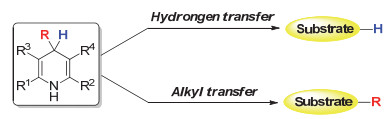
图式 1.
1, 4-二氢吡啶4-位的氢或烷基迁移
Scheme 1.
Hydrongen and alkyl transfer of DHPs

具有1, 4-二氢吡啶结构的汉斯酯(Hantzsch Esters (HEs))在1881年首次被德国化学家Hantzsch[1]合成以来, 不仅在药物化学领域受到广泛关注[2], 同时在有机合成领域中也获得了广泛的应用.由于这类化合物的1, 4-二氢吡啶(DHPs)结构在热力学上有趋芳构化的优势, 因此这类化合物在早期的应用主要是通过氧化剂对其的氧化脱氢反应来制备一些具有不同取代基的吡啶类化合物[3].汉斯酯这种脱氢芳构化的驱动力也表明其具有提供还原性氢的能力, 1955年, Mauzerall和Westheimer[4]首次发现利用1, 4-二氢吡啶化合物可以还原孔雀绿(Malachite Green)得到相应的加氢产物.在此之后, 利用汉斯酯1, 4-二氢吡啶类化合物作为还原剂的加氢还原反应被大量发现并报道[5].同时, 汉斯酯这类1, 4-二氢吡啶类化合物也是辅酶NADH(还原型烟酰胺腺嘌呤二核苷酸)的模型分子, 对研究辅酶NADH在生物体中的氧化还原过程具有不可替代的作用.
汉斯酯参与的氢化还原反应主要是通过1, 4-二氢吡啶结构的4-位的氢迁移来实现, 同时, 4-位非氢取代(如烷基)的汉斯酯在氧化剂作用下也很容易发生非氢取代基的脱除[6].这表明4-位烷基取代的汉斯酯在一定的催化条件下可以通过碳碳键的断裂发生烷基的迁移, 并与相应的原料反应生成烷基取代的产物(Scheme 1).因此, 4-位烷基取代的1, 4-二氢吡啶化合物可以作为一类很好的烷基化试剂.相比于应用广泛的金属烷基化试剂, 用4-位烷基取代的1, 4-二氢吡啶化合物作为烷基化试剂具有以下优点: (1)避免使用价格高昂并且毒性大的烷基金属试剂; (2)相对于烷基金属试剂对水的敏感性, 汉斯酯类化合物具有易操作的优点; (3)由于汉斯酯具有1, 4-二氢吡啶这一仿生骨架, 因此这类烷基化试剂将有助于研究生命体内发生的烷基化反应; (4)这类烷基化试剂简单易得, 很容易通过醛、酮和胺的三组份缩合反应制得[7].虽然4-位烷基取代的1, 4-二氢吡啶化合物作为烷基化试剂具有很多优点, 但是这类烷基迁移反应也具有很大的挑战, 例如烷基迁移与氢迁移的竞争等.
近几年, 大量利用汉斯酯作为烷基化试剂的反应被发现和报道, 科学家发现这类烷基化反应的历程主要是烷基自由基的迁移历程.随着近年来自由基化学的快速发展[8], 特别是光催化领域的发展[9], 为自由基反应提供了温和的自由基激发条件, 使得该类烷基自由基迁移反应的多样化得以成功实现.本文将按照汉斯酯作为烷基化试剂参与的不同反应类型进行介绍.
2013年, Tang小组[10]报道了首例通过路易斯酸催化的烷基汉斯酯的烷基迁移对亚胺的烷基加成反应.在这个反应中, 烷基化的迁移被认为是一个间接协同迁移过程, 在该报道中, 一级和二级的烷基均能很好地对亚胺进行加成反应得到饱和的胺类化合物(Scheme 2).
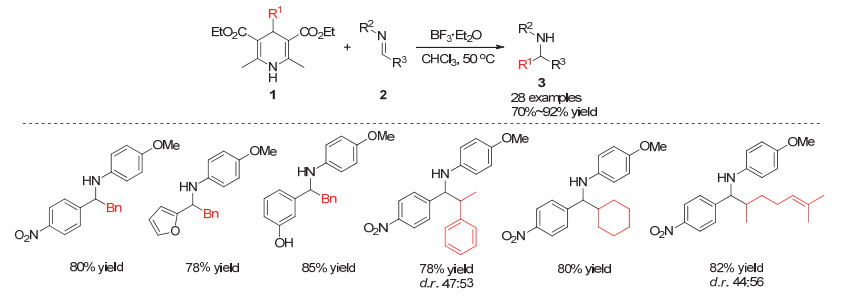
Yu小组[11]在2017年成功实现了光催化下烷基汉斯酯的烷基自由基迁移对亚胺的加成反应.该反应在光催化剂Ir(bpy)2(dtbbpy)PF6或Rh(bpy)3Cl2与苯甲酸共催化作用下能够高效合成含有季碳中心的胺类化合物.研究发现烷基取代的汉斯酯能够与激发态的光催化剂进行单电子转移而形成1, 4-二氢吡啶化合物的自由基正离子, 该中间体进一步发生4-位的碳碳键断裂得到吡啶阳离子6和烷基自由基7, 烷基自由基7可以通过对亚胺的自由基插入或胺自由基9的自由基偶联反应两种路径得到烷基化的胺类化合物5 (Scheme 3).在该体系中, 不同类型的杂环亚胺以及一级或二级的烷基均能很好地兼容.
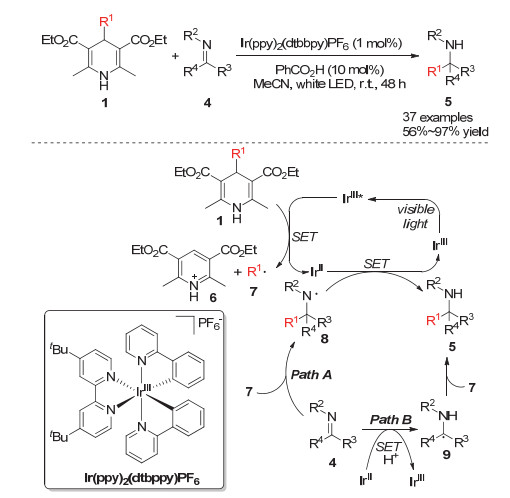
Cheng小组[12]在2017年实现了光催化下烷基汉斯酯的烷基自由基迁移对烯烃的加成反应, 在该体系中使用过渡金属Ir作为光敏催化剂.研究者认为烷基取代的汉斯酯类化合物10可以被光激发并发生4-位的碳碳键断裂形成烷基自由基和DHP自由基15, 烷基自由基可以与底物烯烃进行自由基插入而得到自由基中间体18.该中间体可以通过两种方式得到目标产物:一种是经过Ir催化循环的单电子还原过程得到自由基负离子19, 再进一步质子化得到目标产物; 另一种是与体系中产生的氢自由基进行自由基偶联直接得到目标分子.当烯烃底物是3-位芳基取代的丙烯酸衍生物时, 烷基的加成发生在丙烯酸衍生物的2-号位, 这主要是由于芳基的共轭效应可以稳定烷基自由基的加成中间体.作者同时还发现当使用氰基取代的烷基汉斯酯作为烷基来源时, 可以实现叔碳烷基的迁移加成反应(Scheme 4).
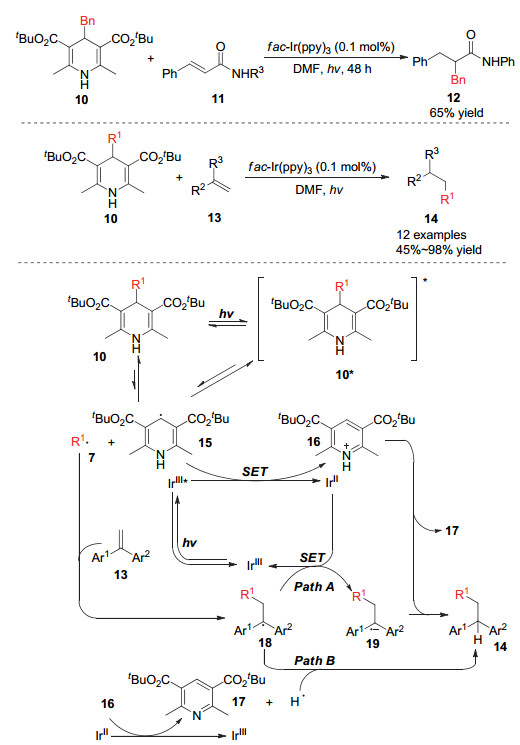
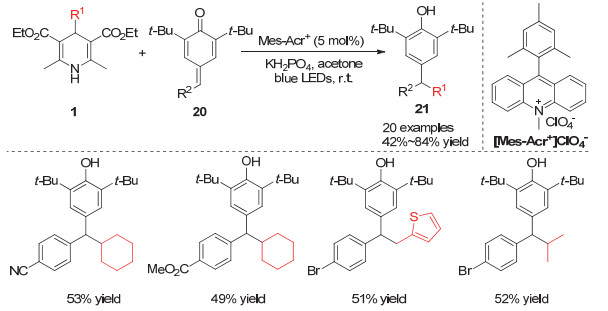
2018年Liu和Ao[13]发展了利用有机光敏催化剂催化的烷基取代汉斯酯对不饱和醌类化合物的烷基加成反应.通过此方法可以高效合成大位阻的酚类化合物21, 研究发现, 当使用氰基取代的汉斯酯作为烷基来源时, 可实现叔碳烷基自由基的迁移加成, 构建对位含有一个季碳中心的大位阻苯酚化合物(Scheme 5).
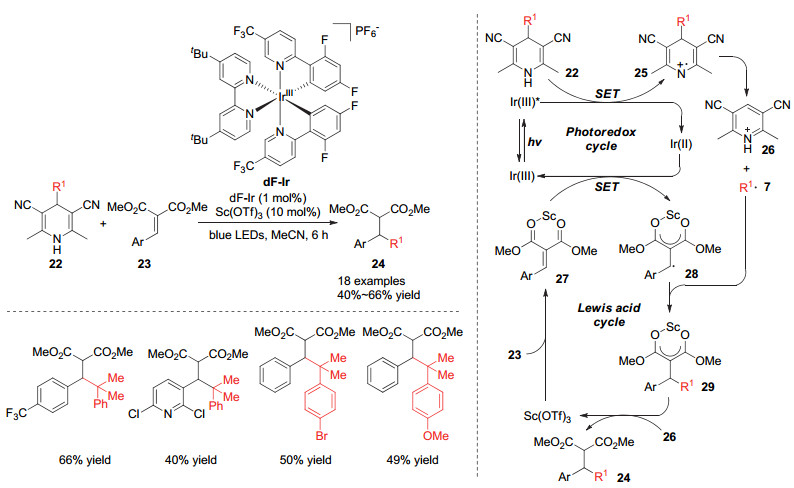
2018年, Scheidt小组[14]利用氰基取代的1, 4-二氢吡啶化合物22作为烷基来源, 通过叔碳中心的烷基对贫电子烯烃的加成, 从而实现了季碳中心的构建.该反应是通过光敏催化剂和路易斯酸共催化的作用实现的, 其催化历程包含光催化循环和路易斯酸催化循环. 1, 4-二氢吡啶化合物22与激发态的光敏催化剂发生单电子转移, 氧化成1, 4-二氢吡啶自由基阳离子25, 该自由基阳离子会在4-位发生碳碳键断裂形成烷基自由基和吡啶正离子.随后还原态的光敏催化剂与路易斯酸活化的烯烃发生单电子转移, 烯烃被还原成为自由基中间体28, 该中间体进一步与1, 4-二氢吡啶产生的自由基发生自由基偶联, 得到烷基加成产物24.这一方法也是利用DHP类底物作为烷基来源构建季碳中心的高效方法(Scheme 6).
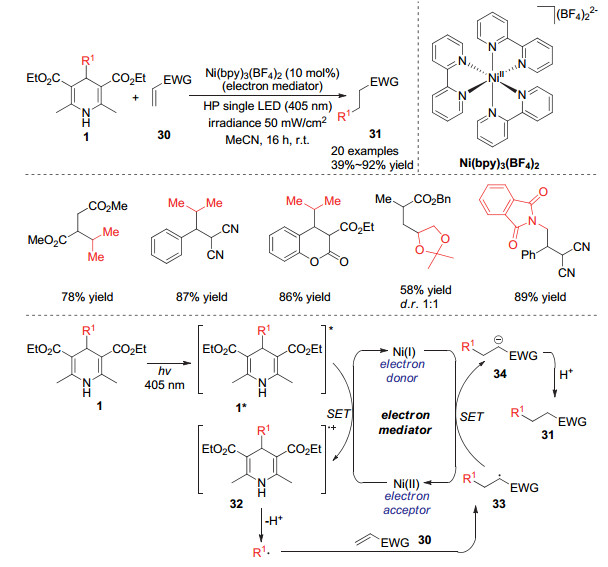
汉斯酯由于其能吸收特定波长的光子而形成激发态的1, 4-二氢吡啶, 该激发态的分子很容易与氧化剂发生单电子转移而被氧化成自由基正离子, 经过碳碳键断裂从而产生烷基自由基. Melchiorre小组[15]在2019年报道了通过光直接激发4-位烷基取代的汉斯酯产生烷基自由基对贫电子烯烃的加成反应.在该体系中需要加入过渡金属Ni作为电子转移媒介, 研究发现4-烷基取代的汉斯酯可以在波长405 nm的光照下形成激发态的DHPs, 该激发态的分子可以将体系中的Ni(Ⅱ)还原为Ni(Ⅰ), 而自身被氧化成自由基正离子32.自由基正离子经过4-位的碳碳键断裂形成的烷基自由基可以对贫电子烯烃进行双键的插入得到自由基中间体33.最后中间体33与体系中被还原的Ni(Ⅰ)发生单电子转移而再生Ni(Ⅱ)和负离子中间体34, 负离子中间体34再进一步质子化得到烷基加成产物31.过渡金属镍在体系中起着传递电子的作用, 从而促进反应中的还原历程.同时Melchiorre小组还发现利用4-位叔丁基取代的氰基1, 4-二氢吡啶作为烷基来源时, 在该反应条件下不能发生烷基的迁移反应, 但是将光源的波长调节至365 nm时, 可以以80%的收率得到叔丁基迁移加成的产物.这也表明该反应历程是通过特定波长的光对1, 4-二氢吡啶类化合物的激发而开始的, 针对不同结构的1, 4-二氢吡啶需要不同波长的光源来进行激发(Scheme 7).
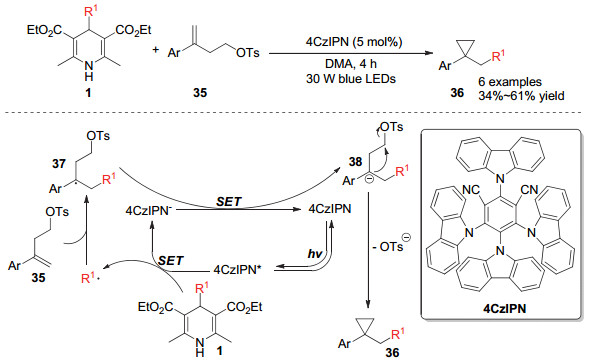
2018年, Kelly和Molander小组[16]报道了利用4-烷基取代的汉斯酯对烯烃进行自由基加成并串联分子内环化合成环丙烷类化合物的方法.该方法利用有机小分子4CzIPN作为光敏催化剂, 激发态的光敏催化剂通过单电子转移被汉斯酯还原形成还原态的光敏催化剂.被氧化的汉斯酯通过碳碳键断裂产生烷基自由基, 该自由基对底物的双键进行自由基插入得到自由基中间体37, 中间体37进一步与还原态的光敏剂发生单电子转移而形成中间体38, 中间体38容易发生分子内环化并离去一份子的对甲苯磺酸负离子而得到目标产物36 (Scheme 8).
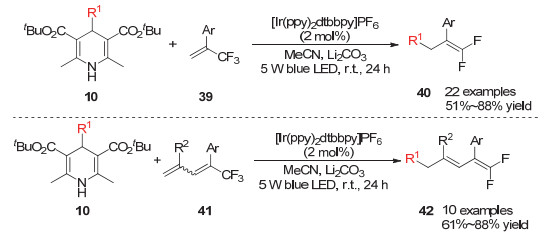
Zhou小组[17]在2019年发展了利用烷基取代的1, 4-二氢吡啶在光敏催化剂作用下产生的烷基自由基对三氟甲基取代的烯烃进行自由基加成后串联脱氟过程的反应, 从而得到端位双氟取代的烯烃产物.该反应使用过渡金属Ir的络合物作为光敏催化剂, 1, 4-二氢吡啶化合物10与激发的光敏剂发生单电子转移后会产生烷基自由基, 该自由基与底物烯烃的双键加成后形成的自由基中间体再经过还原态的光敏催化剂的单电子还原得到负离子中间体, 最后经过三氟甲基上的氟负离子离去得到端位双氟取代的烷基加成烯烃.当使用三氟甲基取代的1, 3-丁二烯类化合物作为底物时, 烷基自由基对双键加成后, 该自由基中间体会经过1, 3-自由基迁移最终得到产物42 (Scheme 9).
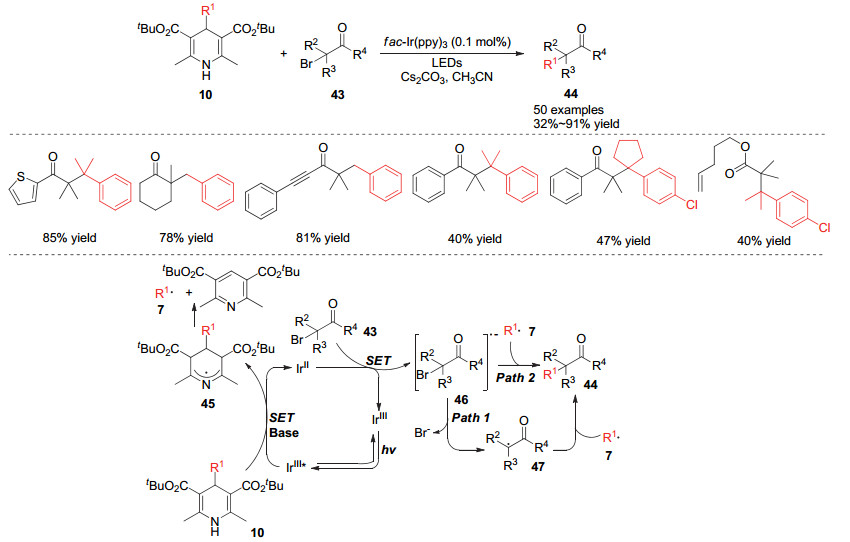
4-烷基取代的1, 4-二氢吡啶类化合物作为烷基化试剂产生的自由基同样可以与卤代物发生碳卤键的取代(偶联)反应. 2016年, Ma和Cheng小组[18]报道了利用4-烷基取代的汉斯酯类化合物作为烷基化试剂, 与α-溴代的酮羰基化合物在光敏催化剂作用下高效构建α-烷基化的酮羰基化合物的方法.该方法利用α-双取代的溴代羰基化合物作为原料可以实现酮羰基的α位的季碳中心的构建, 并且当使用氰基取代的汉斯酯类化合物作为烷基化试剂时, 可以实现叔碳烷基的迁移, 从而合成α、β位均为季碳中心的酮羰基化合物, 一步反应可以同时构建两个季碳中心.该反应历程中, 激发态的光敏催化剂被汉斯酯经过单电子还原后形成还原态光敏催化剂, 该还原态的光敏催化剂与溴代羰基化合物发生单电子转移将溴代羰基化合物还原成自由基负离子46.该活性中间体通过离去溴负离子得到自由基中间体47, 最后与烷基自由基发生自由基偶联得到最终产物.同时研究人员认为自由基负离子46也可以与烷基自由基发生自由基取代而得到最终产物(Scheme 10).
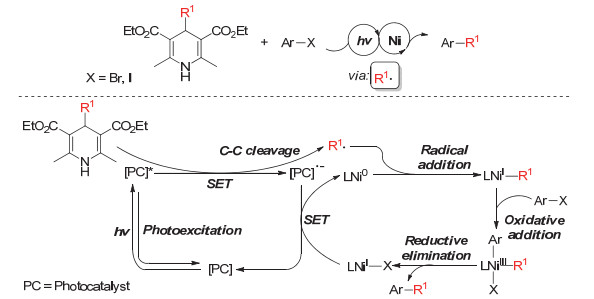
4-烷基取代的汉斯酯类化合物与芳基卤代物的偶联烷基化反应可以通过光敏催化剂与过渡金属Ni共催化实现.其中光敏催化剂的作用主要是: (1)与1, 4-二氢吡啶类化合物进行单电子转移, 通过氧化态的1, 4-二氢吡啶化合物的碳碳键断裂形成烷基自由基; (2)还原态的光敏催化剂单电子还原体系中的Ni(Ⅰ)为Ni(0), 从而完成催化循环.在这类反应中包含光催化氧化还原循环和过渡金属Ni的有机金属氧化还原循环(Scheme 11).
2016年, Nishibayashi小组[19]成功实现了光敏催化剂Ir和过渡金属Ni共催化的芳基碘代物与4-烷基取代的汉斯酯的偶联反应(Scheme 12, Reaction 1).几乎在同时, Molander小组[20]也报道了光敏催化剂4-CzIPN和过渡金属Ni共催化的芳基溴代物与4-烷基取代的汉斯酯的偶联反应(Scheme 12, Reaction 2).在底物适用性方面, 多种杂环溴化物和二级烷基取代的汉斯酯均能很好地转化得到目标的偶联产物.

Molander小组[21]在2018年利用这一策略实现了不同芳基取代的糖类化合物的合成, 这也表明这类方法对于多羟基化合物的兼容性是非常好的. 4-烷基取代的汉斯酯类化合物可以很方便地通过相应的烷基醛来合成, 因此与传统合成芳基糖类化合物方法相比, 使用此方法可以避免使用毒性较大的有机金属试剂, 并且大大拓展了底物官能团的适用性, 为芳基取代的糖类化合物的多样性合成提供了有效方法(Scheme 13).
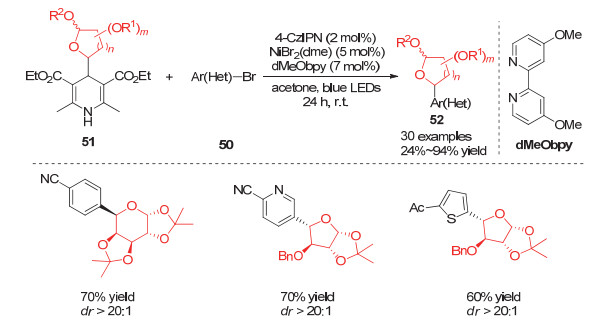
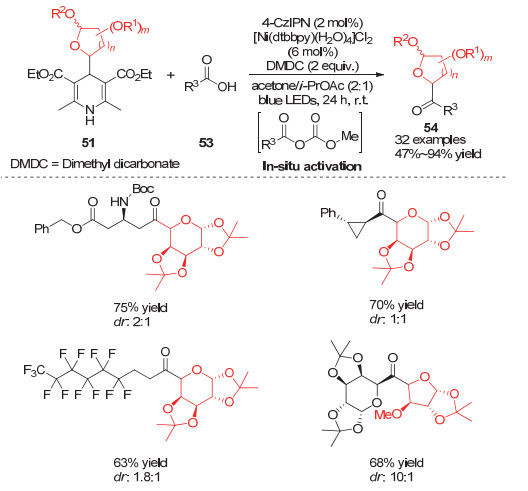
与此同时, Molander小组[22]利用相同策略实现了酰基取代的糖类化合物的合成.他们利用DMDC (Dimethyl dicarbonate)与各种羧酸原位形成相应的活化酸酐, 利用该活性酸酐可以与Ni(Ⅰ)中间体发生类似芳香卤代物的氧化加成, 从而实现各种糖类化合物的酰基化反应.该方法避免了传统合成酰基取代糖类化合物需要加入氧化剂的弊端, 大大拓展了底物取代基的适用性(Scheme 14).
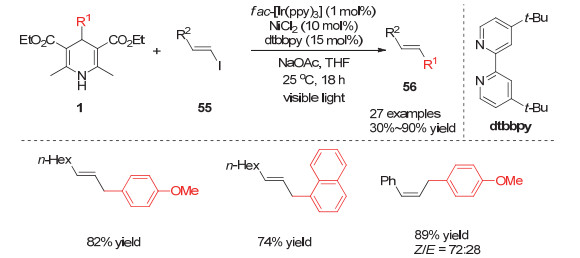
2018年, Nishibayashi小组[23]将他们的催化体系拓展到烯烃碘代物与4-烷基取代的汉斯酯的偶联反应.他们发现当使用烷基取代的反式烯烃碘代物参与反应时可以得到单一反式构型的烷基化偶联产物, 但是当使用芳基取代的反式烯烃碘代物参与反应时, 会得到顺反异构体混合的烯烃产物.进一步机理验证实验表明, 芳基取代的烯烃在光敏催化剂作用下能够发生顺反异构体的转化, 从而得到顺反结构混合的产物(Scheme 15).
Melchiorre小组[24]在2017年发展了无光敏催化剂参与的过渡金属Ni催化的芳基溴化物与4-烷基取代的汉斯酯的烷基化偶联反应.他们通过研究4-位苄基取代的汉斯酯的发射光谱发现, 在405 nm波长的光照下, 在乙腈溶剂中, 该汉斯酯会发生4-位碳碳键的断裂, 并形成苄基自由基.利用这一发现, 他们成功实现了通过特定波长的光对汉斯酯化合物的激发并发生单电子转移从而得到烷基自由基, 在过渡金属Ni的催化作用下与溴代芳烃发生烷基化反应得到偶联产物(Scheme 16).
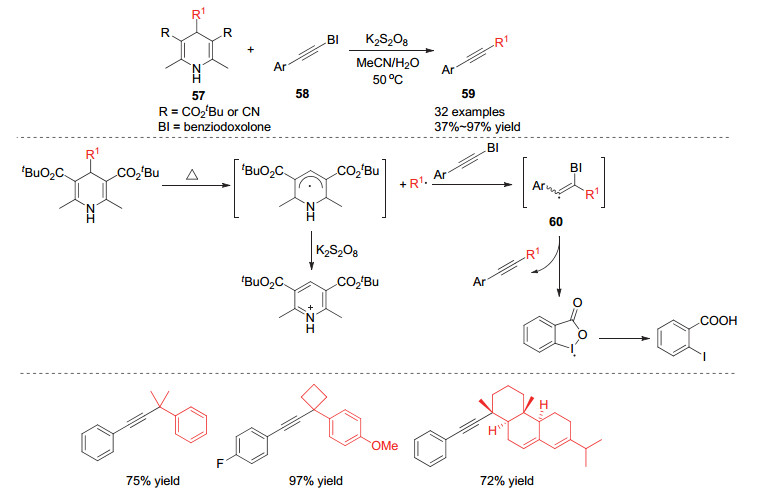
炔烃的官能团化在有机合成中是比较重要的方向[25], 利用烷基取代的汉斯酯同样可以对炔烃进行烷基化. 2018年Cheng小组[26]实现了利用4-烷基取代的汉斯酯类化合物作为烷基化试剂对高价碘Benziodoxolone活化的炔基化合物的烷基化反应, 实现了双取代炔烃的合成.在该反应中, 汉斯酯类化合物在加热条件下产生烷基自由基, 该自由基通过对炔烃三键的自由基插入得到自由基中间体60, 该自由基中间体进一步发生碘自由基的消除离去从而得到烷基化的炔烃产物59.他们还发现当使用氰基取代的汉斯酯作为烷基化试剂时, 能够实现叔碳烷基自由基的迁移, 从而构建一端是季碳中心的炔烃产物(Scheme 17).
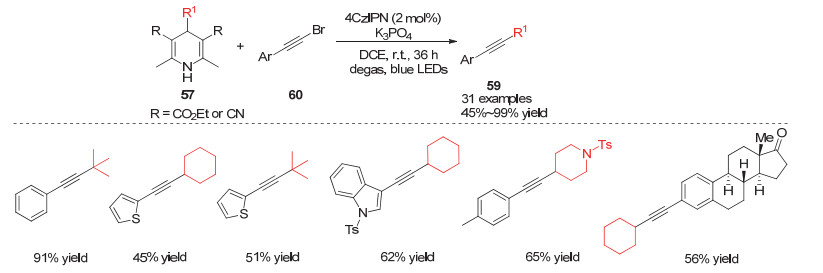
Ye小组[27]在2019年实现了光促进的4-烷基取代的汉斯酯类化合物与炔基溴的烷基化偶联反应.他们利用有机光敏催化剂4CzIPN作为光敏催化剂.在该反应条件下多种杂环炔烃均能很好地参与反应, 同样当要合成季碳中心取代的炔烃时, 可使用氰基取代的汉斯酯化合物作为叔碳烷基来源(Scheme 18).
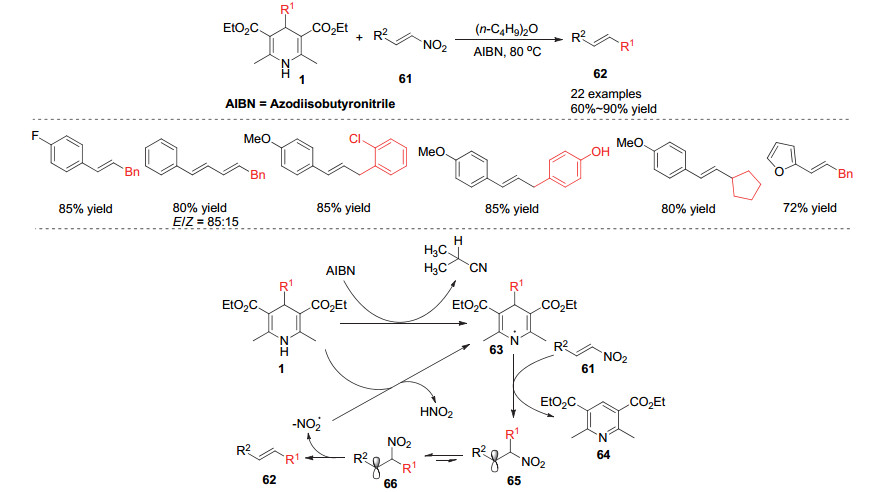
2014年, Tang小组[28]在之前烷基汉斯酯对亚胺加成的工作基础上, 尝试发展烷基汉斯酯对贫电子烯烃的加成反应, 他们发现利用烷基取代的汉斯酯1作为烷基化试剂与硝基烯烃61在AIBN(偶氮二异丁腈)自由基引发作用下可以得到硝基被烷基取代的反式烯烃.进一步研究发现这是通过烷基自由基对烯烃的加成消除历程实现的.烷基取代的汉斯酯1在AIBN的作用下得到DHP(1, 4-二氢吡啶)自由基63, 该自由基在芳构化的驱动力下发生烷基自由基对硝基烯烃61的加成形成自由基中间体65和66, 最后发生硝基自由基的离去形成热力学稳定的反式烯烃.值得注意的是烷基自由基对硝基烯烃的加成是烷基加成在硝基一侧, 这也印证了在该体系中硝基烯烃底物中烯烃另一侧需是富电子共轭结构以稳定自由基中间体65或66 (Scheme 19).
Sakata和Nishibayashi小组在2016年报道了利用烷基取代的汉斯酯作为烷基来源对芳基腈化物的烷基取代反应[29].该反应中光激发态的光敏催化剂与汉斯酯发生单电子转移, 汉斯酯4-位发生碳碳键断裂而形成烷基自由基.同时还原态的光敏催化剂与芳基腈化物发生单电子转移而得到自由基负离子70, 该负离子可以与烷基自由基发生自由基偶联得到对位加成或邻位加成的负离子中间体71或72, 最后通过离去氰基负离子而得到对位氰基或邻位氰基被烷基取代的产物.在这一转化中经历了汉斯酯产生烷基自由基和氰基离去的两次碳碳键断裂, 针对反应历程, 研究者也做了相应的理论DFT计算.结果表明, 烷基取代发生在邻位的自由能垒远高于烷基取代发生在对位的自由基能垒, 这也说明了产物均以对位取代为主的原因.并且研究者发现添加剂醋酸钠虽然能大幅提高反应的产率, 但同时也有助于邻位取代产物的形成(Scheme 20).
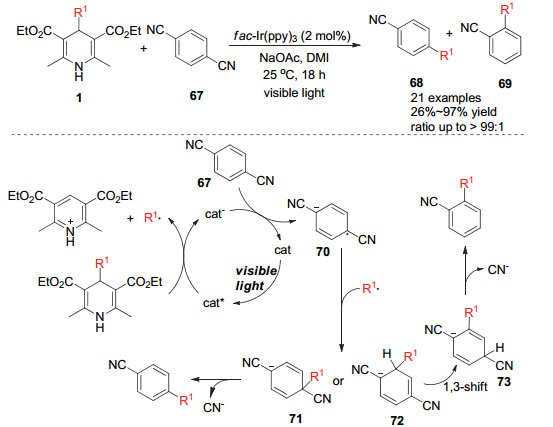
2019年, Molander小组[30]报道了光促进的氮杂环砜类化合物的烷基取代反应. 4-烷基取代的汉斯酯在光敏催化剂作用下产生烷基自由基, 该自由基可以对2-磺酰基取代的氮杂环进行自由基插入形成自由基中间体76, 再进一步被还原态的光敏催化剂单电子还原并离去磺酰基负离子而得到烷基取代的氮杂环化合物75(Scheme 21).
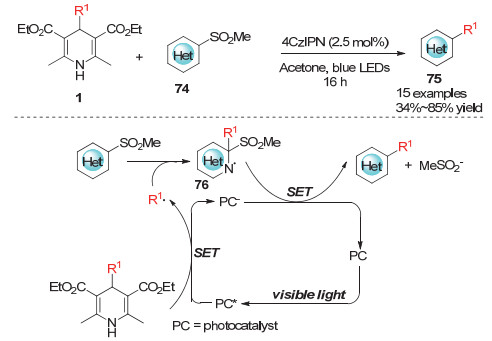
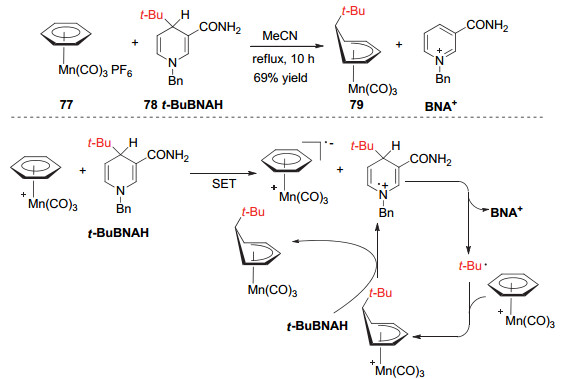
Cao小组[31]在2014年实现了利用1, 4-二氢吡啶化合物78作为烷基化试剂对过渡金属锰化合物77的烷基化反应.反应利用有机金属化合物77与1, 4-二氢吡啶的单电子转移促使1, 4-二氢吡啶发生碳碳键断裂形成烷基自由基, 该自由基进一步与有机金属化合物77上的芳环发生自由基取代得到烷基化的有机金属化合物79(Scheme 22).
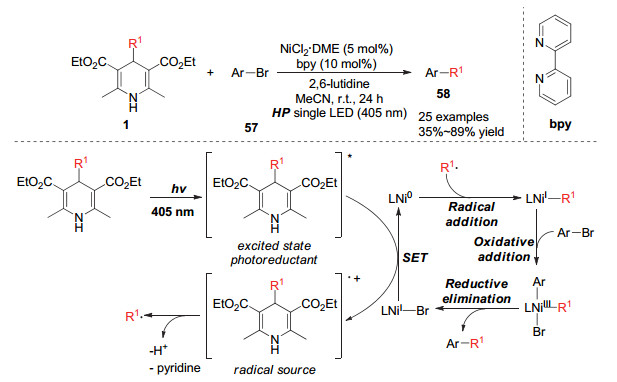
利用亲核性官能团对氮杂环上氮原子的邻位的取代反应是构建2-位官能团化的吡啶或喹啉类化合物的有效方法[32]. 2017年, Molander小组[33]实现了汉斯酯作为烷基化试剂与氮杂环的碳氢氧化偶联反应.该体系利用过硫酸钾作为氧化剂, 汉斯酯被单电子氧化产生烷基自由基, 该自由基可以与氮杂环的碳氮双键进行插入而得到杂环自由基中间体, 该中间体进一步发生氧化脱氢可以得到目标烷基化偶联产物.该方法对底物的适用性非常优秀, 多种官能团化的杂环底物均能很好地参与反应(Scheme 23).
Gutierrez和Molander小组[34]在2018年发展了烷基取代汉斯酯对烯烃取代的环丙烷的烯丙基加成反应, 得到了烷基化的丙烯醇产物.在该反应历程中, 有机光敏催化剂单电子氧化汉斯酯得到烷基自由基, 过渡金属镍会与烯基环丙烷得到烯丙基镍中间体, 该中间体与自由基会发生自由基加成而得到产目标产物.研究者同时还对反应机理进行理论DFT计算, 结果表明该反应的反应历程不同于传统的镍催化的自由基偶联以及Tsuji-Trost反应.反应历程是一个Ni(0)-Ni(Ⅱ)的催化循环, 在与烷基自由基反应之前, 零价镍中间体已经被氧化加成Ni(Ⅱ)中间体, 这与之前光催化剂与过渡金属Ni共催化的烷基自由基偶联反应的Ni(0)-Ni(Ⅰ)的历程是不相同的(Scheme 24).
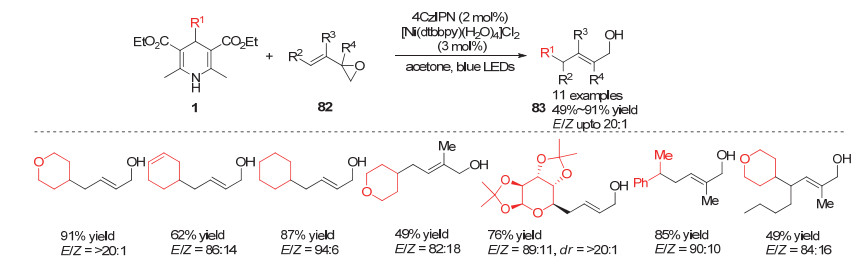
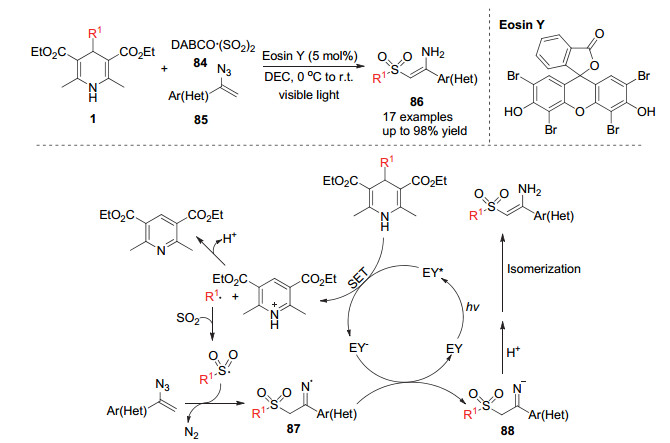
Wu小组[35]在近发展了很多自由基对二氧化硫的插入反应构建磺酰化合物的方法.在2019年, Wu小组[36]报道了在光促进作用下利用4-烷基取代的汉斯酯产生烷基自由基对二氧化硫的插入反应来合成砜类化合物的方法.取代汉斯酯在光催化作用下产生烷基自由基, 该自由基对二氧化硫插入后形成磺酰自由基, 该自由基进一步与叠氮烯烃85插入得到氮自由基中间体87, 该中间体被还原态的光敏催化剂单电子还原可以得到亚胺负离子, 再进一步质子化异构化得到砜类化合物86 (Scheme 25).利用类似策略, Wu小组[37]在2019年利用烷基自由基对二氧化硫插入产生的磺酰自由基对贫电子烯烃的加成得到砜类化合物90 (Scheme 26).
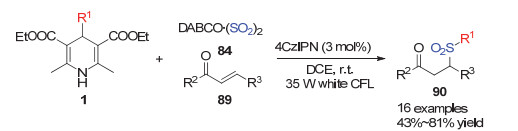
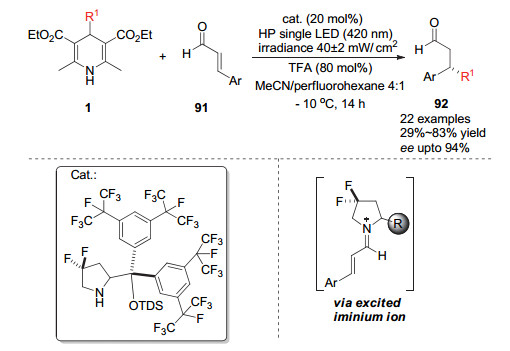
2018年Melchiorre小组[38]利用手性脯氨醇衍生物作为催化剂活化丙烯醛类底物91, 利用烷基取代的汉斯酯作为烷基自由基来源对活性亚胺离子发生自由基偶联得到手性的烷基加成产物.他们利用波长为420 nm的光源激发汉斯酯产生烷基自由基, 同时该光源可激发底物与催化剂形成的亚胺离子过渡态, 最后利用烷基自由基与含有手性催化剂的亚胺离子中间体的自由基不对称偶联得到对映选择性的加成产物92 (Scheme 27).
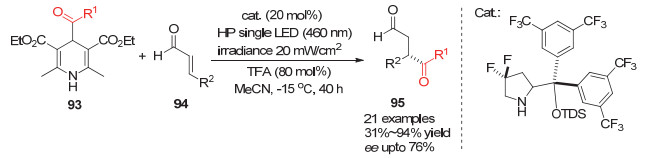
2019年, Melchiorre小组将这一体系拓展到酰基自由基对丙烯醛的不对称加成反应中, 他们用4-位酰基取代的汉斯酯作为酰基自由基的来源, 用手性脯氨醇衍生物做手性催化剂, 在460 nm波长的光照下实现了酰基自由基对丙烯醇类化合物的不对称加成[39](Scheme 28).
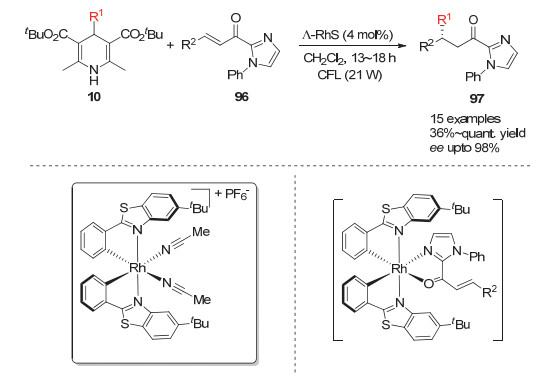
Meggers小组[40]在2018年利用手性光敏催化剂催化贫电子烯烃的不对称烷基化.其中手性光敏催化剂不仅起着诱发4-烷基取代的汉斯酯产生烷基自由基的作用, 同时也作为手性路易斯酸通过与底物的双配位起到底物活化作用.该反应的α, β-不饱和烯烃上需要有额外的杂原子参与催化剂的配位以达到手性空间的诱导作用(Scheme 29).
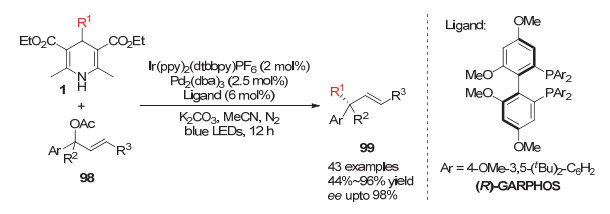
2018年, Yu小组[41]报道了光敏催化剂与过渡金属钯共催化的烯丙基不对称烷基化反应. 4-烷基取代的汉斯酯在光敏催化剂的诱导下产生烷基自由基, 手性钯催化剂生成的烯丙基钯中间体被该自由基中间体发生氧化加成为Pd(Ⅲ)中间体, 再进一步还原消除得到手性的烯丙基产物.含有叔碳的烷基自由基在该体系中也可以很好地参与反应形成带有季碳中心的手性烯丙基产物(Scheme 30).
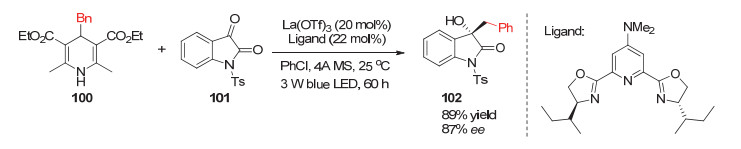
2019年, Jiang小组[42]利用4-苄基取代的汉斯酯作为烷基化试剂, 实现了过渡金属镧催化的苄基对吲哚酮的碳氧双键的不对称加成反应.在该反应中使用的配体是噁唑啉类的手性三齿配体, 产物的对映选择性可以达到89% (Scheme 31).
利用4-取代的汉斯酯类化合物作为烷基化试剂已经发展了多种反应, 并在近年得到快速的进展.与传统烷基化试剂相比具有毒性小、官能团兼容性好等优点, 同时这类烷基化试剂可以快速构建多样性的季碳中心, 并实现多种不对称烷基化反应.并且随着近年自由基化学以及光催化化学的快速发展, 相信会有更多成果被报道出来.但是利用这类烷基化试剂还存在一些挑战, 比如目前报道的4-取代基的迁移几乎是烷基的迁移, sp2碳、sp碳或其它类型取代基的迁移的报道还非常少.另一方面, 使用汉斯酯类化合物作为烷基化试剂时, 离去的吡啶基团的再利用也是目前需要解决的问题.同时, 利用该类试剂参与的不对称反应类型还主要是加成反应, 拓展新的不对称反应类型也是需要进一步研究的.
Hantzsch, A. Ber. Dtsch. Chem. Ges. 1881, 14, 1637. doi: 10.1002/cber.18810140214
(a) Janis, R. A.; Triggle, D. J. J. Med. Chem. 1983, 25, 775. (b) Bocker, R. H.; Guengerich, F. P. J. Med. Chem. 1986, 29, 1596. (c) Xie, W.; Wu, Y.; Zhang, J.; Mei, Q.; Zhang, Y.; Zhu, N.; Liu, R.; Zhang, H. Eur. J. Med. Chem. 2018, 145, 35. (d) Xie, W.; Zhang, H.; He, J.; Zhang, J.; Yu, Q.; Luo, C.; Li, S. Bioorg. Med. Chem. Lett. 2017, 27, 530.
Bergstrom, F. W. Chem. Rev. 1944, 35, 77. doi: 10.1021/cr60111a001
Mauzerall, D.; Westheimer, F. H. J. Am. Chem. Soc. 1955, 77, 2261. doi: 10.1021/ja01613a070
For selected reviews see: (a) Ouellet, S. G.; Walji, A. M.; Macmillan, D. W. C. Acc. Chem. Res. 2007, 40, 1327. (b) de Vries, J. G.; Mrsic, N. Catal. Sci. Technol. 2011, 1, 727. (c) Zheng, C.; You, S.-L. Chem. Soc. Rev. 2012, 41, 2498. (d) Huang, W.; Cheng, X. Synlett 2017, 28, 148. (e) Li, X.; Meng, Y.; Yi, P.; Stepień, M.; Chmielewski, P. J. Angew. Chem., Int. Ed. 2017, 56, 10810.
Loev, B.; Snader, K. M. J. Org. Chem. 1965, 30, 1914. doi: 10.1021/jo01017a048
魏振中, 李江飞, 王泽云, 李品华, 王永秋, 有机化学, 2017, 37, 1835. http://www.cnki.com.cn/Article/CJFDTOTAL-YJHU201707024.htmWei, Z.; Li, J.; Wang, Z.; Li, P.; Wang, Y. Chin. J. Org. Chem. 2017, 37, 1835(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-YJHU201707024.htm
For selected examples see: (a) Zou, Y.-Q.; Hörmann, F. M.; Bach, T. Chem. Soc. Rev. 2018, 47, 278. (b) Wang, F.; Chen, P.; Liu, G. Acc. Chem. Res. 2018, 51, 2036. (c) Wang, K.; Kong, W. Chin. J. Chem. 2018, 36, 247. (d) Qiu, S.; Wang, C.; Xie, S.; Huang, X.; Chen, L.; Zhao, Y.; Zeng, Z. Chem. Commun. 2018, 54, 11383. (e) Xie, L.-Y.; Peng, S.; Liu, F.; Chen, G.-R.; Xia, W.; Yu, X.; Li, W.-F.; Cao, Z.; He, W.-M. Org. Chem. Front. 2018, 5, 2604. (f) Lu, L.-H.; Zhou, S.-J.; He, W.-B.; Xia, W.; Chen, P.; Yu, X.; Xu, X.; He, W.-M. Org. Biomol. Chem. 2018, 16, 9064. (g) Zheng, Y.; Liu, M.; Qiu, G.; Xie, W.; Wu, J. Tetrahedron 2019, 75, 1663. (h) Liu, K.-J.; Jiang, S.; Lu, L.-H.; Tang, L.-L.; Tang, S.-S.; Tang, H.-S.; Tang, Z.; He, W.-M.; Xu, X. Green Chem. 2018, 20, 3038. (i) Xie, L.-Y.; Peng, S.; Liu, F.; Yi, J.-Y.; Wang, M.; Tang, Z.; Xu, X.; He, W.-M. Adv. Synth. Catal. 2018, 360, 4259. (j) Xie, L.-Y.; Peng, S.; Liu, F.; Chen, G.-R.; Xia, W.; Yu, X.; Li, W.-F.; Cao, Z.; He, W.-M. Org. Chem. Front. 2018, 5, 2604. (k) Guo, T.; Wei, X.-N.; Liu, Y.; Zhang, P.-K.; Zhao, Y.-H. Org. Chem. Front. 2019, 6, 1414.
For selected examples see: (a) Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527. (b) Teplý, F. Collect. Czech. Chem. Commun. 2011, 76, 859. (c) Narayanam, J. M.; Stephenson, C. R. Chem. Soc. Rev. 2011, 40, 102. (d) Xuan, J.; Xiao, W. J. Angew. Chem., Int. Ed. 2012, 51, 6828. (e) Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687. (f) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. Chem. Rev. 2013, 113, 5322. (g) Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387. (h) Xuan, J.; Lu, L. Q.; Chen, J. R.; Xiao, W. J. Eur. J. Org. Chem. 2013, 6755. (i) Hari, D. P.; König, B. Angew. Chem., Int. Ed. 2013, 52, 4734. (j) Hopkinson, M. N.; Sahoo, B.; Li, J. L.; Glorius, F. Chem. Eur. J. 2014, 20, 3874. (k) Peñ-López, M.; Rosas-Hernández, A.; Beller, M. Angew. Chem., Int. Ed. 2015, 54, 5006. (l) Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898. (m) Wang, D.; Zhang, L.; Luo, S. Acta Chim. Sinica 2017, 75, 22(in Chinese). (王德红, 张龙, 罗三中, 化学学报, 2017, 75, 22.)
Li, G.; Chen, R.; Wu, L.; Fu, Q.; Zhang, X.; Tang, Z. Angew. Chem., Int. Ed. 2013, 52, 8432. doi: 10.1002/anie.201303696
Zhang, H.-H.; Yu, S. J. Org. Chem. 2017, 82, 9995. doi: 10.1021/acs.joc.7b01425
Gu, F.; Huang, W.; Liu, X.; Chen, W.; Cheng, X. Adv. Synth. Catal. 2017, 360, 925.
Wu, Q.-Y.; Min, Q.-Q.; Ao, G.-Z.; Liu, F. Org. Biomol. Chem. 2018, 16, 6391. doi: 10.1039/C8OB01641K
Mcdonald, B. R.; Scheidt, K. A. Org. Lett. 2018, 20, 6881.
Van Leeuwen, T.; Buzzetti, L.; Perego, L. A.; Melchiorre, P. Angew. Chem., Int. Ed. 2019, 58, 4953. doi: 10.1002/anie.201814497
Milligan, J. A.; Phelan, J. P.; Polites, V. C.; Kelly, C. B.; Molander, G. A. Org. Lett. 2018, 20, 6840. doi: 10.1021/acs.orglett.8b02968
Chen, H.; Anand, D.; Zhou, L. Asian J. Org. Chem. 2019, 8, 661. doi: 10.1002/ajoc.201900026
Chen, W.; Liu, Z.; Tian, J.; Li, J.; Ma, J.; Cheng, X.; Li, G. J. Am. Chem. Soc. 2016, 138, 12312. doi: 10.1021/jacs.6b06379
Nakajima, K.; Nojima, S.; Nishibayashi, Y. Angew. Chem., Int. Ed. 2016, 55, 14106. doi: 10.1002/anie.201606513
Gutiérrez-Bonet, Á.; Tellis, J. C.; Matsui, J. K.; Vara, B. A.; Molander, G. A. ACS Catal. 2016, 6, 8004. doi: 10.1021/acscatal.6b02786
Dumoulin, A.; Matsui, J. K.; Gutiérrez-Bonet, Á.; Molander, G. A. Angew. Chem., Int. Ed. 2018, 57, 6614. doi: 10.1002/anie.201802282
Badir, S. O.; Dumoulin, A.; Matsui, J. K.; Molander, G. A. Angew. Chem., Int. Ed. 2018, 57, 6610. doi: 10.1002/anie.201800701
Nakajima, K.; Guo, X.; Nishibayashi, Y. Chem. Asian J. 2018, 13, 3653. doi: 10.1002/asia.201801542
Buzzetti, L.; Prieto, A.; Roy, S. R.; Melchiorre, P. Angew. Chem., Int. Ed. 2017, 56, 15039. doi: 10.1002/anie.201709571
For selected examples see: (a) Wu, C.; Lu, L.-H.; Peng, A.-Z.; Jia, G.-K.; Peng, C.; Cao, Z.; Tang, Z.; He, W.-M.; Xu, X. Green Chem. 2018, 20, 3683. (b) Lu, L.-H.; Zhou, S.-J.; Sun, M.; Chen, J.-L.; Xia, W.; Yu, X.; Xu, X.; He, W.-M. ACS Sustainable Chem. Eng. 2019, 7, 1574. (c) Wu, C.; Xiao, H.-J.; Wang, S.-W.; Tang, M.-S.; Tang, Z.-L.; Xia, W.; Li, W.-F.; Zhong, C.; He, W.-M. ACS Sustainable Chem. Eng. 2019, 7, 2169. (d) Wu, C.; Wang, Z.; Hu, Z.; Zeng, F.; Zhang, X.-Y.; Cao, Z.; Tang, Z.; He, W.-M.; Xu, X. Org. Biomol. Chem. 2018, 16, 3177. (e) Wang, Z.; Yang, L.; Liu, H.-L.; Tan, Y.-Z.; Bao, W.-H.; Wang, M.; Tang, Z.; He, W.-M. Chin. J. Org. Chem. 2018, 38, 2639(in Chinese). (王峥, 杨柳, 刘慧兰, 谭英芝, 包文虎, 汪明, 唐子龙, 何卫民, 有机化学, 2018, 38, 2639.)
Liu, X.; Liu, R.; Dai, J.; Cheng, X.; Li, G. Org. Lett. 2018, 20, 6906. doi: 10.1021/acs.orglett.8b03050
Song, Z.-Y.; Zhang, C.-L.; Ye, S. Org. Biomol. Chem. 2019, 17, 181. doi: 10.1039/C8OB02912A
Li, G.; Wu, L.; Lv, G.; Liu, H.; Fu, Q.; Zhang, X.; Tang, Z. Chem. Commun. 2014, 50, 6246. doi: 10.1039/C4CC01119H
Nakajima, K.; Nojima, S.; Sakata, K.; Nishibayashi, Y. ChemCatChem 2016, 8, 1028. doi: 10.1002/cctc.201600037
Wang, Z.-J.; Zheng, S.; Matsui, J. K.; Liu, Z.; Molander, G. A. Chem. Sci. 2019, 10, 4389. doi: 10.1039/C9SC00776H
Cao, L.; Zheng, L.; Huang, Q. J. Organomet. Chem. 2014, 768, 56. doi: 10.1016/j.jorganchem.2014.06.021
For selected examples see: (a) Xie, L.-Y.; Peng, S.; Tan, J.-X.; Sun, R.-X.; Yu, X.; Dai, N.-N.; Tang, Z.-L.; Xu, X.; He, W.-M. ACS Sustainable Chem. Eng. 2018, 6, 16976. (b) Xie, L.-Y.; Peng, S.; Lu, L.-H.; Hu, J.; Bao, W.-H.; Zeng, F.; Tang, Z.; Xu, X.; He, W.-M. ACS Sustainable Chem. Eng. 2018, 6, 7989. (c) Xie, L.-Y.; Peng, S.; Jiang, L.-L.; Peng, X.; Xia, W.; Yu, X.; Wang, X.-X.; Cao, Z.; He, W.-M. Org. Chem. Front. 2019, 6, 167.
Gutiérrez-Bonet, Á.; Remeur, C.; Matsui, J. K.; Molander, G. A. J. Am. Chem. Soc. 2017, 139, 12251. doi: 10.1021/jacs.7b05899
Matsui, J. K.; Gutiérrez-Bonet, Á.; Rotella, M.; Alam, R.; Gutierrez, O.; Molander, G. A. Angew. Chem., Int. Ed. 2018, 57, 15847. doi: 10.1002/anie.201809919
For selected examples see: (a) Gong, X.; Wang, M.; Ye, S.; Wu, J. Org. Lett. 2019, 21, 1156. (b) Ye, S.; Qiu, G.; Wu, J. Chem. Commun. 2019, 55, 1013. (c) Ye, S.; Zheng, D.; Wu, J.; Qiu, G. Chem. Commun. 2019, 55, 2214. (d) Ye, S.; Li, Y.; Wu, J.; Li, Z. Chem. Commun. 2019, 55, 2489. (e) Gong, X.; Li, X.; Xie, W.; Wu, J.; Ye, S. Org. Chem. Front. 2019, 6, 1863. (f) Zhang, J.; Xie, W.; Ye, S.; Wu, J. Org. Chem. Front. 2019, 6, 2254. (g) Ye, S.; Xiang, T.; Li, X.; Wu, J. Org. Chem. Front. 2019, 6, 2183. (h) Ye, S.; Li, X.; Xie, W.; Wu, J. Asian J. Org. Chem. 2019, 8, 893. (i) Ye, S.; Li, X.; Xie, W.; Wu, J. Eur. J. Org. Chem. 2019, 10.1002/ejoc.201900396. (j) Zhang, J.; Li, X.; Xie, W.; Ye, S.; Wu, J. Org. Lett. 2019, 21, DOI: 10.1021/acs.orglett.9b01323.(k)Zong,Y.;Lang,Y.;Yang,M.;Li,X.;Fan,X.;Wu,J.Org.Lett.2019,21,1935.
Wang, X.; Li, H.; Qiu, G.; Wu, J. Chem. Commun. 2019, 55, 2062. doi: 10.1039/C8CC10246E
Wang, X.; Yang, M.; Xie, W.; Fan, X.; Wu, J. Chem. Commun. 2019, 55, 6010. doi: 10.1039/C9CC03004B
Verrier, C.; Alandini, N.; Pezzetta, C.; Moliterno, M.; Buzzetti, L.; Hepburn, H. B.; Vega-Penaloza, A.; Silvi, M.; Melchiorre, P. ACS Catal. 2018, 8, 1062. doi: 10.1021/acscatal.7b03788
Goti, G.; Bieszczad, B.; Vega-Penaloza, A.; Melchiorre, P. Angew. Chem., Int. Ed. 2019, 58, 1213. doi: 10.1002/anie.201810798
de Assis, F. F.; Huang, X.; Akiyama, M.; Pilli, R. A.; Meggers, E. J. Org. Chem. 2018, 83, 10922. doi: 10.1021/acs.joc.8b01588
Zhang, H.-H.; Zhao, J.-J.; Yu, S. J. Am. Chem. Soc. 2018, 140, 16914. doi: 10.1021/jacs.8b10766
Li, F.; Tian, D.; Fan, Y.; Lee, R.; Lu, G.; Yin, Y.; Qiao, B.; Zhao, X.; Xiao, Z.; Jiang, Z. Nat. Commun. 2019, DOI: 10.1038/s41467-019-09857-9.

图式 5 有机光敏催化剂催化的烷基自由基对烯烃的加成
Scheme 5 Alkylation of olefins catalyzed by organo-photocatalyst

图式 6 路易斯酸和有机光敏催化剂共催化的烷基自由基对烯烃的加成
Scheme 6 Alkylation of olefins co-catalyzed by Lewis acid and organo-photocatalyst

图式 7 通过光直接激发1, 4-二氢吡啶产生烷基自由基
Scheme 7 Generation of alkyl radicals from direct excitation of DHPs

图式 9 烷基自由基对三氟甲基取代烯烃的脱氟烷基化反应
Scheme 9 Defluorinative alkylation of 1-trifluoromethyl alkenes with alkyl radicals

图式 12 光敏催化剂与过渡金属Ni共催化的sp2碳卤键断裂的烷基化偶联反应
Scheme 12 Photoredox/Nickel co-catalyzed alkylation of aryl halides

图式 13 通过光敏催化剂与过渡金属Ni共催化来合成芳基取代的糖类化合物
Scheme 13 Synthesis of arylated saccharides through Nickel and Photoredox dual catalysis

图式 14 通过光敏催化剂与过渡金属Ni共催化来合成羰基取代的糖类化合物
Scheme 14 Synthesis of acylated saccharides through Nickel and Photoredox dual catalysis

图式 15 通过光敏催化剂与过渡金属Ni共催化的卤代烯烃的烷基化偶联反应
Scheme 15 Cross-coupling of alkenyl halides through Nickel and Photoredox dual catalysis

图式 16 过渡金属Ni参与的芳基溴代物的烷基化偶联反应
Scheme 16 Cross-coupling of aryl bromide through Nickel catalysis

图式 18 光促进的烷基自由基的炔基化反应
Scheme 18 Visible light promoted alkynylation with alkyl radicals

图式 20 烷基自由基对芳环上氰基的取代反应
Scheme 20 Aromatic substitution reactions of cyanoarenes with alkyl radicals

图式 22 η6-芳基取代的羰基锰化合物的碳氢烷基化反应
Scheme 22 Alkylation of (η6-arene)tricarbonylmanganese complexes

图式 24 烷基自由基参与的单电子Tsuji-Trost反应
Scheme 24 Single-electron Tsuji-Trost reaction with alkyl radicals

图式 26 烷基自由基对二氧化硫和贫电子烯烃的插入反应
Scheme 26 Insertion of sulfur dioxide and electron-deficient alkenes with alkyl readicals

图式 29 手性光敏催化剂催化的烷基自由基对贫电子烯烃的手性加成
Scheme 29 Enantioselective β-alkylation electron-deficient alkenes catalyzed by chiral photoredox catalyst

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:















 下载:
下载: