Department of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China
Received Date:
30 April 2019 Available Online:
15 September 2019
Fund Project:
Project supported by the National Key Research and Development Program of China (No. 2017YFA0207302), the National Natural Science Foundation of China (Nos. 21672175, 91856110, 21332007, 21472153), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education, China
Abstract:
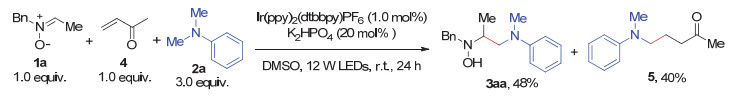
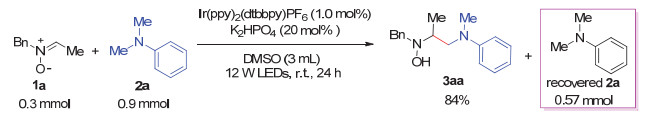
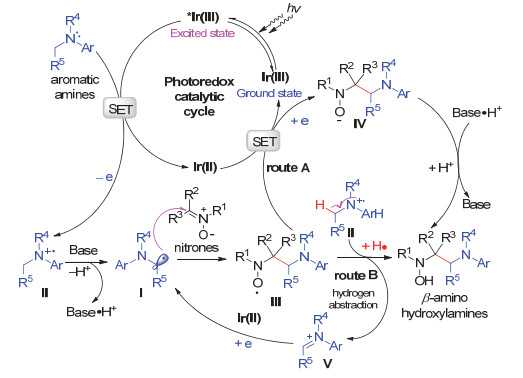
Carbon-carbon bond formation at α-amino carbon based on α-aminoalkyl radicals is an essential transformation in the synthesis of nitrogen-containing compounds. Recently, some novel photoredox catalytic protocols for this goal have been developed, in which α-aminoalkyl radicals were generated from a sequential oxidation/α-deprotonation of aromatic tertiary amines. Inspired by these studies and based on our previous works, we have developed the cross-coupling reaction of nitrones with aromatic tertiary amines via visible light photoredox catalysis. This method features a radical addition of α-aminoalkyl radicals to nitrones with advantages of simple operation, mild conditions, atom economy, a broad scope of nitrone substrates; and allows for an easy access to β-amino hydroxylamines, which could be readily converted into vicinal diamines. Compared with the UV-excited organophotosensitizer-promoted coupling reaction of nitrones with tertiary amines, visible light is a more safe and convenient light source, the photo-excited electron transfer (PET) by 1 mol% of Ir-photocatalyst is more efficient. In addition, nitrones exclusively server as excellent radical acceptors thus with a broader range of structures. A general procedure of this coupling reaction is as follows:To a 25 mL Schlenk tube equipped with a magnetic stir bar were added a nitrone (0.30 mmol), a tertiary amine (0.90 mmol), Ir(ppy)2(dtbbpy)PF6 (0.003 mmol, 1.0 mol%) and K2HPO4 (0.06 mmol, 20 mol%). After being evacuated and backfilled with argon for three times, DMSO (3 mL) was added to the tube. Then the tube was placed approximately 7 cm away from a 12 W blue LEDs, and the reaction mixture was stirred at r.t. under an argon atmosphere for 24 h. The reaction was quenched with saturated aqueous NaHCO3 (25 mL), and the mixture was extracted with dichloromethane (DCM, 20 mL×3). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to afford the desired cross-coupling product β-amino hydroxylamine.
(a) Renaud, P.; Giraud, L. Synthesis1996, 913; (b) Hart, D. in Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P. Eds. Weinheim, Germany: Wiley-VCH, 2001, Vol. 2: pp 279; (c) Aurrecoechea, J. M.; Suero, R. Arkivoc2004, partxiv, 10, at www.arkat-usa.org; (d) Nakajima, K.; Miyake, Y.; Nishibayashi, Y. Acc. Chem. Res.2016, 49, 1946.
[2]
For some selected reviews on visible light photocatalysis, see: (a) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322; (b) Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035; (c) Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Chem. Rev. 2007, 107, 2725; (d) Beatty, J. B.; Stephenson, C. R. J. Acc.Chem. Res. 2015, 48, 1474; (e) Tellis, J. C.; Kelly, C. B.; Primer, D. N.; Jouffroy, M.; Patel, N. R.; Molander, G. A. Acc. Chem. Res.2016, 49, 1429; (f) Ghosh, L.; Marzo, L.; Das, A.; Shaikh, R.; König, B. Acc. Chem. Res. 2016, 49, 1566; (g) Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Acc. Chem. Res. 2016, 49, 1911; (h) Arora, A.; Weaver, J. D. Acc. Chem. Res. 2016, 49, 2273; (i) Li, W.; Zheng, X.-Y.; Zeng, J.-G.; Cheng, P. Chin. J. Org. Chem.2017, 37, 1(in Chinese); (刘薇, 郑昕宇, 曾建国, 程辟, 有机化学, Chin. J. Org. Chem.2017, 37, 1; (j) Wang, D.-H.; Zhang, L.; Luo, S.-Z. Acta Chim. Sinica2017, 75, 22(in Chinese); (王德红, 张龙, 罗三中, 化学学报, 2017, 75, 22); (k) Song, H.; Liu, X.-Y.; Qin, Y. Acta Chim. Sinica2017, 75, 1137(in Chinese); (宋颢, 刘小宇, 秦勇, 化学学报, 2017, 75, 1137); (l) Cheng, X.-K.; Hu, X.-G.; Lu, Z. Chin. J. Org. Chem.2017, 37, 251(in Chinese); (程骁恺, 胡新根, 陆展, 有机化学, 2017, 37, 251; (m) Wu, J.; Li, J.-W.; Li, H.; Zhu, C.-Y. Chin. J. Org. Chem.2017, 37, 2203(in Chinese); (吴江, 李嘉雯, 李昊, 朱纯银, 有机化学, 2017, 37, 2203); (n) Liu, Y.-H.; Dong, W. Chin. J. Chem.2017, 35, 1491; (o) Ruan, L.-H.; Dong, Z.-C.; Chen, C.-X.; Wu, S.; Sun, J. Chin. J. Org. Chem.2017, 37, 2544(in Chinese); (阮利衡, 董振诚, 陈春欣, 吴爽, 孙京, 有机化学, 2017, 37, 2544); (p) Yi, H.; Zhang, G.-T.; Wang, H.-M.; Huang, Z.-Y.; Wang, J.; Singh, A. K.; Lei, A.-W. Chem. Rev.2017, 117, 9016; (q) Ye, H.; Xiao, C.; Lu, L.-Q. Chin. J. Org. Chem.2018, 38, 1897(in Chinese); (叶辉, 肖聪, 陆良秋, 有机化学, 2018, 38, 1897); (r) Xu, W.-X.; Dai, X.-Q.; Xu, H.-J.; Weng, J.-Q. Chin. J. Org. Chem.2018, 38, 2807(in Chinese); (徐雯秀, 戴小强, 徐涵靖, 翁建全, 有机化学, 2018, 38, 2807).
[3]
(a) McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science2011, 334, 1114; (b) Prier, C. K.; MacMillan, D. W. C. Chem. Sci.2014, 5, 4173.
[4]
(a) Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc.2012, 134, 3338; (b) Kohls, P.; Jadhav, K.; Pandey, G.; Reiser, O. Org. Lett., 2012, 14, 672; (c) Espelt, L. R.; Wiensch, E. R.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107; (d) Lin, S.-X.; Sun, G.-J.; Kang, Q. Chem. Commun.2017, 53, 7665.
[5]
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem. Eur. J. 2012, 18, 16473. doi: 10.1002/chem.201203066
[6]
(a) Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. J. Am. Chem. Soc.2015, 137, 13768; (b) Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Angew. Chem. Int. Ed.2016, 55, 6776.
For reviews on vicinal diamines, see: (a) Lucet, D.; Gall, T. L.; Mioskowski, C. Angew. Chem. Int. Ed.1998, 37, 2580; (b) Visa, A.; Pradilla, R. F.; Garcίa, A.; Flores, A. Chem. Rev. 2005, 105, 3167; (c) Kotti, S. R. S. S.; Timmons, C.; Li, G. Chem. Biol. Drug. Des.2006, 67, 101; (d) Cardona, F.; Goti, A. Nat. Chem.2009, 1, 269; (e) Grygorenko, O. O.; Radchenko, D. S.; Volochnyuk, D. M.; Tolmachev, A. A.; Komarov, I. V. Chem. Rev. 2011, 111, 5506; (f) Zhu, Y.-G.; Cornwall, R. G.; Du, H.-F.; Zhao, B.-G.; Shi, Y. Acc. Chem. Res.2014, 47, 3665.
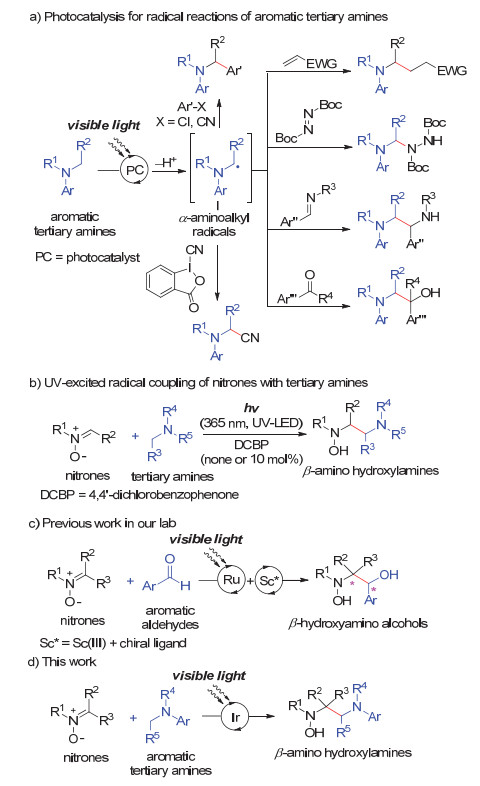
图式 1
可见光催化氧化还原体系中硝酮与芳香叔胺的自由基偶联反应设计
Scheme 1
The design of cross-coupling of nitrones with aromatic tertiary amines via visible light photoredox catalysis

 下载:
下载:






 下载:
下载:





 下载:
下载:
