1, 3-烯炔的双键部分加成也是产生炔丙基自由基的重要途径. 2015年, Loh等[16]实现了铜或钴催化的1, 3-烯炔类化合物烯烃片段的双官能化反应(图 9).在该反应中, 铜或钴起到催化产生叔丁氧基自由基以及叔丁过氧自由基的作用.叔丁氧基自由基通过攫取醇的氧α位氢原子产生氧α位碳自由基43, 该自由基对1, 3-烯炔39的烯烃部分加成后产生炔丙基自由基44.随后, 该自由基与叔丁过氧自由基发生偶联并生成最终的烯烃双官能化产物42.反应所得到的β-过氧醇可以通过简单的转化得到β-羟基酮以及1, 3-炔丙二醇化合物.

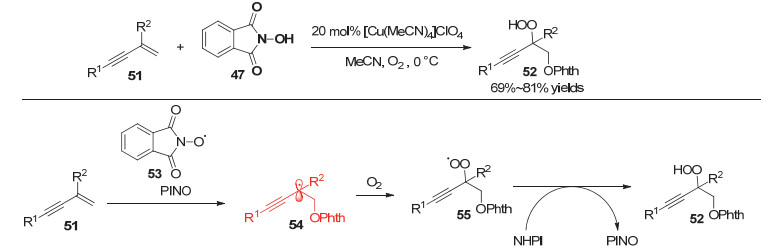
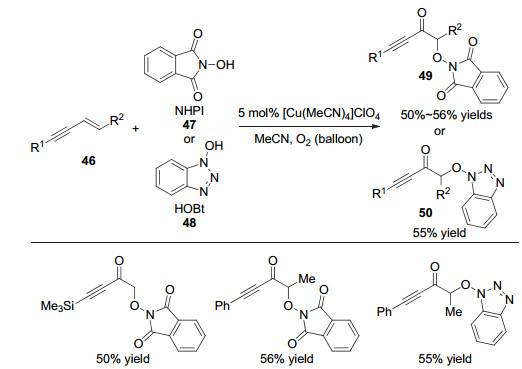
2015年, Woerpel等[17]通过铜催化实现了1, 3-烯炔烯烃片段的氧化反应.该反应以氧气作为绿色氧化剂, N-羟基邻苯二甲酰亚胺(NHPI)和N-羟基苯并三唑(HOBt)作为自由基前体, 能够以中等的收率给出α氧代炔丙酮类化合物(图 10).
2016年, Woerpel等[18]又运用类似的策略实现了炔丙位的过氧化反应, 并提出了以下可能的机理:首先, NHPI转变为氮氧自由基(PINO)后对1, 3-烯炔的烯烃片段进行加成并生成炔丙位自由基54, 随后被氧气捕获生成过氧自由基55; 该物种攫取NHPI中的氢原子生成最终产物并且产生一分子PINO继续参与后续反应(图 11).
2017年, Loh等[19]应用类似的策略实现了铁催化的1, 3-烯炔类化合物烯烃片段的过氧化-氨甲酰化反应(图 12).值得注意的是, 该反应不仅能够适用于1, 3-烯炔类化合物, 也能够适用于普通的芳基烯烃和1, 3-二烯类化合物的烯烃双官能化, 具有较好的底物普适性.
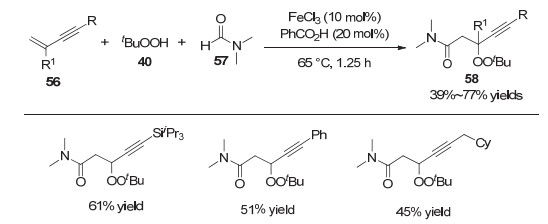
当炔丙基自由基异构成为联烯基自由基后, 其参与的偶联反应便能实现另一类重要不饱和化合物——联烯的合成[20]. 1993年, Blechert等[21a]实现了从炔丙基自由基到联烯自由基的转化, 并将其运用到环化反应当中.在该反应中, AIBN和Bu3SnH联用可以生成锡自由基, 进而攫取炔丙位的溴原子并产生炔丙基自由基60; 随后, 该自由基异构为联烯自由基61后发生5-exe-trig环化, 最终通过氢原子攫取后生成目标产物62a和62b(图 13). 2006年, Zard等[21b]也运用类似的策略构建了带有联烯结构的并环化合物.
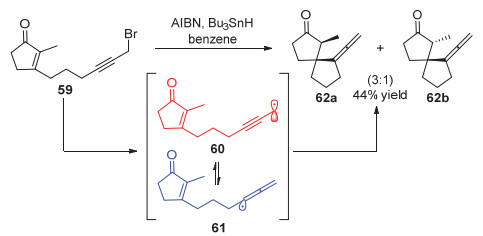
2016年, Cárdenas等[22]运用镍催化的方法实现了炔丙基溴代物与烷基格氏试剂的偶联反应.与Fu等工作不同的是, 作者使用菲罗啉类配体L4时, 能够选择性地得到联烯类产物(图 14).
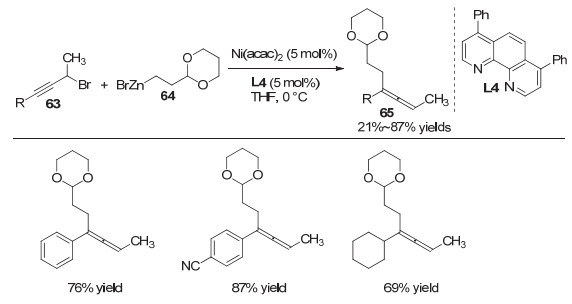
2017年, 刘国生等[23]率先实现了1, 3-烯炔类化合物的三氟甲基氰基化反应, 以高收率和高区域选择性得到了联烯腈类化合物.该反应具有广泛的底物适用范围以及良好的官能团兼容性(图 15, a).有意思的是, 当使用配体L5和L6时, 可以分别得到1, 3-烯炔的1, 2位官能化和1, 4位官能化的产物.对此, 作者通过详细的机理研究, 阐述了生成两种不同产物所经历的可能路径.与此同时, 作者也对该反应的催化不对称的过程进行了初步探究.当使用手性的双噁唑啉配体L5时, 能够以78%的收率和73%的ee值得到手性的联烯腈类化合物(图 15, b).为了进一步了解反应的机理, 作者将含有三元环结构的1, 3-烯炔底物72投入到反应中, 发现当使用双噁唑啉配体L5时, 生成的三元环开环产物74多于联烯腈产物73, 而当使用菲罗啉类配体L6时, 反应则主要得到未开环的联烯腈产物.基于该实验, 作者认为该反应存在炔丙基自由基到联烯自由基的异构过程, 并且烷基自由基更倾向与(Box)CuⅡ(CN)2中间体作用, 而联烯自由基则更倾向与(phen)CuⅡ(CN)2中间体作用并最终生成联烯腈产物.作者也通过理论计算为该反应路径提供了理论支持(图 15, c).
2019年, 鲍红丽等[24]运用铜催化的自由基加成/偶联策略实现了1, 3-烯炔向联烯腈类化合物的转化(图 16).作者发展了一种生成联烯自由基的通用方法并实现了其在分子间1, 3-烯炔的1, 4-碳氰化和1, 4-亚磺酰亚胺化反应中的应用.这类多取代的联烯可以通过简单的后期转化得到许多其它具有重要合成价值的化合物, 如:氟化乙烯基氰化物、内酯、官能化的联烯酰胺、1-氨基萘等.此外, 作者通过TEMPO捕获实验、自由基Clock实验验证了反应途径中联烯自由基的存在, 并且在DFT理论计算等手段的辅助下提出了合理的反应途径.首先, 自由基前体80被一价铜单电子还原得到自由基物种81和二价铜, 该自由基物种对1, 3-烯炔75进行加成得到联烯自由基84; 然后, 二价铜物种与TMSCN发生转移金属化得到二价的异氰铜物种82, 该异氰铜物种随后与联烯自由基作用得到联烯腈类化合物79.
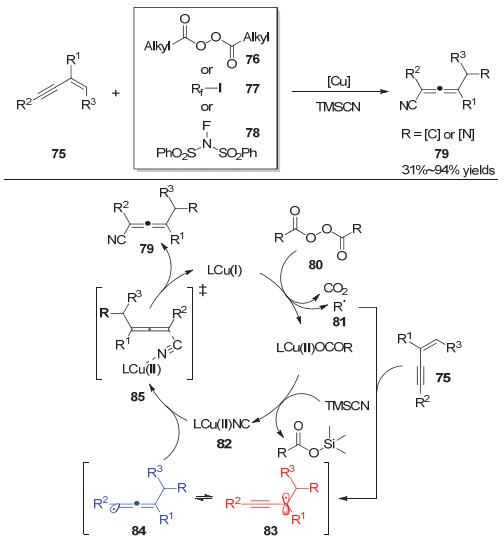
同年, 鲍红丽等[25]还实现了1, 3烯炔的1, 4-烷基芳基化反应(图 17).该反应通过使用过氧化月桂酰类化合物(LPO)作为烷基自由基前体, 芳基硼酸作为芳基亲核试剂, 在铜催化下以中等到较好的收率得到四取代联烯类化合物.
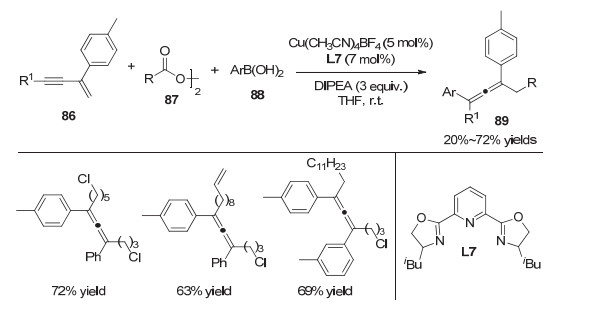
2019年, 王细胜等[26]报道了镍催化的1, 3-烯炔的碳氟烷基化反应(图 18).该反应利用简单易得的1, 3-烯炔和二氟烷基卤代物, 合成了二氟烷基取代的联烯类化合物, 具有条件温和、底物范围广泛、官能团兼容性好等优势.该反应机理如下:一价镍催化剂首先与芳基硼酸发生转移金属化生成一价芳基镍物种93, 该物种随后将二氟烷基卤代物还原得到二氟烷基碳自由基95以及二价的芳基镍物种94; 随后, 二氟烷基碳自由基对1, 3-烯炔加成产生炔丙位自由基96, 然后经过自由基迁移生成联烯自由基97, 而该自由基中间体对二价芳基镍物种加成并通过还原消除得到最终的产物.作者通过TEMPO捕获实验以及自由基Clock反应验证了反应过程中二氟烷基自由基的存在.
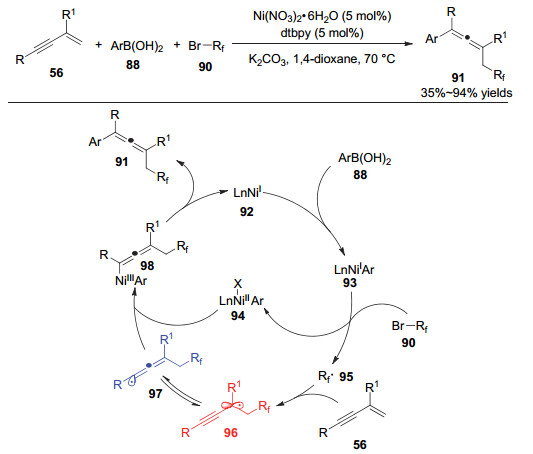
炔丙基自由基除了可以直接对金属络合物加成或异构为联烯自由基后再对金属络合物加成外, 还可以进一步被氧化为炔丙基正离子中间体, 从而进一步脱质子形成取代的1, 3-烯炔类化合物. 2018年, 鲍红丽等[27]实现了炔丙醇与二烷基过氧化物的脱水脱羧偶联反应(图 19).该反应从简单的炔丙醇出发, 在铁催化的条件下实现了多种取代的1, 3-烯炔类化合物的合成.同时, 该反应表现出较好的底物兼容性, 芳基炔丙醇以及烷基炔丙醇都能很好地参与到反应中, 且具有较好的官能团兼容性, 带有烯烃、炔烃、羰基、卤素的烷基过氧化物都能很好地参与反应.铁催化剂被认为既起到了单电子催化的作用, 又起到了路易斯酸催化的作用.首先, 炔丙醇99在三价铁的活化下脱去一分子水生成1, 3-烯炔中间体103, 过氧化物被二价铁还原后同时产生叔丁氧基负离子和羧基自由基; 后者脱去一分子二氧化碳生成烷基自由基, 该自由基对1, 3-烯炔103加成生成炔丙位自由基104.然后, 该自由基被三价铁氧化成炔丙基正离子105, 并被叔丁氧基负离子攫取质子后生成最终的1, 3-烯炔类产物.
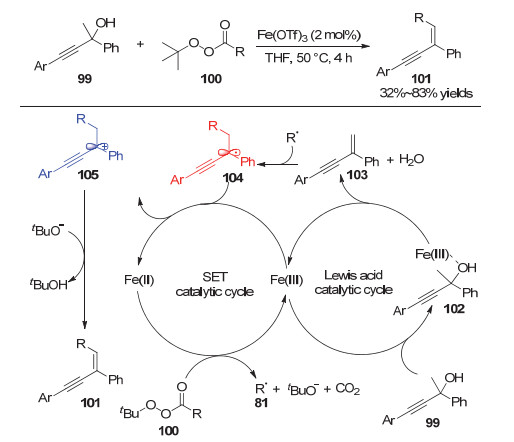
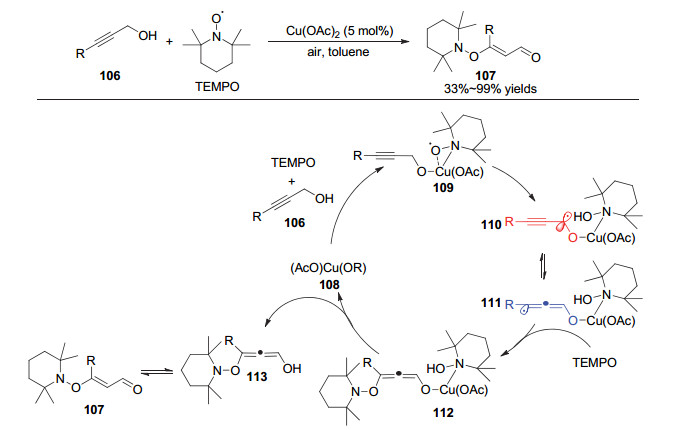
2019年, Jang等[28]通过铜催化实现了四甲基哌啶氮氧化物(TEMPO)对炔丙醇的加成反应, 合成了乙烯基烷氧胺类化合物(图 20).在该反应中, 芳基或烷基取代的炔丙醇都能够较好地参与到反应中.作者通过控制实验排除了经历炔醛中间体的反应过程, 并且验证了底物中羟基以及炔烃官能团的重要性.具体机理如图所示:首先, Cu(OAc)2, 炔丙醇底物和TEMPO共同作用生成三中心两电子的Cu(Ⅱ)-TEMPO自由基加和物109; 随后, TEMPO攫取炔丙醇氧α位的氢原子后生成炔丙基自由基中间体110, 并异构为联烯自由基111; 随后, 另一分子TEMPO与联烯自由基111偶联生成联烯醇铜中间体112, 解离后经过烯醇互变生成乙烯基烷氧胺类产物.
综上所述, 近年来炔丙基自由基参与的化学转化得到了较好的发展.不仅通过自由基途径实现了非端炔类化合物的炔丙位官能化反应, 有效合成了新的炔烃类化合物, 而且也为联烯、1, 3-烯炔等其它不饱和化合物的合成提供了有效的方法.
在今后的研究中, 这一领域仍有三个方面值得努力: (1)与炔丙基自由基偶联的组分或自由基途径炔丙位官能化的类型还有待进一步拓展, 目前局限于碳碳键的形成; (2)炔丙位官能化的原子经济性或步骤经济性有待进一步改善, 可以考虑过氢原子转移的方式产生炔丙基自由基[29]; (3)利用炔丙基自由基合成手性联烯类化合物方面, 对映选择性控制还有待进一步改进.相信通过合成化学家们的持续努力, 炔丙位自由基参与的反应一定会更加成熟, 为有机合成的发展助力!
(a) Trost, B. M.; Li, C.-J. Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley-VCH, New York, 2014. (b) Trotuş, I. T.; Zimmermann, T.; Schüth, F. Chem. Rev. 2014, 114, 1761. (c) Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev. 2016, 116, 3086. (d) Huang, D.; Liu, Y.; Qin, A.-J.; Tang, B.-Z. Polym. Chem. 2018, 9, 2853.
Ding, C.-H.; Hou, X.-L. Chem. Rev. 2011, 111, 1914. doi: 10.1021/cr100284m
(a) Nicholas, K. M.; Pettit, R. Tetrahedron Lett. 1971, 37, 3475. (b) Nicholas, K. M.; Pettit, R. J. Organomet. Chem. 1972, 44, 21.
Melikyan, G. G. Acc. Chem. Res. 2015, 48, 1065. doi: 10.1021/ar500365v
Geri, R.; Oilizzi, C.; Lardicci, L.; Caporusso, A. M. Gazz. Chim. Ital. 1994, 124, 241.
(a) Miyake, Y.; Uemura, S.; Nishibayashi, Y. ChemCatChem 2009, 1, 342. (b) Zhang, D.-Y.; Hu, X.-P. Tetrahedron Lett. 2015, 56, 283. (c) Xiao, Y.-L.; Pan, Q.; Zhang, X.-G. Acta Chim. Sinica 2015, 73, 383(in Chinese). (肖玉兰, 潘强, 张新刚, 化学学报, 2015, 73, 383).
Bruneau, C.; Dixneuf, P. H. Metal Vinylidenes and Allenylidenes in Catalysis, Wiley-VCH, Weinheim, 2008.
(a) Kropf, H.; SchrÖder, R.; FÖlsing, R. Synthesis 1977, 894. (b) Alvarez, L. X.; Christ, M. L.; Sorokin, A. B. Appl. Catal. A: Gen. 2007, 325, 303.
Smith, S. W.; Fu, G. C. J. Am. Chem. Soc. 2008, 130, 12645. doi: 10.1021/ja805165y
Oelke, A. J.; Sun, J.-W.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 2966. doi: 10.1021/ja300031w
Pelphrey, P. M.; Popov, V. M.; Joska, T. M.; Beierlein, J. M.; Bolstad, E. S. D.; Fillingham, Y. A.; Wright, D. L.; Anderson, A. C. J. Med. Chem. 2007, 50, 940. doi: 10.1021/jm061027h
Schley, N. D.; Fu, G. C. J. Am. Chem. Soc. 2014, 136, 16588. doi: 10.1021/ja508718m
Domingo-Legarda, P.; Soler-Yanes, R.; Quirós-López, M. T.; Buñuel, E.; Cárdenas, D. J. Eur. J. Org. Chem. 2018, 35, 4900.
安伦, 童非非, 张新刚, 化学学报, 2018, 76, 977. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346924.shtmlAn, L.; Tong, F.-F.; Zhang, X.-G. Acta Chim. Sinica 2018, 76, 977(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346924.shtml
Lu, F.-D.; Liu, D.; Zhu, L.; Lu, L.-Q.; Yang, Q.; Zhou, Q.-Q.; Wei, Y.; Lan, Y.; Xiao, W.-J. J. Am. Chem. Soc. 2019, 141, 6167. doi: 10.1021/jacs.9b02338
Cheng, J.-K.; Loh, T.-P. J. Am. Chem. Soc. 2015, 137, 42. doi: 10.1021/ja510635k
Andia, A. A.; Miner, M. R.; Woerpel, K. A. Org. Lett. 2015, 17, 2704. doi: 10.1021/acs.orglett.5b01120
Miner, M. R.; Woerpel, K. A. Eur. J. Org. Chem. 2016, 1860.
Cheng, J.-K.; Shen, L.; Wu, L.-H.; Hu, X.-H.; Loh, T.-P. Chem. Commun. 2017, 53, 12830. doi: 10.1039/C7CC08074C
(a) Chen, Z.-Y.; Zhou, D.-S.; Zhou, J.-Y.; Wu, S.-H. Acta Chim. Sinica 1997, 55, 1138(in Chinese). (陈招银, 周大顺, 周景尧, 吴世晖, 化学学报, 1997, 55, 1138). (b) Ma, S.-M. Chem. Rev. 2005, 105, 2829. (c) Lo, V. K.; Wong, M.; Che, C. Org. Lett. 2008, 10, 517. (d) Yang, L.-J.; Ma, J.-A. Acta Chim. Sinica 2016, 74, 130(in Chinese). (杨丽军, 马军安, 化学学报, 2016, 74, 130). (e) Huang, X.; Ma, S.-M. Acc. Chem. Res. 2019, 52, 1301.
(a) Wartenberg, F.-H.; Junga, H.; Blechert, S. Tetrahedron Lett. 1993, 34, 5251. (b) Alameda-Angulo, C.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 2006, 47, 913.
Soler-Yanes, R.; Arribas-Álvarez, I.; Guisán-Ceinos, M.; Buñuel, E.; Cárdenas, D. J. Chem. Eur. J. 2017, 23, 1584. doi: 10.1002/chem.201603758
Wang, F.; Wang, D.-H.; Zhou, Y.; Liang, L.; Lu, R.-H.; Chen, P.-H.; Lin, Z.-Y.; Liu, G.-S. Angew. Chem., Int. Ed. 2018, 57, 7140. doi: 10.1002/anie.201803668
Zhu, X.; Deng, W.; Chiou, M.-F.; Ye, C.; Jian, W.; Zeng, Y.; Jiao, Y.; Ge, L.; Li, Y.; Zhang, X.; Bao, H. J. Am. Chem. Soc. 2019, 141, 548. doi: 10.1021/jacs.8b11499
Ye, C.-Q.; Li, Y.-J.; Zhu, X.-T.; Hu, S.-M.; Yuan, D.-Q.; Bao, H.-L. Chem. Sci. 2019, 10, 3632. doi: 10.1039/C8SC05689G
Zhang, K.-F.; Bian, K.-J.; Li, C.; Sheng, J.; Li, Y.; Wang, X.-S. Angew. Chem. Int. Ed. 2019, 58, 5069. doi: 10.1002/anie.201813184
Ye, C.-Q.; Qian, B.; Li, Y.-J.; Su, M.; Li, D.-L.; Bao, H.-L. Org. Lett. 2018, 20, 3202. doi: 10.1021/acs.orglett.8b01043
Kang, Y.-W.; Choi, Y.-J.; Jang, H.-Y. Org. Lett. 2014, 16, 4842. doi: 10.1021/ol502341f
Horn, E. J.; Rosen, B. R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M. D.; Baran, P. S. Nature 2016, 533, 77. doi: 10.1038/nature17431

图 1 炔丙基自由的转化(FG:官能团)
Figure 1 Transformations of propargylic radicals (FG: functional group)

图 4 二氢叶酸还原酶抑制剂13的催化不对称合成
Figure 4 Catalytic asymmetric synthesis of a potent dihydrofolate reductase inhibitor

图 8 可见光催化和铜催化协同作用的不对称炔丙位氰基化反应
Figure 8 Asymmetric propargylic cyanation enabled by synergetic photoredox and copper catalysis

图 9 铜催化和钴催化的1, 3-烯炔的过氧化-氧烷基化反应
Figure 9 Copper- and cobalt-catalyzed peroxidation-oxyalkylation reactions of 1, 3-enynes

图 11 铜催化1, 3-烯炔的过氧化反应
Figure 11 Copper-catalyzed peroxidation reactions of 1, 3-enynes

图 12 铁催化1, 3-烯炔的过氧化-氨甲酰化反应
Figure 12 Iron-catalyzed peroxidation-carbamylation reactions of 1, 3-enynes

图 13 AIBN引发的炔丙位自由基到联烯自由基的异构
Figure 13 Isomerization of propargylic radical to allene radical initiated by AIBN

图 14 镍催化1, 3-烯炔的1, 4-双官能化反应
Figure 14 Nickel-catalyzed 1, 4-difunctionalization of 1, 3-enynes

图 15 铜催化1, 3-烯炔的1, 2-和1, 4-双官能化反应
Figure 15 Copper-catalyzed 1, 2- and 1, 4-difunctionalization of 1, 3-enynes

图 16 铜催化1, 3-烯炔的1, 4-双官能化反应
Figure 16 Copper-catalyzed 1, 4-difunctionalization of 1, 3-enynes

图 17 铜催化1, 3-烯炔的1, 4-烷基芳基化反应
Figure 17 Copper-catalyzed 1, 4-alkylarylation of 1, 3-enynes

图 18 镍催化1, 3-烯炔的1, 4-双官能化反应
Figure 18 Nickel-catalyzed 1, 4-difunctionalization of 1, 3-enynes

图 19 铁催化的炔丙醇脱水烷基化反应
Figure 19 Iron-catalyzed dehydrative alkylation of propargyl alcohols

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:







 下载:
下载: