图 1.
钯催化的电化学氯代反应
Figure 1.
Palladium-catalyzed electrochemical chlorination

芳香或芳香杂环氯代物广泛存在于许多天然产物、合成染料、农药、香料以及药物合成中间体中[1].芳香和芳香杂环氯代物可以通过还原(氢化脱除)、亲核取代(丁基锂置换)、亲核加成(Heck反应、插羰反应、Suzuki偶联反应)等化学反应将氯原子转变为其他官能团, 从而被广泛应用于具有生理、药理活性的药物分子, 复杂天然产物和有机功能高分子材料的构筑中[2].因此发展高效通用的策略来合成(杂)芳香族氯代物具有十分重要的科学意义和应用价值.
常见的传统的制备(杂)芳香族氯代物的方法包括直接的锂卤交换[3], 苯胺重氮化再氯代(Sandmeyer反应)[4]以及富电子芳香化合物的亲电取代[5].尽管上述传统方法已经在有机合成领域中取得了极大的应用, 然而依然存在反应条件苛刻, 官能团兼容性差, 底物局限于活化的芳烃, 选择性不易控制, 产生大量的金属盐废弃物等诸多缺点.
近年来, 过渡金属催化的C—H键活化反应作为一种原子经济性和绿色高效的策略被越来越多的化学家们用来合成官能团化的有机分子[6].其中, 利用导向基团的策略, 过渡金属钯[7]、铜[8]、铑[9]、钌[10]、钴[11]等催化的C—H键选择性卤代反应取得了长足的进步和发展.然而, 这些反应都需要使用当量甚至过量的有毒、昂贵、容易导致副产物的氧化剂(Cl2, NCS, PhI(OAc)2, TBHP, DDQ等)来促进反应的发生或是回收催化剂.因此, 发展更加绿色环保的氧化体系代替相关反应中当量的化学氧化剂将会是更为理想的选择.
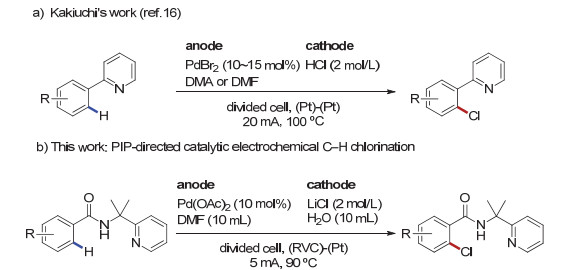
有机电合成已经发展成为现代合成方法学中一个充满活力和极具发展前景的研究领域[12, 13].有机电合成利用电能驱动化学反应, 避免了化学氧化剂或者还原剂的使用, 能从化学转化的源头上控制污染; 更为重要的是通过调节电流、电位可以控制反应的速率以及化学选择性, 从而实现传统化学反应不易实现的转化.基于这些优势, 该领域的发展日新月异, 相关研究成果突飞猛进.其中, 结合电化学以及过渡金属催化的金属有机电化学也取得了重要的研究进展[14, 15]. 2009年, Kakiuchi课题组[16]首次通过电化学的方法实现了以2-苯基吡啶为底物的芳烃邻位C(sp2)—H键的卤化反应.然而, 吡啶导向基团不易脱除以及转化, 极大地限制了该反应的应用(图 1a).近年来, 我们课题组发展了一系列电氧化促进的过渡金属催化的碳氢键官能团化反应[17].在本工作中, 我们使用容易脱除的PIP胺(2-(吡啶基)异丙基胺)作为导向基, 通过电化学条件下钯催化的C(sp2)—H的氯代反应, 实现芳基氯代物和杂芳基氯代物的高效高选择性合成(图 1b)[18].
我们选用PIP胺为导向基的苯甲酸衍生物1a作为模板底物, 采用网状玻碳电极(RVC)为工作电极, 在用阴离子交换膜分隔的H型电解槽中进行恒电流电解.经过仔细的条件优化确定的最佳反应条件为:在10 mol% Pd(OAc)2的存在下, 以LiCl作为氯源, DMF作为溶剂, 在90 ℃的温度下以5.0 mA恒电流电解12 h, 能以92%的核磁收率得到氯代产物(Entry 1, Table 1). PdCl2或Pd(OCOCF3)2也显示了相似的反应活性(Entries 2~3).通过考察阳极的溶剂发现, 反应在N, N-二甲基乙酰胺(DMA), 六甲基磷酰三胺(HMPA), 二甲基亚砜(DMSO)以及水中均能顺利进行, 收率相对较差(Entries 4~7).以HCl, NaCl和NH4Cl等作为氯源, 反应也能进行, 收率总体不如LiCl做氯源好(Entries 8~10).当温度降低到80 ℃时, 收率略微下降; 当温度升高到100 ℃, 收率没有明显的提高.将反应电流由5 mA降低到2.5 mA时, 收率变化不大, 但是反应时间需要延长一倍.在通电量不变的情况下, 将电流由5 mA增加到10 mA时, 原料不能完全转化, 氯代产率下降到67%(Entries 13~14).控制实验表明, 没有钯催化剂和不通电的情况下, 氯代反应不能发生(Entries 15~16).
 下载:
导出CSV
下载:
导出CSV
 |
||
| Entry | Variation from standard conditions above | Yieldb/% |
| 1 | none | 92 (85)c |
| 2 | PdCl2 instead of Pd(OAc)2 | 89 |
| 3 | Pd(OCOCF3)2 instead of Pd(OAc)2 | 91 |
| 4 | DMA instead of DMF (anode) | 80 |
| 5 | HMPA instead of DMF (anode) | 63 |
| 6 | DMSO instead of DMF (anode) | 12 |
| 7 | H2O instead of DMF (anode) | 34 |
| 8 | HCl instead of LiCl | 72 |
| 9 | NaCl instead of LiCl | 71 |
| 10 | NH4Cl instead of LiCl | 65 |
| 11 | 80 ℃ instead of 90 ℃ | 90 |
| 12 | 100 ℃ instead of 90 ℃ | 92 |
| 13 | 2.5 mA instead of 5 mA (24 h) | 88 |
| 14 | 10 mA instead of 5 mA (6 h) | 67 |
| 15 | no Pd(OAc)2 | nr |
| 16 | no electric current | nr |
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃, 12 h (9.0 F/mol). b Yield was determined by 1H NMR analysis with CH2Br2 as the internal standard. c Isolated yield in parentheses. | ||
确定最优条件之后, 我们对电化学条件下钯催化的芳烃邻位C(sp2)—H键氯代反应的底物普适性进行了考察(Table 2).该电化学氯代体系对氟、氯、溴等卤素, 烷基、酯基、烷氧基、硝基、磺酰基、三氟甲基等都有很好的兼容性, 各种取代的苯甲酸衍生物都能以良好的收率得到相应的氯代产物.对于邻位有强供电子取代基的底物(2d, 2e)和对位有强供电子取代基的底物(2c, 2v), 产物都是导向基邻位C—Cl键的形成, 说明导向的策略可以克服亲电取代背景反应的干扰.苯环对位是供电子基团时, 能够以优异的收率得到单一的邻位双氯代的产物(2s~2x); 间位是三氟甲基、磺酰基、硝基等吸电子取代基时, 位阻更小的邻位C—H键更容易发生氯代反应, 产物以单氯代为主, 单双取代产物的比例大于10:1 (2l~2n).当间位是溴和甲氧基时, 氯代反应会同时产生单双取代产物, 其中单氯代的产物占主导地位(2k, 2o).值得高兴的是, 我们发展的电化学条件下钯催化的C(sp2)—H键氯代反应策略, 不仅适用于普通苯甲酸衍生物, 还适用于在合成化学和生物活性中具有重要应用的杂环底物.噻吩类底物能以优异的收率得到相应的氯代产物(2p, 2q).
下载:
导出CSV
 |
 |
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃. |
连续C—H官能团化策略在有机合成中具有重要的应用价值.通过连续碳氢键官能团化的策略能在同一个分子中分步引入多个不同或相同的官能团, 从而合成高度复杂的多种官能团化的有机分子.为了证明电化学卤代反应的应用前景, 我们计划从间位是甲基、甲氧基和氰基的底物出发, 首先经过我们课题组发展的电氧化促进的碳氢键溴代反应[17a]得到邻位单溴化的产物(3a~3c), 接下来继续衍生再进行电化学氯代反应.令我们高兴的是, 反应能够顺利进行, 以中等的收率得到了邻位C(sp2)—H分别被溴代和氯代的产物, 从而经过两步碳氢官能团化反应得到了三个不同官能团取代的复杂苯甲酰胺类衍生物(4a~4c)(表 3).
下载:
导出CSV
 |
 |
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃. |
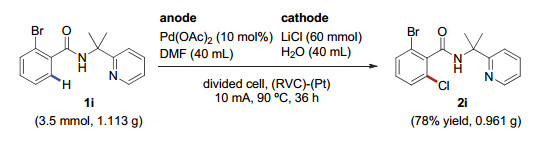
为了体现电化学氯化反应在合成上的实用性, 我们将反应规模由毫克级别放大到克级别, 如图 2所示. 3.5 mmol (1.113 g)的1i在90 ℃的温度下, 以10 mA的恒电流电解36 h, 能以78%的分离收率得到相应的氯代产物2i, 展现了该方法潜在的工业应用前景.
为了阐明电化学条件下钯催化的碳氢键氯化反应的机理, 我们开展了一系列的探究实验.
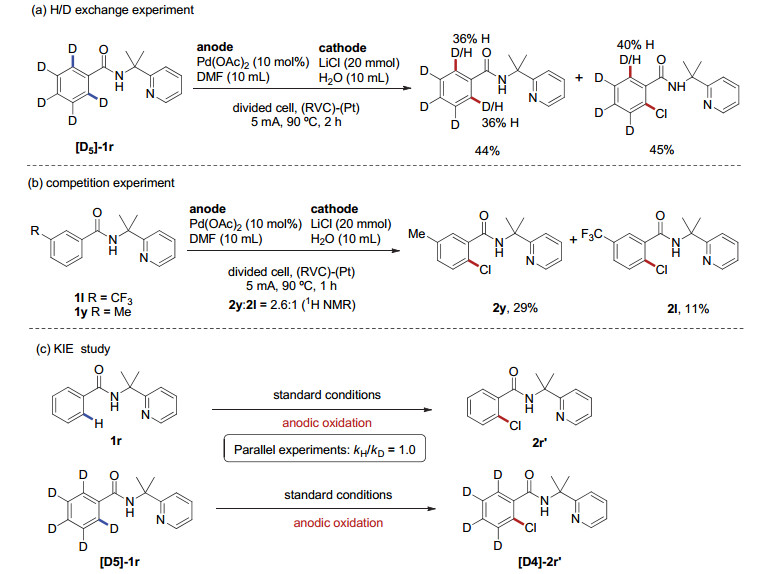
将同位素标记的苯甲酰胺底物[D5]-1r置于在电化学氯代反应条件下电解2 h后, 在回收的原料和生成的氯代产物中均观察到了明显的H/D交换现象, 这表明在催化循环过程中Pd物种对C(sp2)—H键的插入是可逆的(图 3a).
为了研究取代基对底物反应活性的影响, 我们进行了分子间的竞争反应实验(图 3b).反应通过等量混合的间甲基取代底物1y和间三氟甲基取代底物1l, 在标准条件下反应1 h, 在低转化率情况下对比两种产物的比例.通过粗核磁分析, 有29%的甲基底物转化为相应的氯代产物, 而只检测到11%的三氟甲基底物转化为目标产物.实验结果表明:在该转化中, 富电子底物展示了比缺电子底物更高的反应活性.
我们还做了分子间平行氘代实验(图 3c).实验结果显示动力学同位素效应KIE=1.0, 这说明电化学氯代反应中C—H键活化这一步不是决速步骤.
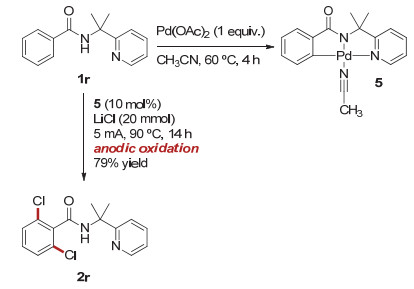
为了获得反应机理的详细信息, 我们根据文献报道[17a]合成并研究了反应中与催化剂相关的中间体(图 4).将化学计量的醋酸钯和等当量的底物1r在乙腈溶剂中室温反应4 h后, 我们成功分离得到了稳定的Pd(Ⅱ)络合物5; 用络合物5代替标准反应条件中的催化剂, 加入到电化学氯代体系中进行反应, 能以79%的分离收率得到氯代产物2r, 因此, 我们推断该反应中可能生成单核的环钯中间体.
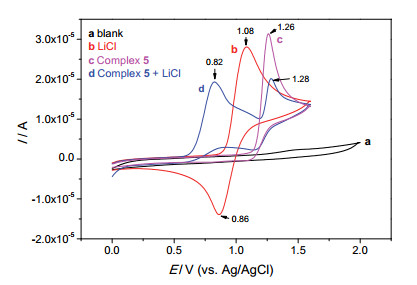
电化学氯代反应机理的研究相对不足, 为了分析氯代反应中微量的电活性物质, 进一步深入了解反应机理, 我们做了相关循环伏安实验(图 5).从曲线b可知, LiCl中的Cl-在CH3CN/n-Bu4NPF6体系中有一个氧化峰(Ep=1.08 V vs. Ag/AgCl)和一个还原峰(Ep=0.86 V).结合文献报道[19], 我们认为Cl-可能被阳极氧化产生Cl3-, 也不能排除Cl2, Cl·或者Cl+的生成[16].而钯络合物5的氧化电位(Ep=1.26 V)明显高于Cl-的氧化电位(曲线c).当向LiCl中加入络合物5后, LiCl的还原峰消失, 络合物5的氧化峰减弱(Ep=0.82 V, 曲线d).这可能说明络合物5和Cl-的氧化产物Cl3-或者Cl·有相互作用.
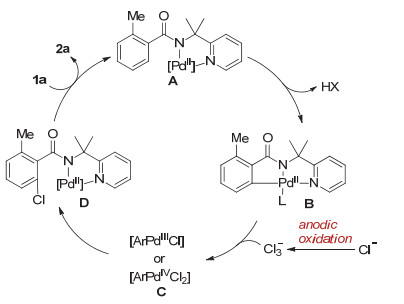
基于以上实验结果和之前的研究基础, 我们提出了电氧化促进的钯催化的碳氢键氯代反应的可能机理(图 6).醋酸钯首先与底物发生配位, 促使金属中心选择性的靠近邻位碳氢键; 随后发生Pd对C—H键的活化形成五元环钯金属中间体B.接下来, 环钯中间体B与阳极氧化生成的Cl3-反应生成Pd(Ⅲ)或Pd(Ⅳ)的络合物C, 络合物C发生还原消除转化为中间体D; 最后, 中间体D发生配体交换得到氯代产物并实现催化剂的再生, 从而完成催化循环.
我们以PIP胺为双齿导向基团, 以廉价易得的LiCl为氯化试剂, 实现了电化学条件下钯催化的苯甲酰胺类化合物的邻位C(sp2)—H键氯代反应.该反应体系具有良好的官能团兼容性, 底物普适性广, 产率优异, 后处理简单.我们还可以从简单的酰胺底物出发, 采用连续碳氢键卤化反应的策略, 实现高度复杂的多官能团取代的苯甲酰胺类化合物的构建, 体现了该方法在合成和药物化学领域中潜在的应用价值.
所有的电解均在H型分隔电解槽中进行, 阴阳两极用阴离子交换膜隔开.使用网状玻碳电极(RVC 10 mm×10 mm×12 mm)作为阳极, 铂片(10 mm×10 mm×0.2 mm)作为阴极.在阳极池中依次加入Pd(OAc)2 (5.6 mg, 0.025 mmol, 10 mol%), 苯甲酰胺衍生物(0.25 mmol)和DMF (10 mL).在阴极池中依次加入LiCl (847.8 mg, 20.0 mmol)和去离子水(10 mL).然后将反应装置置于90 ℃的油浴锅中, 以5 mA的恒定电流进行电解反应, 用TLC或1H NMR判断原料检测反应进程.反应结束后, 加入EtOAc (50 mL)稀释后用水(20 mL×3)洗涤, 再用盐水(20 mL)洗涤.有机相并用无水Na2SO4干燥, 过滤, 滤液减压浓缩后经柱层析分离纯化得到氯代产物.注意:由于氯离子需要从阴极经阴离子交换膜缓慢进入阳极, 所以电解达到恒流状态大概需要5 min.
(a) Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937; (b) Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew Chem., Int. Ed. 2005, 44, 4442.
For selected reviews, see: (a) Hassan, J.; Se'vignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359; (b) Littke, A. F.; Fu, G. C. Angew Chem., Int. Ed. 2002, 41, 4176; (c) Corbet, J. P.; Mignani, G. Chem. Rev. 2006, 106, 2651; (d) Yin, L.-X.; Liebscher, J. Chem. Rev. 2007, 107, 133.
For a review on an ortho-lithiation approach, see: Snieckus, V. Chem. Rev. 1990, 90, 879.
Hodgson, H. H. Chem. Rev. 1947, 40, 251. doi: 10.1021/cr60126a003
De La Mare, P. B. D. Electrophilic Halogenation, Cambridge University Press, New York, 1976.
For selected reviews on transition-metal-catalyzed C-H functionalization, see: (a) Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074; (b) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew Chem., Int. Ed. 2009, 48, 5094; (c) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242; (d) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147; (e) Arockiam, P. B.; Bruneau, C.; Dixneuf, P. H. Chem. Rev. 2012, 112, 5879; (f) Ackermann, L. C. Acc. Chem. Res. 2014, 47, 281; (g) Pei, P.; Zhang, F.; Yi, H.; Lei, A. Acta Chim. Sinica 2017, 75, 15(in Chinese). (裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15); (h) Du, J.-Y.; Xia, C.-G.; Sun, W. Acta Chim. Sinica 2018, 76, 329(in Chinese). (杜俊毅, 夏春谷, 孙伟, 化学学报, 2018, 76, 329).
For selected examples of palladium-catalyzed direct halogenation of C-H bonds, see: (a) Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300; (b) Wan, X. B.; Ma, Z. X.; Li, B. J.; Zhang, K. Y.; Cao, S. K.; Zhang, S. W.; Shi, Z. J. J. Am. Chem. Soc. 2006, 128, 7416; (c) Zhao, X.; Dimitrijevic, E.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 3466; (d) Wang, X.-C.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 10326; (e) Gao, D.; Gu, Q.; You, S.-L. ACS Catal. 2014, 4, 2741; (f) Chu, L.; Xiao, K.-J.; Yu, J.-Q. Science 2014, 346, 451; (g) Zhao, K.; Yang, L.; Liu, J.-H.; Xia, C.-G. Chin. J. Org. Chem. 2018, 38, 2833(in Chinese). (赵康, 杨磊, 刘建华, 夏春谷, 有机化学, 2018, 38, 2833).
(a) Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790; (b) Wang, W.; Pan, C.; Chen, F.; Cheng, J. Chem. Commun. 2011, 47, 3978; (c) Mo, S.; Zhu, Y.; Shen, Z. Org. Biomol. Chem. 2013, 11, 2756; (d) Du, Z.-J.; Gao, L.-X.; Lin, Y.-J.; Han, F.-S. ChemCatChem 2014, 6, 123; (e) Hufman, L. M.; Stahl, S. S. J. Am. Chem. Soc. 2008, 130, 9196; (f) King, A. E.; Huffman, L. M.; Casitas, A.; Costas, M.; Ribas, X.; Stahl, S. S. J. Am. Chem. Soc. 2010, 132, 12068; (g) Wang, Z.-L.; Zhao, L.; Wang, M.-X. Org. Lett. 2011, 13, 6560; (h) Wang, Z.-L.; Zhao, L.; Wang, M.-X. Org. Lett. 2012, 14, 1472; (i) Casitas, A.; Ribas, X. Chem. Sci. 2013, 4, 2301; (j) Zhang, H.; Yao, B.; Zhao, L.; Wang, D.-X.; Xu, B.-Q.; Wang, M.-X. J. Am. Chem. Soc. 2014, 136, 6326; (k) Truong, T.; Klimovica, K.; Daugulis, O. J. Am. Chem. Soc. 2013, 135, 9342; (l) Suess, A. M.; Ertem, M. Z. C.; Cramer, J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797; (m) Zhang, Q.; Yin, X.-S.; Zhao, S.; Fang, S.-L.; Shi, B.-F. Chem. Commun. 2014, 50, 8353.
For selected examples of rhodium-catalyzed direct halogenation of C-H bonds, see: (a) Schroder, N.; Wencel-Delord, J.; Glorius, F. J. Am. Chem. Soc. 2012, 134, 8298; (b) Hwang, H.; Kim, J.; Jeong, J.; Chang, S. J. Am. Chem. Soc. 2014, 136, 10770; (c) Qian, G.; Hong, X.; Liu, B.; Mao, H.; Xu, B. Org. Lett. 2014, 16, 5294.
For an example of ruthenilum-catalyzed ortho-halogenation, see: Wang, L.-H.; Ackermann, L. Chem. Commun. 2014, 50, 1083.
For an example of Co-catalyzed ortho-halogenation, see: (a) Yu, D.-G.; Gensch, T.; de Azambuja, F.; Vásquez-Céspedes, S.; Glorius, F. J. Am. Chem. Soc. 2014, 136, 17722; (b) Gu, Z.-Y., Ji, S.-J. Acta Chim. Sinica 2018, 76, 347(in Chinese). (顾正洋, 纪顺俊, 化学学报, 2018, 76, 347).
For recent reviews on organic electrochemistry, see: (a) Yuan, Y.; Cao, Y.; Qiao, J.; Lin, Y.; Jiang, X.; Weng, Y.; Tang, S.; Lei, A. Chin. J. Chem. 2019, 37, 49; (b) Tang, S.; Liu, Y.; Lei, A. Chem 2018, 4, 27; (c) Liu, K.; Song, C.; Lei, A. Org. Biomol. Chem. 2018, 16, 2375; (d) Sauer, G. S.; Lin, S. ACS Catal. 2018, 8, 5175; (e) Parry, J.; Fu, N.; Lin, S. Synlett 2018, 29, 257; (f) Nutting, J. E.; Rafiee, M.; Stahl, S. S. Chem. Rev. 2018, 118, 4834; (g) Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485; (h) Waldvogel, S. R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C. Chem. Rev. 2018, 118, 6706; (i) Moeller, K. D. Chem. Rev. 2018, 118, 4817; (j) Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338; (k) Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230; (l) Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302; (m) Hou, Z.-W.; Mao, Z.-Y.; Xu, H.-C. Synlett 2017, 28, 1867; (n) Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492.
For recent examples on organic electrochemistry, see: (a) Yuan, Y.; Yao, A.; Zheng, Y.; Gao, M.; Zhou, Z.; Qiao, J.; Hu, J.; Ye, B.; Zhao, J.; Wen, H.; Lei, A. iScience 2019, 12, 293; (b) Wang, P.; Tang, S.; Huang, P. F.; Lei, A. W. Angew. Chem., Int. Ed. 2017, 56, 3009; (c) Zhang, Z.; Zhang, L.; Cao, Y.; Li, F.; Bai, G.; Liu, G.; Yang, Y.; Mo, F. Org. Lett. 2019, 21, 762; (d) Yan, H.; Hou, Z.-W.; Xu, H.-C. Angew. Chem., Int. Ed. 2019, 58, 4592; (e) Hou, Z.-W.; Mao, Z.-Y.; Zhao, H.-B.; Melcamu, Y. Y.; Lu, X.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2016, 55, 9168; (f) Rafiee, M.; Wang, F.; Hruszkewycz, D. P.; Stahl, S. S. J. Am. Chem. Soc. 2018, 140, 22; (g) Wang, H.; Zhang, J.; Tan, J.; Xin, L.; Li, Y.; Zhang, S.; Xu, K. Org. Lett. 2018, 20, 2505; (h) Lin, D. Z.; Huang, J. M. Org. Lett. 2018, 20, 2112; (i) Ye, Z.; Ding, M.; Wu, Y.; Li, Y.; Hua, W.; Zhang, F. Green Chem. 2018, 20, 1732; (j) Wang, Q.-Q.; Xu, K.; Jiang, Y.-Y.; Liu, Y.-G.; Sun, B.-G.; Zeng, C.-C. Org. Lett. 2017, 19, 5517; (k) Wiebe, A.; Lips, S.; Schollmeyer, D.; Franke, R.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2017, 56, 14727; (l) Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.-H.; Chen, J.; Starr, J.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 7448; (m) Horn, E. J.; Rosen, B. R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M. D.; Baran, P. S. Nature 2016, 533, 77.
For selected reviews on transition-metal-catalyzed electrochemical C-H functionalization, see: (a) Sauermann, N.; Meyer, T. H.; Qiu, Y.; Ackermann, L. ACS Catal. 2018, 8, 7086; (b) Sauermann, N.; Meyer, T. H.; Ackermann, L. Chem.-Eur. J. 2018, 24, 16209; (c) Ma, C.; Fang, P.; Mei, T.-S. ACS Catal. 2018, 8, 7179; (d) Jiao, K.-J.; Zhao, C.-Q.; Fang, P.; Mei, T.-S. Tetrahedron Lett. 2017, 58, 797; (e) Wu, Y.-X.; Xi, Y.-C.; Zhao, M.; Wang, S.-Y. Chin. J. Org. Chem. 2018, 38, 2590(in Chinese). (吴亚星, 席亚超, 赵明, 王思懿, 有机化学, 2018, 38, 2590).
For selected examples on transition-metal-catalyzed electrochemical C-H functionalization, see: (a) Qiu, Y.; Stangier, M.; Meyer, T. H.; Oliveira, J. C. A.; Ackermann, L. Angew. Chem. Int. Ed. 2018, 57, 14179; (b) Sauermann, N.; Mei, R.; Ackermann, L. Angew. Chem. Int. Ed. 2018, 57, 5090; (c) Gao, X.; Wang, P.; Zeng, L.; Tang, S.; Lei, A. J. Am. Chem. Soc. 2018, 140, 4195; (d) Tang, S.; Wang, D.; Liu, Y.; Liu, L.; Lei, A. Nature Commun. 2018, 9, 798; (e) Xu, F.; Li, Y.-J.; Huang, C.; Xu, H.-C. ACS Catal. 2018, 8, 3820; (f) Shrestha, A.; Lee, M.; Dunn, A. L.; Sanford, M. S. Org. Lett. 2018, 20, 204; (g) Grayaznova, T. V.; Dudkina, Y. B.; Islamov, D. R.; Kataeva, O. N.; Sinyashin, O. G.; Vicic, D. A.; Budnikova, Y. Н. J. Organomet. Chem. 2015, 785, 68; (h) Amatore, C.; Cammoun, C.; Jutand, A. Adv. Synth. Catal. 2007, 349, 292; (i) Freund, M. S.; Labinger, J. A.; Lewis, N. S.; Bercaw, J. E. J. Mol. Catal. 1994, 87, L11.
Kakiuchi, F.; Kochi, T.; Mutsutani, H.; Kobayashi, N.; Urano, S.; Sato, M.; Nishiyama, S.; Tanabe, T. J. Am. Chem. Soc. 2009, 131, 11310. doi: 10.1021/ja9049228
(a) Yang, Q.-L.; Wang, X.-Y.; Wang, T.-L.; Yang, X.; Liu, D.; Tong, X.; Wu, X.-Y.; Mei, T.-S. Org. Lett. 2019, 21, 2645; (b) Yang, Q.-L.; Li, C.-Z.; Zhang, L.-W.; Li, Y.-Y.; Tong, X.; Wu, X.-Y.; Mei, T.-S. Organometallics 2019, 38, 1208; (c) Yang, Q.-L.; Wang, X.-Y.; Lu, J.-Y.; Zhang, L.-P.; Fang, P.; Mei, T.-S. J. Am. Chem. Soc. 2018, 140, 11487; (d) Li, Y.-Q.; Yang, Q.-L.; Fang, P.; Mei, T.-S.; Zhang, D. Org. Lett. 2017, 19, 2905; (e) Ma, C.; Zhao, C.-Q.; Li, Y.-Q.; Zhang, L.-P.; Xu, X.; Zhang, K.; Mei, T.-S. Chem. Commun. 2017, 53, 12189; (f) Yang, Q.-L.; Li, Y.-Q.; Ma, C.; Fang, P.; Zhang, X.-J.; Mei, T.-S. J. Am. Chem. Soc. 2017, 139, 3293.
During this manuscript preparation, Kakiuchi reported similar work using benzamide derivatives: Konishi, M.; Tsuchida, K.; Sano, K.; Kochi, T.; Kakiuchi, F. J. Org. Chem. 2017, 82, 8716. However, the work was independently carried out. The reaction conditions and directing groups used in these two studies are different.
(a) Sun, H.; Yu, L.; Jin, X.; Hu, X.; Wang, D.; Chen, G. Z. Electrochem. Commun. 2005, 7, 685; (b) Yu, L.; Jin, X.; Chen, G. Z. J. Electroanal. Chem. 2013, 688, 371.

图 5 循环伏安实验
Figure 5 Cyclic voltammograms recorded on a Pt electrode (area=0.03 cm2): (a) MeCN containing 0.1 mol/L n-Bu4NPF6; (b) solution (a) after addition of 5 mmol/L LiCl; (c) solution (a) after addition of 2 mmol/L palladacycle 5; (d) solution (b) after addition of 2 mmol/L palladacycle 5
表 1 氯代反应条件的优化a
Table 1. Optimization of chlorination conditions a
|
|
||
| Entry | Variation from standard conditions above | Yieldb/% |
| 1 | none | 92 (85)c |
| 2 | PdCl2 instead of Pd(OAc)2 | 89 |
| 3 | Pd(OCOCF3)2 instead of Pd(OAc)2 | 91 |
| 4 | DMA instead of DMF (anode) | 80 |
| 5 | HMPA instead of DMF (anode) | 63 |
| 6 | DMSO instead of DMF (anode) | 12 |
| 7 | H2O instead of DMF (anode) | 34 |
| 8 | HCl instead of LiCl | 72 |
| 9 | NaCl instead of LiCl | 71 |
| 10 | NH4Cl instead of LiCl | 65 |
| 11 | 80 ℃ instead of 90 ℃ | 90 |
| 12 | 100 ℃ instead of 90 ℃ | 92 |
| 13 | 2.5 mA instead of 5 mA (24 h) | 88 |
| 14 | 10 mA instead of 5 mA (6 h) | 67 |
| 15 | no Pd(OAc)2 | nr |
| 16 | no electric current | nr |
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃, 12 h (9.0 F/mol). b Yield was determined by 1H NMR analysis with CH2Br2 as the internal standard. c Isolated yield in parentheses. | ||
 下载: 导出CSV
下载: 导出CSV
表 2 底物拓展a
Table 2. Substrate scopea
|
|
|
|
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃. |
下载: 导出CSV
表 3 连续C—H键卤化反应a
Table 3. Sequential double C—H activationa
|
|
|
|
| a Reaction conditions: 1a (0.25 mmol), Pd(OAc)2 (10 mol%), DMF (10 mL) (anode), and LiCl (20 mmol), H2O (10 mL) (cathode) in H-type divided cell with RVC anode (10 mm×10 mm×12 mm), platinum plate cathode (10 mm×10 mm×0.2 mm) and a anion-exchange membrane, 5 mA, 90 ℃. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: