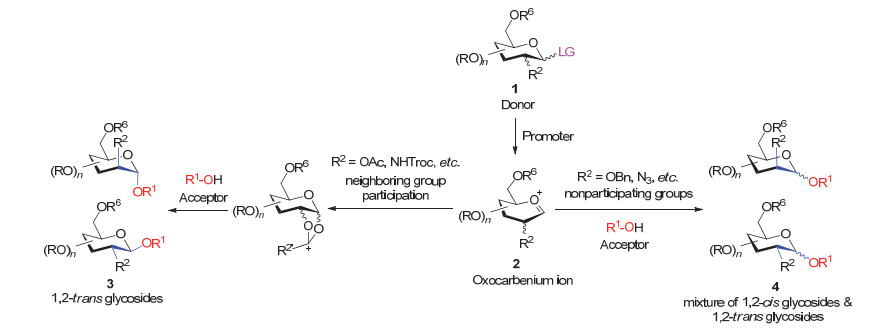
图 1.
糖基化的一般反应过程
Figure 1.
General mechanism of glycosylation reactions

糖类化合物(Carbohydrates)是自然界中分布最为广泛的一类生物分子, 与核酸、蛋白质和脂类共同构成了细胞的基本组成.长久以来, 糖类主要被视为生物体的结构物质(如纤维素)和能量物质(如淀粉和糖原).然而随着糖生物学研究的不断深入, 人们越来越意识到糖类是一类重要的信息分子, 广泛参与病原体感染、免疫应答、细胞信号转导、细胞分裂和分化、炎症反应、精卵识别和肿瘤转移等生命过程[1, 2].另一方面, 糖类也是重要的药用分子, 比如被广泛应用于疾病治疗和预防的抗凝药肝素、氨基糖苷类抗生素以及细菌多糖结合疫苗等[3, 4].此外, 糖类还常作为“手性池”起始原料, 用于合成具有重要生物活性及应用价值的天然产物或医药中间体[5].
聚糖的基本结构单元为单糖, 单糖与单糖之间以糖苷键相连, 形成线性或者分支的聚糖结构.按照所含单糖单元数目的多少, 聚糖又可分为寡聚糖(寡糖)和多聚糖(多糖).寡糖和多糖之间并没有明显的界限, 在20世纪70年代, 一般超过10个单糖单元组成的聚糖结构就可以被称为多糖; 而随着糖合成技术的进步, 合成的糖越来越大, 目前一般可以将超过20个单糖单元所组成的聚糖称为多糖.
天然存在的多糖(如糖胺聚糖、细菌荚膜多糖等)大多结构复杂, 且存在微观不均一性, 这使得人们难以通过分离纯化的方式得到足量纯净的多糖样品.另外, 多糖的生物合成没有固定的模板, 不由基因直接编码, 因此也无法像核酸或者蛋白质那样通过PCR或者基因表达的方式获取样品[6].多糖样品的获得性问题成为限制其生物学功能和构效关系研究的瓶颈.
化学合成是获取均一结构多糖的重要途径.但由于多糖的结构十分复杂, 且受限于糖合成的水平, 人们通常选择短的糖片段进行合成.在某些情况下, 合成短的糖链就能够满足研究需要, 然而在分子尺寸依赖的生物识别过程中[7], 短的糖链往往难以模拟天然多糖的复杂结构.例如Takahashi等[8]发现β-1, 3连接的葡聚糖与受体Dectin-1的结合力随糖链长度增加而增强, 但即便是十七糖, 其结合力也比天然的多糖弱很多; 另外, Petitou等[9]在研究合成的小分子肝素抑制凝血酶活性时发现, 合成的十五糖(约5.5 kD)比天然肝素(约15 kD)的IC50仍小一个数量级.此外, 细菌的抗原性多糖成分(或其蛋白结合物)往往能够刺激机体产生强的免疫反应, 但有时使用合成的短的糖链所引起的抗体滴度却很低[10].
合成分子尺寸和复杂性与天然多糖相当的聚糖化合物有助于从分子水平理解多糖的生物学功能及构效关系.近几十年来, 糖化学家们发展了大量新颖的糖基化方法, 发展了高效的糖链组装策略, 并且开始运用这些方法和策略进行复杂结构多糖的化学合成.目前, 已经有多篇综述性文章对糖基化方法和糖链组装策略进行了详细的总结[11], 本文将择其代表性的方法和策略进行简介, 重点对其在复杂多糖合成中的应用进行综述.
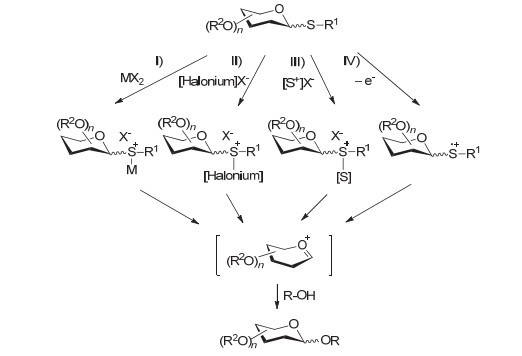
聚糖化学合成中最重要的反应是糖单元与糖单元之间的偶联反应——糖基化反应(Glycosylation reaction).通常来讲, 糖基化反应的底物包括两种:其一为端基带某种离去基团(Leaving group, LG)的糖基供体(Donor), 其二为带有亲核反应基团的糖基受体(Acceptor).根据受体亲核反应基团的不同, 糖基化反应可以分为氧-糖基化反应、氮-糖基化反应和碳-糖基化反应等, 相应的糖基化反应产物被称作氧苷、氮苷和碳苷等.
以氧-糖基化为例, 一般的反应过程如图 1所示, 供体1被促进剂(Promoter)活化后, 生成氧鎓离子(Oxocarbenium ion)中间体2.当C-2位存在邻基参与基团(Participating groups)时, 受体(R1-OH)从邻基参与基团的反方向进攻端基碳, 从而生成1, 2-反式糖苷3 (1, 2-trans glycoside).常见的邻基参与基团主要为酯型保护基, 如乙酰基和苯甲酰基等.
而当C-2位不存在邻基参与基团时, 则受体可以从糖环的α和β面进攻端基碳, 从而生成1, 2-反式和1, 2-顺式糖苷(1, 2-cis glycoside)混合物4.反式和顺式的比例受很多因素影响, 比如端基效应、离去基、保护基、受体性质和反应条件(如活化方式、添加剂、溶剂、压力、温度、浓度)等.
最早的糖基化反应可以追溯到19世纪末期. 1879年, Michael等[12]首次报道了使用氯/溴代糖作为糖基供体, 与亲核性较强的苯酚钾盐发生糖基化反应合成简单酚苷的例子.自那以后, 人们在发展糖基化方法方面做了大量工作, 并陆续报道了数十种新颖的糖基供体, 如卤代糖供体[13]、半缩醛糖基供体[14]、三氯乙酰亚胺酯供体[15]、硫苷供体[16]、戊烯苷供体[17]、糖烯供体[18]、糖基磷酸酯供体[19]、邻炔基苯甲酸酯糖基供体[20]等.随着这些糖基供体的出现, 还发展出各式各样与之匹配的活化体系; 这些糖基化方法有各自的适用范围, 在聚糖的化学合成中各显神通.
在众多的糖基供体中, 最常用的是卤代糖[13]、糖基三氯乙酰亚胺酯[15]和硫苷[16].
卤代糖(Glycosyl halide)是糖的端基为卤素取代的一类糖基供体, 包括氟代糖、氯代糖、溴代糖和碘代糖.其中最早得到广泛应用的是溴代糖和氯代糖.
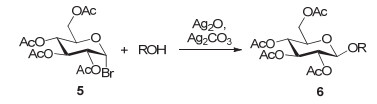
1901年, Koenigs和Knorr[21]使用全乙酰葡萄糖溴代物5为供体, Ag2CO3/Ag2O作为活化剂, 成功实现了与醇的偶联反应(图 2).这是糖基化方法发展过程中至关重要的一步, 该方法被命名为Koenigs-Knorr反应.
除了溴代糖, Koenigs-Knorr反应也可以使用氯代糖为供体.早期的时候Koenigs-Knorr反应只能使用简单的醇做受体, 而以亲核性较弱的糖作受体时, 反应效率不高.随后人们发现换用活性更高的重金属盐可以提高氯/溴代糖端基离去基的离去能力, 进而提高反应效率, 比如Zemplén和Helferich等分别于1930年和1949年报道了二价的汞盐Hg(OAc)2[22]和Hg(CN)2/HgBr2[23]能更好地替代Ag2CO3.后续其它重金属盐类活化剂也被发展起来, 如银盐AgClO4[24]、AgOTf[25]、AgNO3[26]、铜盐Cu(OTf)2[27]、镉盐CdCO3[28]和有机锌盐Zn(p-tBuC6H4CO2)2[29], 其中AgOTf被证明是最有效的活化剂之一.
氟代糖是卤代糖中化学性质最稳定的糖基供体, 然而其早期却很少被应用于糖合成中.主要原因在于缺少高效的活化剂, 直到1981年, Mukaiyama小组才首次报道了使用SnCl2/AgClO4[30]活化氟代糖的例子.随后其他一些研究者引入了一系列的活化剂, 使得氟代糖成为受欢迎的糖基供体之一.其它一些常用的活化剂包括SnCl2/TrClO4[31], 二(环戊二烯基)金属衍生物Cp2MCl2[32], SiF4[33], TfOH[34]等.
相比氟/氯/溴代糖, 碘代糖是一种活性很高的糖基供体, 不过它也是最不稳定的, 这在很大程度上限制了碘代糖的应用.直到20世纪末期, 随着制备方式的进步, 碘代糖作为糖基供体才得到复兴, 有时其高活性会带来反应时间缩短、效率提高和立体选择性改善等优点[35].通常α-碘代糖在碱性条件下可以高效生成β-糖苷[36], 或者使用“卤代物催化”, 在TBAI (四丁基碘化铵)和DIPEA (N, N-二异丙基乙胺)条件下, 原位异构为β-碘代糖, 用于合成α-糖苷[37].
卤代糖作为一类经典的糖基供体, 其使用有时也受到限制, 例如可能会生成原酸酯副产物, 高活性卤代糖不适于长时间存储(如碘代糖一般原位制备), 严重依赖重金属盐, 等等.近期我们研究小组及其他研究者使用有机小分子成功实现了氯/溴代糖活化[38, 39], 克服了其依赖重金属盐的缺点, 这些方法在糖合成中的价值有待进一步检验.如何使卤代糖糖基化更加绿色环保仍然值得继续探索.
Schmidt小组[15]于1980年首次报道了一种很受欢迎的糖基供体——糖基三氯乙酰亚胺酯(Glycosyl trichloroacetimidate, TCAI), 该供体也被称作Schmidt供体.
Schmidt供体可以从半缩醛糖和Cl3CCN在碱性条件下制备, 使用K2CO3有利于生成β-型异构体, 而使用NaH、Cs2CO3和DBU (1, 8-二氮杂二环[5.4.0]十一碳-7-烯)则倾向于生成α-型异构体(图 3a).该供体在储藏时有一定的稳定性, 容易被催化量的路易斯酸(如TMSOTf或BF3•Et2O)所活化, 糖基化反应的效率通常很高(图 3b).正是由于这些优点, 糖基三氯乙酰亚胺酯在复杂聚糖及糖缀合物的化学合成中应用得十分广泛.
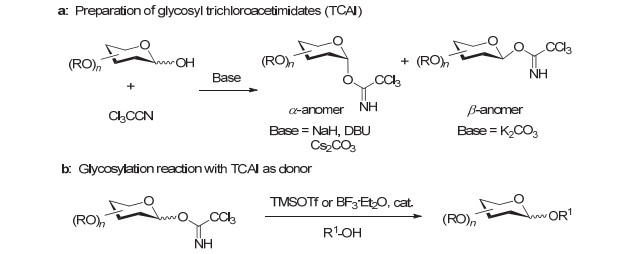
然而, 糖基三氯乙酰亚胺酯也不是完美的糖基供体, 比如当与活性很低的受体反应时, 会重排为N-糖基三氯乙酰胺.使用反加操作(Inverse procedure)[40]——即把预先混合的受体与活化剂加至供体中, 有时可以较好地解决这一问题.值得注意的是, 某些糖基三氯乙酰亚胺酯的衍生型供体在某些糖基化反应中表现出更高的效率, 例如Yu小组[41]报道的O-糖基N-苯基三氟乙酰亚胺酯(O-Glycosyl N-phenyltrifluoroacetimidate, PTFA)供体, 近期也有比较多的合成应用.
硫苷(Thioglycoside)是聚糖合成中另一类明星糖基供体, 最常用的硫苷为苯硫苷和乙硫苷.最早使用硫苷进行糖基化反应可以追溯到1973年, Ferrier小组[42]以HgSO4为活化剂, 实现了全苄基葡萄糖苯硫苷与糖受体之间的偶联反应.
之后, 一系列新的硫苷活化剂被开发出来.它们主要可以被分为四类, 如图 4所示.
第一类是金属盐类活化剂.早期的例子主要是汞盐、铜盐和银盐, 这类活化剂使用时通常需要比较苛刻的条件, 现已很少使用.
第二类是类卤鎓离子亲电试剂.其中最常用的是van Boom和Fraser-Reid分别报道的NIS/TfOH[43]和NIS/AgOTf[44], 这两种活化剂都可以直接买到, 活化能力很强并且副反应较少, 因而在硫苷活化中用得很广泛.
第三类是有机硫试剂, 这类活化剂有Garegg首先使用的二甲基甲硫基硫鎓三氟甲磺酸盐(DMTST)[45], Whitesides发展的PhSCl/AgOTf[46], Crich发展的苯基哌啶基亚砜(BSP)[47]及类似结构的二苯亚砜(Ph2SO)[48]、苯基吗啉基亚砜(BSM)[49]与三氟甲磺酸酐的组合(BSP/Tf2O, Ph2SO/Tf2O, BSM/Tf2O)等.
第四类是单电子转移试剂及光、电活化方法, 如Sina 发展的六氯锑酸三(4-溴苯基)铵(TBPA)[50]、电化学法[51]及近年来不断兴起的光化学法[52]等.
硫苷的活化一般需要当量的活化剂, 不过近期也有研究者报道亚当量的活化剂也能完全活化硫苷. 2013年, Pohl小组[53]发现使用Ph3Bi(OTf)2可以在室温下活化硫苷, 该活化剂具有溶解性好、对空气/光稳定并且半衰期长等优点, 并且有些例子只需要0.5 equiv.活化剂即可完全活化硫苷供体.
总之, 由于硫苷制备非常方便, 在许多功能基转化条件下也足够稳定, 而且可以很容易被软亲电试剂活化, 因此使用硫苷进行糖基化反应一直受到重视, 不仅新的活化剂或活化方式不断地被报道, 而且有关硫苷用于复杂糖类化合物合成的工作也大量发表.更重要的是, 硫苷既可以作为端基保护基、又可以作为优良离去基的性质, 赋予了它在聚糖类化合物合成中独特的价值, 使它尤其适用于聚糖的液相一釜多步合成.
使用现有的糖基化方法, 探索如何高效地将糖砌块组装在一起, 进而形成复杂的聚糖结构, 这是糖化学中另一个重要的问题, 即聚糖的组装策略问题.
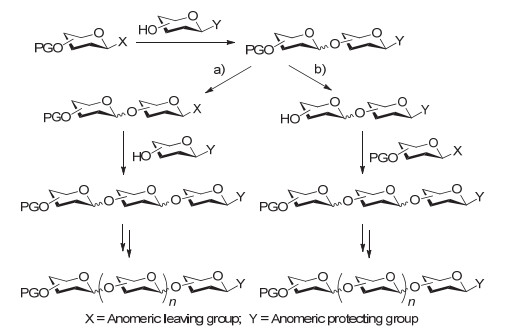
传统的聚糖组装策略主要有两种类型.第一种是线性合成, 按照糖链合成的延伸方向, 又可以分为从非还原端到还原端(图 5a)和从还原端到非还原端(图 5b)两种类型; 第二种是汇聚式合成, 一般是指先通过线性合成得到一些短的糖片段, 最后再进行糖片段之间的偶联.汇聚式合成可以实现分子尺寸的快速增加.
无论是线性合成还是汇聚式合成, 通常都会涉及到端基取代基的不断调整或糖上临时保护基的反复脱除, 这就使得整体合成步骤一般较长, 需要对中间体分离纯化, 最终降低了聚糖合成的效率.
为了提高聚糖组装的效率, 近三十年来研究者们发展了不少新策略.其中最具代表性的工作包括正交选择性的液相一釜合成策略[54]、基于糖基供体活性的程序化液相一釜合成策略[55]、预活化液相一釜合成策略[56]和自动化固相合成策略[57].
液相一釜合成策略一般是在同一个反应容器中, 实现多个糖砌块从非还原端向还原端的有序偶联, 最终通过一次纯化步骤即可快速实现目标聚糖链的组装.这类策略避免了传统聚糖链合成中反复的保护/去保护操作, 也不需要对中间体进行分离, 从而使聚糖合成步骤大大简化.
正交选择性的液相一釜合成策略是通过选用不同的活化剂, 实现多个带有不同离去基的糖砌块的正交选择性活化, 从而在同一个反应容器中完成多个糖苷键的构建(图 6).
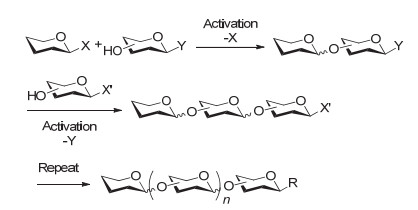
该策略最早由Takahashi小组[54]于1994年提出, 随后被成功应用于多种聚糖如植物抗毒素诱导剂七糖[58]、薯蓣皂苷类化合物[59]、黏蛋白核心分支七糖[60]、糖氨基酸[61]、寡聚唾液酸[62]、糖胺聚糖[63]、甘露聚糖[64]等的化学合成中.然而该策略也存在一定缺陷, 即为了实现正交活化, 至少需要制备两套带不同离去基的糖砌块, 这给原料的制备增加了困难, 而且目前能够实现正交活化的离去基组合也还比较有限.
基于糖基供体活性的程序化液相一釜合成策略是使用一套带相同离去基的糖基模块, 按照其反应活性的高低依次加入到反应瓶中, 使糖基化反应按照设计好的方向有序进行(图 7).其基本原理是只要相邻两个模块活性差别足够大, 活性较高的糖基供体就会与活性较低的糖模块发生糖基化反应, 而不会发生活性较低的糖模块间的自身偶联.
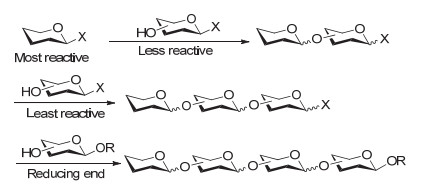
该策略由Wong小组[55]在1999年提出, 目前已经被成功应用于一些肿瘤相关糖抗原Globo-H[65]、Lewis X二聚体[66]、Lewis Y[67]、N3 minor八糖[68]、岩藻糖化的GM1表位[69]以及肝素类五糖[70]、胚胎干细胞表面糖链Lc4和IV2Fuc-Lc4[71]等重要寡糖分子的合成.不过, 由于该策略对糖基模块活性差别的依赖很大, 目前最多实现四个组分的一釜合成; 并且由于糖模块活性需要通过精细的保护基操作来调节, 使得糖模块制备比较繁琐, 这在很大程度上限制了该策略的进一步应用.
预活化液相一釜合成策略由我们小组和Huang小组于2004年共同提出[56], 其基本原理如图 8所示:单糖供体(一般为对甲基苯硫苷)在低温下用促进剂预先活化, 生成活性中间体(Active intermediate), 随后加入带相同离去基的单糖受体进行反应, 即可完成二糖的偶联; 二糖又可以作为供体与下一个带相同离去基的受体偶联, 这样多次重复供体预活化和加入受体进行糖基化的操作, 即可实现目标聚糖链的快速合成.
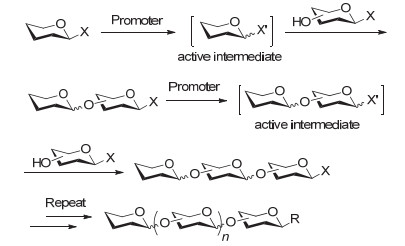
在预活化一釜合成策略中, 所有糖基供体和受体都使用相同的离去基和活化条件, 只通过加料顺序的改变就能实现聚糖链的有序延长, 而且能够实现低活性供体与高活性受体的偶联, 克服了程序化一釜合成策略对糖模块反应活性的依赖.该策略的成功应用包括肿瘤抗原Globo-H[72]和KH-1[73]、透明质酸寡糖[74]、Lewis X[75]、肝素类寡糖[76]、复杂型双天线N-连接十二糖[77]、绿藻衣壳蛋白新型N-glycan[78]以及多种细菌表面糖链[79]的合成等.
不过该策略目前也还存在一些不足, 比如有时候会出现苷元转移, 因此仍然需要发展新的偶联方法来提高单步糖基化反应的偶联效率.
液相一釜合成策略通过在同一个反应瓶中快速完成多步糖基化反应, 最后只进行一次分离纯化, 从而大大提高糖合成效率; 而自动化固相合成策略与之不同, 其反应在固相载体上进行, 通过载体的辅助完成中间体的快速纯化并且使用机器来提高聚糖合成效率.
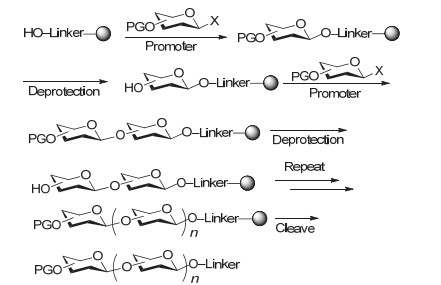
Seeberger小组[57]于2001年首先报道了聚糖的自动化固相合成技术.与多肽或核酸的自动化合成方法类似, 他们通过一个连接臂(Linker)将糖受体连接在树脂上, 每步糖基化反应完成后, 脱除临时保护基再进行下一步反应, 中间体的纯化通过简单的过滤和洗涤即可快速完成, 最后再将目标糖链从树脂上切下来(图 9).
使用自动固相合成组装聚糖链, 一般只需要在最后一步借助柱层析分离, 而且由于每次糖基化可以使用大大过量的糖基供体以确保负载在树脂上的糖基受体尽量反应完全, 因而整体效率通常都比较高.
Seeberger及其他小组将自动化固相合成策略用于多种聚糖结构的合成, 包括植物抗毒素诱导子十二糖[57]、肿瘤相关糖抗原Globo-H[80]和Ley-Lex (KH-1)[81]、N-glycan的核心五糖[82]、β-甘露聚糖[83]、糖胺聚糖[84]以及糖基磷脂酰肌醇(Glycosylphosphatidylinositol, GPI)六糖等[85].
不过, 该策略也存在一些不足之处, 例如: (1)由于糖基受体与树脂相连导致反应活性降低, 因此每步糖基化反应需要加入多达10 equiv.的供体以确保受体尽量完全消耗, 这对于本身就需要多步反应才能制备的糖基模块是极大的消耗, 因而花费较大; (2)对于比较难构建的糖苷键如1, 2-顺式糖苷键, 由于缺乏可靠的制备方法, 因此往往需要使用提前制备好的二糖模块来替代; (3)随着聚糖链的不断延长, 产物与片段缺失的序列很难分离, 因而通常还需要引入额外的分离纯化策略.
在过去几十年里, 糖化学家创造性地发展了一系列新颖的糖基供体和相应的活化方式, 极大地丰富了聚糖化学合成的工具箱; 另一方面, 基于这些糖基化方法以及对糖基化反应理解的不断深入, 发展了一些高效的聚糖组装策略, 这使得聚糖合成效率得到了极大的提高.可以说, 就像小分子天然产物全合成“没有合成不出来的分子”一样, 对于大多数长度适当的寡糖结构, 人们都有办法完成其化学合成.
然而, 结构复杂的长链多糖的化学合成对糖化学家而言仍然是一项富有挑战的任务.事实上, 迄今为止, 人们完成的复杂长链多糖的化学合成工作也为数不多, 接下来我们将结合多糖合成代表性的例子进行介绍.
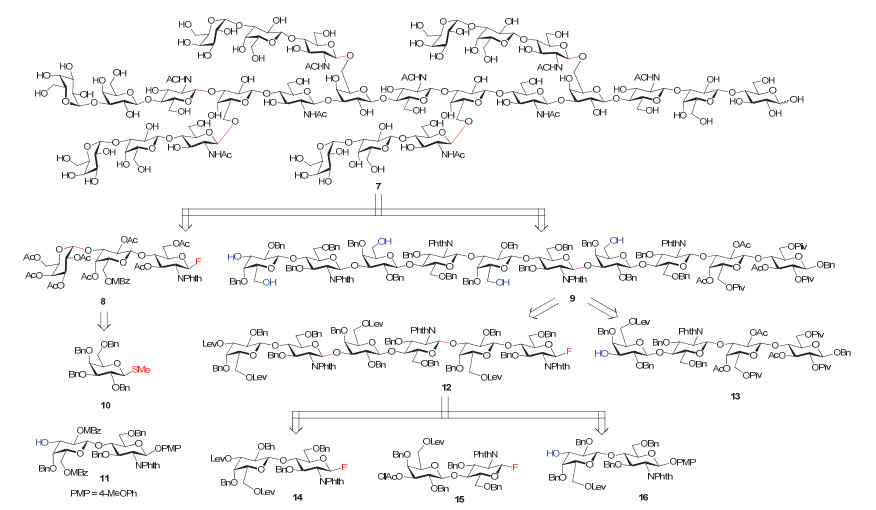
早在1993年, Ogawa小组[86]报道了一例来源于兔红细胞膜的复杂多糖7的化学合成, 该分子由25个单糖单元组成, 是一种含有多个N-乙酰乳糖胺的五天线型多糖(图 10).作者设计了高度汇聚式的合成策略, 希望最后通过一步糖基化反应将5个三糖供体8和一个含5个裸露羟基的十糖受体9偶联在一起.氟代糖供体8可以拆分为硫苷供体10和还原端PMP (4-甲氧基苯基)保护的二糖受体11.十糖受体9可以由氟代糖12与已知寡糖化合物13反应再脱除临时保护基Lev得到; 而化合物12则可以继续拆分为三个二糖模块14, 15和16.在这个工作中, 除了供体10外, 所有的糖偶联反应都以氟代糖为供体.汇聚式合成策略的使用, 使得线性步骤大为缩短.但在糖模块的线性合成过程中, 多次涉及到临时保护基的脱除以及将端基PMP保护基调整为离去基F, 因而总的步骤仍然较长, 这在一定程度上降低了整体合成效率.
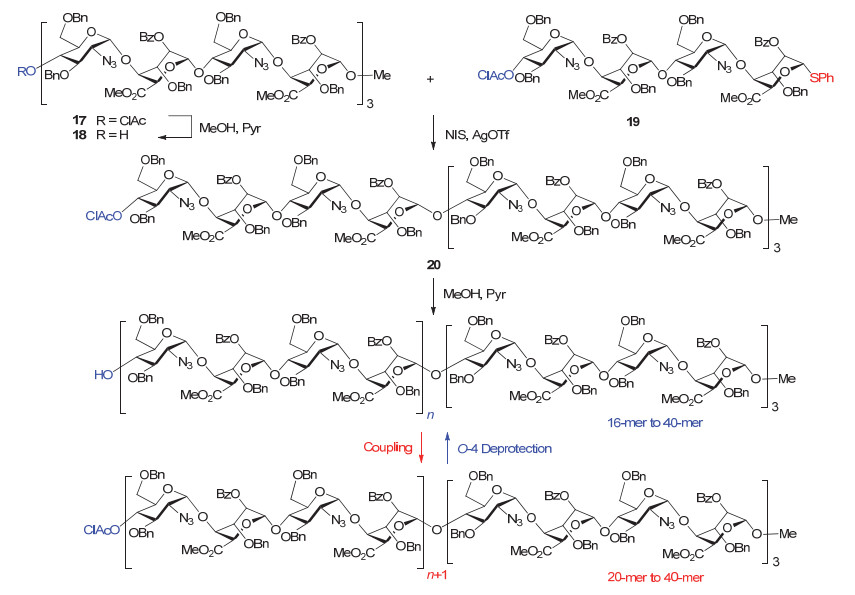
近期, Gardiner小组[87]报道了他们在多糖化学合成中的最新进展.他们以四糖为基本模块, 使用从还原端到非还原端的线性合成策略, 合成了一系列长度从16-mer到40-mer的肝素类多糖.如图 11所示, 他们以带一个临时保护基ClAc的十二糖17为起始原料, 脱除ClAc后得受体18, 再与四糖硫苷供体19在NIS/AgOTf条件下发生偶联, 得到的十六糖20再次脱除ClAc, 继续与四糖硫苷19偶联, 之后不断重复脱除临时保护基/偶联过程, 最终可以得到最长为四十糖的全保护肝素类多糖.
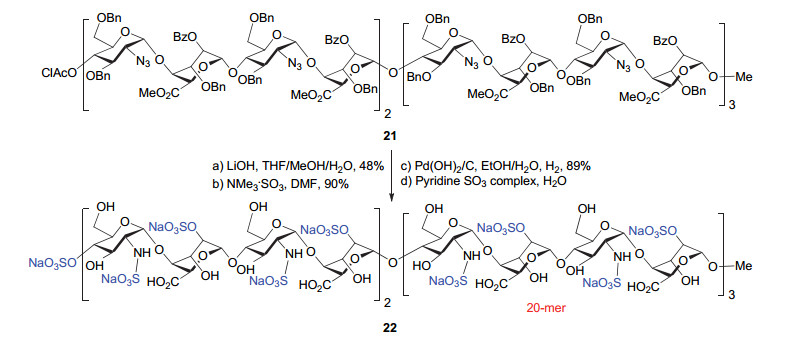
需要说明的是, 在该工作中, 作者只对最长为二十糖的聚糖链21进行了脱保护和官能团化, 得到N-硫酸化和部分O-硫酸化的二十糖22.即便如此, 聚糖22也已经是人工合成的结构均一的最长肝素类多糖(图 12).
2005年, Kong小组[88]报道了一例植物来源多糖23的化学合成工作(图 13).该多糖属于阿拉伯半乳聚糖, 存在于具有免疫调节活性的紫花紫锥菊(Echinacea purpurea)中, 由一条β-1, 6连接的半乳糖主链和若干α-1, 2、α-1, 3连接的L-呋喃阿拉伯糖分支组成.
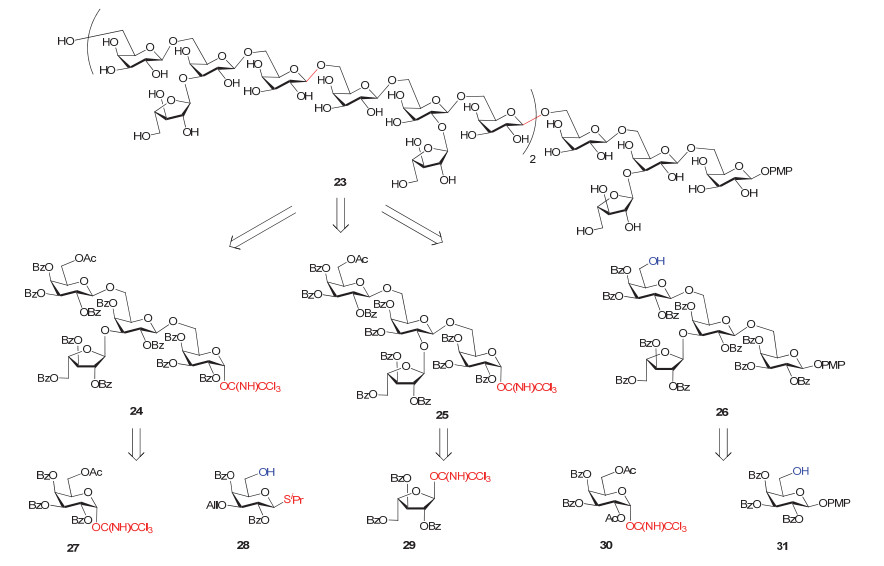
作者设计合成了多个单糖砌块27, 28, 29, 30和31, 这些砌块使用乙酰基(Ac)或者烯丙基(All)作为临时保护基, 其余位置均用Bz保护, 糖苷键的构建主要使用糖基三氯乙酰亚胺酯和硫苷作为供体.首先从单糖砌块出发, 经多步反应合成了三个四糖片段24、25和26; 再以这些四糖为模块, 从还原端向非还原端延长糖链; 最后再进行整体脱保护操作即得目标二十糖23.
细菌细胞壁往往由丰富的多糖结构组成, 这些多糖往往与细菌的存活力和致病力有关, 其生物合成酶往往是一些抗生素的作用靶标.另外, 一些抗原性多糖(或其蛋白缀合物)能刺激机体产生保护性抗体, 常常被开发成预防性疫苗.因此, 细菌来源的多糖吸引了很多糖化学家的合成兴趣.
Pozsgay小组[89]早在20世纪末就致力于痢疾志贺氏菌糖疫苗的研究, 他们于1998年报道了痢疾志贺氏菌内毒素O-抗原四糖重复序列的一个四聚体32的合成.如图 14所示, 作者从四糖受体33出发, 以相同骨架的四糖三氯乙酰亚胺酯34为模块, 通过模块之间的糖基化偶联反应快速延长糖链, 接着脱除临时保护基用于下一步糖基化, 最终高效地合成了十六糖32.值得一提的是, 作者在后续的活性评价中发现, 合成得到的多糖与载体蛋白缀合后, 所引起的血清抗体滴度比天然来源的多糖-蛋白缀合物更高[90].
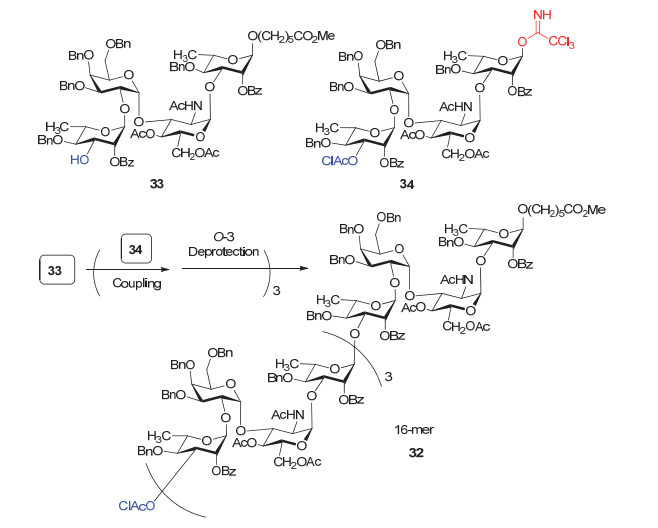
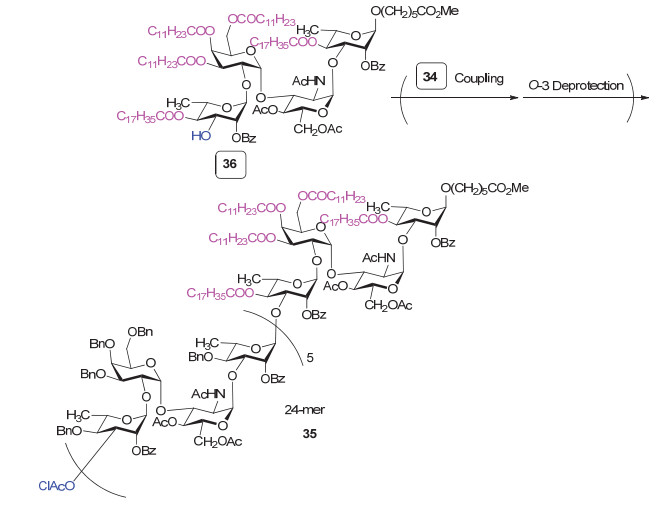
在另一个工作中, Pozsgay等[91]采用类似的“模块化”策略继续延长糖链, 最终完成了一个二十四糖35的合成(图 15).为了方便在糖基化反应完成后快速分离目标产物, 作者在还原端的四糖模块36上引入了多个疏水性长链烷烃, 这样在合成过程中, 只需将产物吸附在反相硅胶上, 之后用廉价低毒的醇类溶剂洗脱就可以完成目标产物的纯化.
结核分枝杆菌细胞壁中富含结构独特的多糖结构, 其中阿拉伯半乳聚糖(Arabinogalactan, AG)得到了较多关注, 该多糖全部由呋喃型糖组成, 这类单糖不存在于任何哺乳动物体内, 而且含有多个合成难度较大的1, 2-顺式呋喃阿拉伯糖苷键.化学合成得到的阿拉伯半乳聚糖(或其片段)可用于阐明结核分枝杆菌细胞壁生物合成途径, 进而有助于研制新型抗结核药物; 同时也有开发成预防结核病的新型糖疫苗的潜力.
2007年和2011年, Lowary[92]和Ito[93]小组分别完成了两条几乎相同的阿拉伯聚糖37和38的化学合成(图 16和图 17).这两个多糖都由22个呋喃阿拉伯糖组成(Araf22), 有3处分支位点, 且非还原端含4个β-1, 2糖苷键.
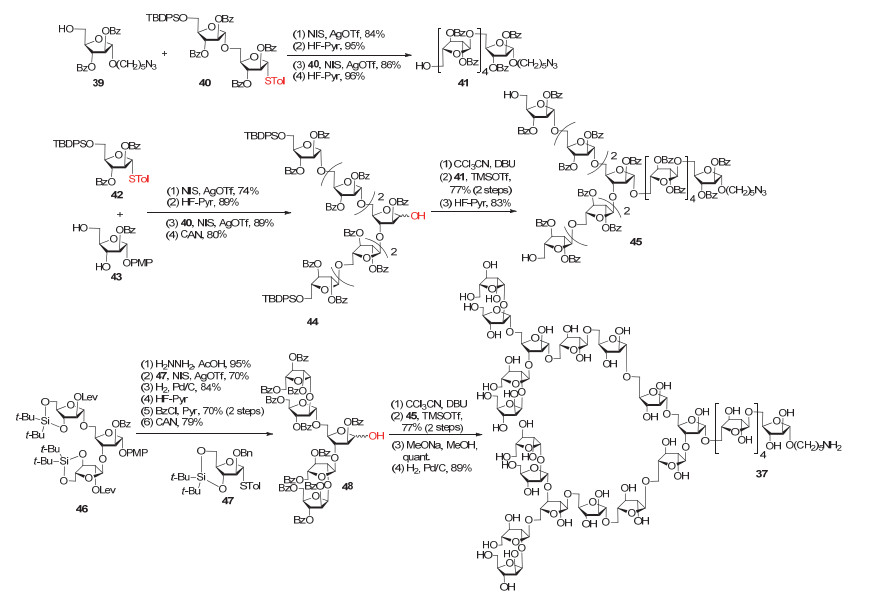
在Lowary小组的全合成工作中, 合成短的糖模块时使用对甲苯硫苷为供体, 而在片段偶联阶段选用糖基三氯乙酰亚胺酯作为供体, 另外, 除临时保护基选用TBDPS或者Lev外, 其余位点都用Bz保护以方便最后一步整体去保护.
作者首先合成了三种主要的糖片段41、45和48.分支五糖48的两个β-1, 2呋喃阿拉伯糖苷键是使用3, 5-O-di-tert-butylsilylidene (DTBS)保护的单糖供体构建的.将这些片段依次拼接在一起, 最后脱除Bz和还原N3, 即得到目标多糖37(图 16).
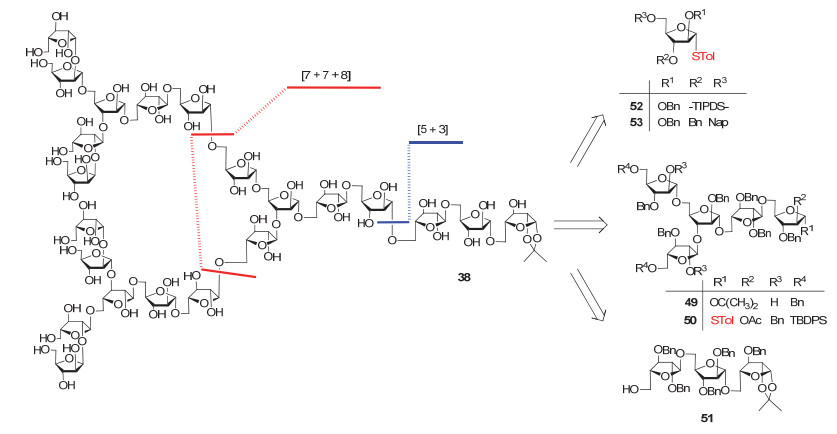
Ito在合成二十二糖38时, Bn是主要的保护基类型, 而且所有糖苷键的生成都是使用对甲苯硫苷为糖基供体(图 17).作者首先将整个分子拆分为三个糖片段49、50和51, 而单糖供体52和53则用于立体选择性构建β-1, 2呋喃阿拉伯糖苷键.使用环状硅保护基TIPDS保护的硫苷52可以与受体49直接糖基化得到β-立体构型为主的产物, 而使用Nap介导的分子内苷元转移(以硫苷53为糖基供体), 则可以得到β-立体构型唯一的产物.与Lowary小组合成多糖37时需多次将端基PMP转化为三氯乙酰亚胺酯不同的是, Ito等的合成是将还原端1, 2-异丙叉基通过“三步法”转化为对甲苯硫苷.
近期, Hotha小组[94]报道了他们在结核分枝杆菌细胞壁阿拉伯半乳聚糖化学合成中的进展.如图 18所示, 目标分子含有两个呋喃半乳糖, 而不含非还原端的β-1, 2糖苷键.同以上几个多糖合成类似, 作者也采用了模块化的汇聚式合成策略.而与Lowary或Ito使用糖基三氯乙酰亚胺酯或者硫苷为供体不同的是, 该工作中所有糖苷键的构建均采用Hotha小组自己发展的AuCl3/AgOTf共催化的以炔丙基原酸酯为供体的糖基化方法.最终以47步反应, 0.09%的总收率完成了二十一糖52的全合成.
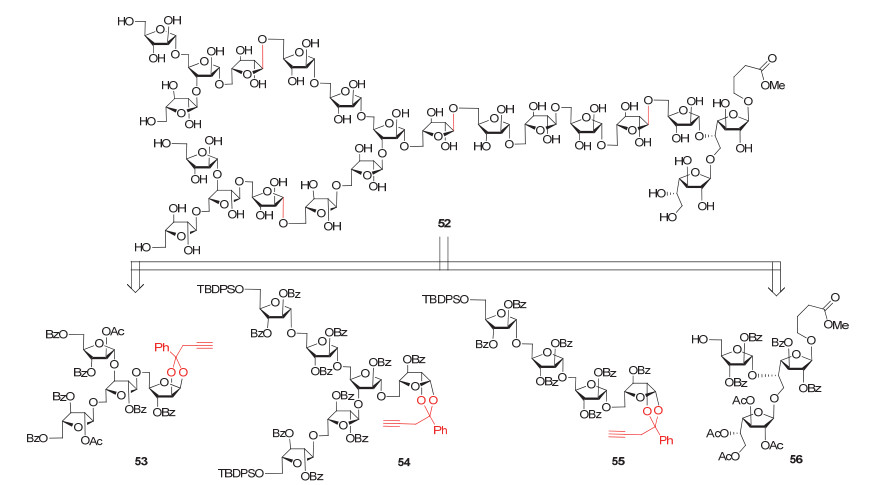
另外, Hotha小组[95]最近还报道了一例阿拉伯半乳聚糖中二十五糖片段的合成, 其结构与52比较类似, 唯一的差别在于非还原端含有4个β-1, 2连接的单糖残基, 并且使用的是该组近期发展的糖基碳酸酯[96]为供体.
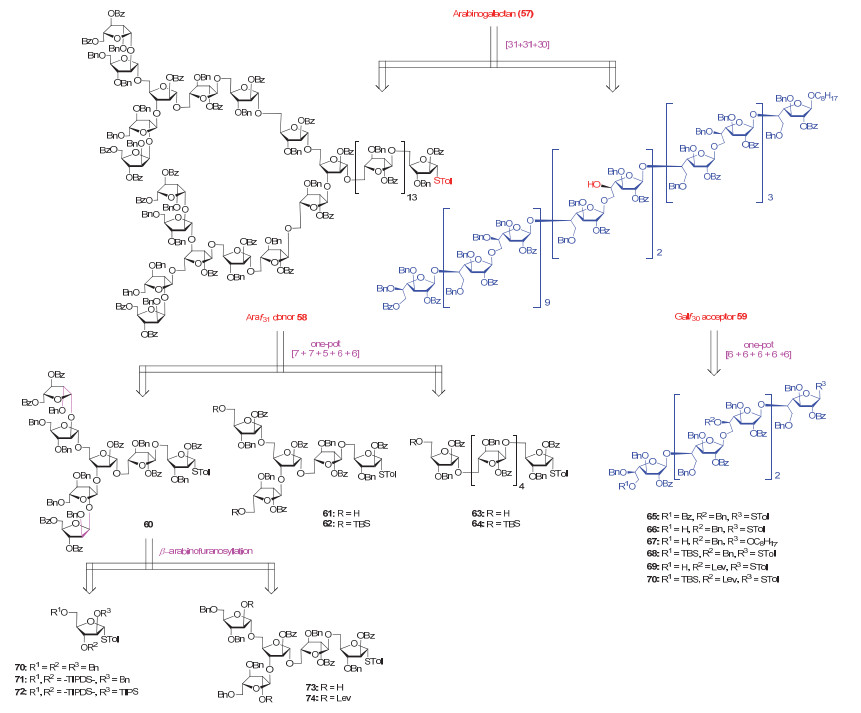
结核分枝杆菌细胞壁多糖Arabinogalactan (57, 图 19)的首次全合成是由我们小组[79e]于2017年完成的.完整的Arabinogalactan由一条含30个呋喃半乳糖的线性主链和两条相邻的含31个呋喃阿拉伯糖的分支侧链组成.该分子共有8处分支位点, 且非还原端含有8个化学上难以构建的β-1, 2呋喃阿拉伯糖苷键.

如图 20所示, Arabinogalactan (57)首先被拆分为两条多糖链, 即Araf31供体58和Galf30受体59. Araf31供体58可以采用预活化液相一釜合成策略, 由寡糖链60, 61和63合成得到.类似地, Galf30受体59可以由寡糖链65, 66, 69和67经过一釜五组分糖基化反应合成.目标分子非还原端的β-1, 2呋喃阿拉伯糖苷键由环状硅保护的硫苷供体71和二醇硫苷受体73在预活化操作下高效构建.所有的寡糖链(62, 64, 68, 70和74)均可由适当保护的单糖硫苷经预活化一釜糖基化反应得到.值得一提的是, 在最后使用Araf31供体58和Galf30受体59的双糖基化反应中, 经过大量的反应条件筛选, 最后发现只有在我们小组发展的BSM/Tf2O活化条件下, 才可以高收率地完成三条多糖链的定点偶联.最后经整体去保护, 便完成了多糖Arabinogalactan (57)的全合成.
结核分枝杆菌细胞壁中其它重要多糖, 如主要的抗原性成分脂阿拉伯甘露聚糖(Lipoarabinomannan, LAM)也有多个片段合成的报道.代表性的例子如Fraser-Reid小组[97]2006年完成的全保护二十八糖和近期Hotha小组[98]报道的阿拉伯甘露二十一糖的合成等.
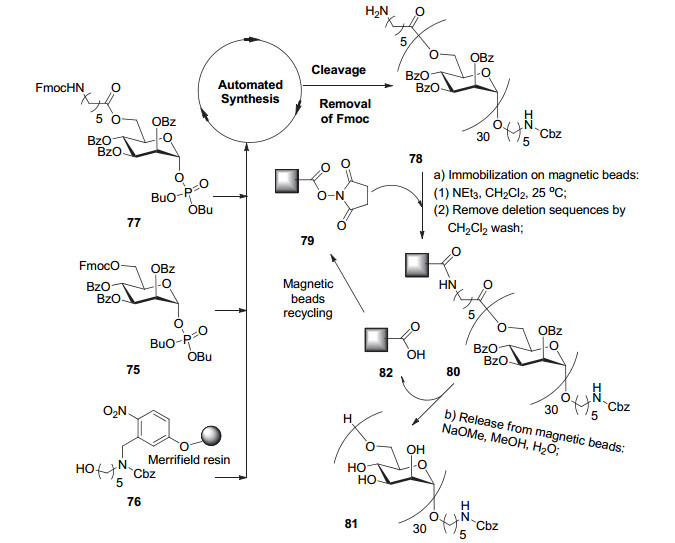
2013年, Seeberger小组[99]报道了他们使用自动化固相合成在多糖合成领域中的重要进展.如图 21所示, 他们以6-位Fmoc保护, 其它位点Bz保护的甘露糖磷酸酯75为糖基供体, 使用带有紫外下可断裂连接臂(Linker)的树脂76作固相载体, 首先经固相合成得到29糖(每步糖基化偶联后使用Pyr/Ac2O封闭未反应完全的受体, 再脱除Fmoc以进行下一步糖基化), 最后一步糖基化使用6-位带有一个功能基团的糖基供体77, 固相合成完成后, 将糖链从树枝上切下来, 再脱除Fmoc得到78.由于固相合成中有一部分受体不会完全反应, 因此从树脂上切下来的是由产物和片段缺失序列组成的混合物.为了将30-mer产物从混合物中分出, 作者发展了一种“捕获-释放”的方法, 即使用丁二酰亚胺活化的磁珠79作为“捕获”试剂, 只有完整的30-mer能与磁珠反应, 再将片段缺失序列通过简单的洗涤除去, 最后在MeONa/MeOH条件下将糖链从磁珠上“释放”下来, 同时脱除Bz, 即得到结构均一的甘露糖三十聚体81.另外, 释放下来的磁珠82可以循环使用.
2017年, Seeberger小组[100]报道了他们使用自动化固相合成更长α-1, 6甘露聚糖的工作.如图 22所示, 作者放弃了上一个工作中所使用的糖基磷酸酯, 而换用更稳定的乙硫苷83作供体, 而且在甘露糖3, 4位使用Bn保护以提高供体反应活性.作者发现随着糖链的不断延长, n-1的片段缺失序列越来越多, 而且与完整长度的糖链在HPLC上的分离度也越来越差.为了解决这个问题, 他们在最后5步糖基化时使用了大大过量的糖基供体(2×6.4 equiv.), 最终耗时250 h, 经过102步反应得到部分保护的50-mer 84 (5%总收率, 平均每步97%).整体去保护即完成甘露糖五十聚体85合成.
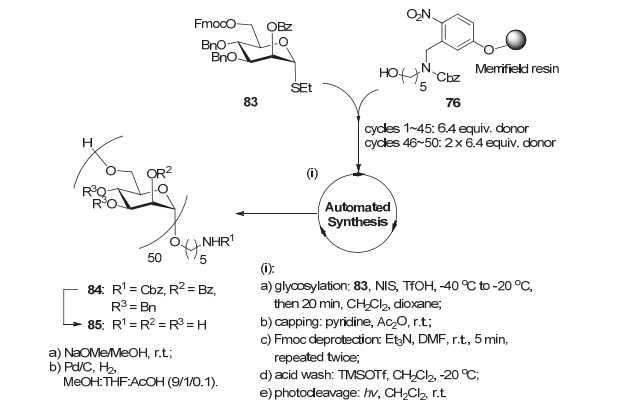
最近, Seeberger小组[101]对自动化固相合成技术中的“加帽(Capping)”反应进行了改进.他们发现使用MsOH/Ac2O/CH2Cl2条件代替原先的Pyr/Ac2O条件, 可以显著提高加帽反应的效率, 缩短合成时间和提高总收率.作者使用改进的自动化固相合成技术重新合成多糖85时, 总耗时可以缩短至4天, 并且总收率提高至22%.
近几十年来, 聚糖的化学合成取得了惊人的进步.许多性质优良的糖基供体、高效新颖的活化方式以及以液相一釜合成和自动固相合成为代表的聚糖链组装新策略的发展和不断成熟, 使得糖化学家们的武器库变得越来越充实, 很多曾经无法通过化学方法得到的聚糖结构现在都能合成甚至大量制备.
然而, 和核酸或多肽相比, 聚糖化学合成的技术水平仍然有待提高.糖合成化学中还有很多难题没有得到有效解决, 例如:糖基化反应的详细机理还有待进一步探索; 某些糖苷键特别是1, 2-顺式糖苷键选择性构建方法还比较有限; 糖基化方法或聚糖链组装策略的普适性还有待提高.正因为如此, 聚糖的化学合成通常需要精心的设计和反复的尝试, 迄今为止还是非常依赖具备专门知识和技能的实验室.
对于自然界广泛存在的多聚糖, 其化学合成、功能阐释和构效关系等方面的研究还处在起步阶段, 这片处女地有待糖科学家们去继续探索, 更多的重要进展和有趣发现值得人们期待.
Varki, A. Glycobiology 1993, 3, 97. doi: 10.1093/glycob/3.2.97
Krasnova, L.; Wong, C.-H. Annu. Rev. Biochem. 2016, 85, 599. doi: 10.1146/annurev-biochem-060614-034420
Boltje, T. J.; Buskas, T.; Boons, G.-J. Nat. Chem. 2009, 1, 611. doi: 10.1038/nchem.399
Seeberger, P. H.; Werz, D. B. Nature 2007, 446, 1046. doi: 10.1038/nature05819
(a) Gao, Y.; Cao, Z.; Han, Z.; Zhang, Q.; Hu, J.; Guo, R.; He, X.; Ding, F.; You, Q.; Zhang, Y. Chin. J. Org. Chem. 2019, 39, 390. (高阳光, 曹周, 韩忠享, 张强, 胡杰, 郭锐, 贺贤然, 丁菲, 尤庆亮, 张勇民, 有机化学, 2019, 39, 390); (b) Guo, Q.; Wang, X.; Huang, C.; Zhang, P.; Li, Y.; Chen, B. Chin. J. Org. Chem. 2018, 38, 940. (邱果, 王新承, 黄崇品, 张璞, 李英霞, 陈标华, 有机化学, 2018, 38, 940); (c) Yuan, W.; Huang, Y.; Wu, C.; Liu, X.; Xia, Y.; Wang, H. Chin. J. Chem. 2017, 35, 1739; (d) Guo, B.; Ye, L.; Tang, G.; Zhang, L.; Yue, B.; Tsang, S. C. E.; He, H. Chin. J. Chem. 2017, 35, 1529; (e) Mao, R.; Sun, L.; Wang, Y.-S.; Zhou, M.-M.; Xiong, D.-C.; Li, Q.; Ye, X.-S. Chin. Chem. Lett. 2018, 29, 61; (f) Tang, S.; Xiong, D.-C.; Jiang, S.; Ye, X.-S. Org. Lett. 2016, 18, 568.
Bertozzi, C. R.; Kiessling, L. L. Science 2001, 291, 2357. doi: 10.1126/science.1059820
Gabius, H.-J. The Sugar Code:Fundamentals of Glycosciences, John Wiley & Sons, New Jersey, 2011.
Tanaka, H.; Kawai, T.; Adachi, Y.; Hanashima, S.; Yamaguchi, Y.; Ohno, N.; Takahashi, T. Bioorg. Med. Chem. 2012, 20, 3898. doi: 10.1016/j.bmc.2012.04.017
Petitou, M.; Duchaussoy, P.; Driguez, P.-A.; Hérault, J.-P.; Lormeau, J.-C.; Herbert, J.-M. Bioorg. Med. Chem. Lett. 1999, 9, 1155. doi: 10.1016/S0960-894X(99)00155-9
Wang, L.; Feng, S.; An, L.; Gu, G.; Guo, Z. J. Org. Chem. 2015, 80, 10060. doi: 10.1021/acs.joc.5b01686
(a) Zhu, X.; Schmidt, R. R. Angew. Chem. Int. Ed. 2009, 48, 1900; (b) Hsu, C.-H.; Hung, S.-C.; Wu, C.-Y.; Wong, C.-H. Angew. Chem. Int. Ed. 2011, 50, 11872; (c) Seeberger, P. H. Acc. Chem. Res. 2015, 48, 1450; (d) Kulkarni, S. S.; Wang, C.-C.; Sabbavarapu, N. M.; Podilapu, A. R.; Liao, P.-H.; Hung, S.-C. Chem. Rev. 2018, 118, 8025.
Michael, A. Am. Chem. J. 1879, 1, 305. doi: 10.1021/ja02151a603
Toshima, K.; Tatsuta, K. Chem. Rev. 1993, 93, 1503. doi: 10.1021/cr00020a006
(a) Garcia, B. A.; Poole, J. L.; Gin, D. Y. J. Am. Chem. Soc. 1997, 119, 7597; (b) Garcia, B. A.; Gin, D. Y. J. Am. Chem. Soc. 2000, 122, 4269.
(a) Schmidt, R. R.; Michel, J. Angew. Chem. Int. Ed. 1980, 19, 731; (b) Schmidt, R. R. Angew. Chem. Int. Ed. 1986, 25, 212.
(a) Codée, J. D. C.; Litjens, R. E. J. N.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Chem. Soc. Rev. 2005, 34, 769; (b) Lian, G.; Zhang, X.; Yu, B. Carbohydr. Res. 2015, 403, 13.
Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583. doi: 10.1021/ja00224a060
Danishefsky, S. J.; Bilodeau, M. T. Angew. Chem. Int. Ed. 1996, 35, 1380. doi: 10.1002/(ISSN)1521-3773
Plante, O. J.; Palmacci, E. R.; Andrade, R. B.; Seeberger, P. H. J. Am. Chem. Soc.2001, 123, 9545. doi: 10.1021/ja016227r
Yu, B. Acc. Chem. Res. 2018, 51, 507. doi: 10.1021/acs.accounts.7b00573
Koenigs, W.; Knorr, E. Ber. Dtsch. Chem. Ges. 1901, 34, 957. doi: 10.1002/(ISSN)1099-0682
Zemplén, G.; Gerecs, A. Ber. Dtsch. Chem. Ges. 1930, 63, 2720. doi: 10.1002/cber.v63:10
Helferich, B.; Wedemeyer, K. F. Justus Liebigs Ann. Chem. 1949, 563, 139. doi: 10.1002/(ISSN)1099-0690
Igarashi, K.; Irisawa, J.; Honma, T. Carbohydr. Res. 1975, 39, 213. doi: 10.1016/S0008-6215(00)86131-5
Kronzer, F. J.; Schuerch, C. Carbohydr. Res. 1973, 27, 379. doi: 10.1016/S0008-6215(00)81320-8
Wulff, G.; Röhle, G.; Krüger, W. Chem. Ber. 1972, 105, 1097. doi: 10.1002/(ISSN)1099-0682
Yamada, H.; Hayashi, T. Carbohydr. Res. 2002, 337, 581. doi: 10.1016/S0008-6215(02)00029-0
Bernstein, S.; Conrow, R. B. J. Org. Chem. 1971, 36, 863. doi: 10.1021/jo00806a001
Nishizawa, M.; Garcia, D. M.; Shin, T.; Yamada, H. Chem. Pharm. Bull. 1993, 41, 784. doi: 10.1248/cpb.41.784
Mukaiyama, T.; Murai, Y.; Shoda, S. Chem. Lett. 1981, 10, 431. doi: 10.1246/cl.1981.431
Mukaiyama, T.; Hashimoto, Y.; Shoda, S. Chem. Lett. 1983, 12, 935. doi: 10.1246/cl.1983.935
Matsumoto, T.; Maeta, H.; Suzuki, K. Tetrahedron Lett. 1988, 29, 3567. doi: 10.1016/0040-4039(88)85294-8
Hashimoto, S.; Hayashi, M.; Noyori, R. Tetrahedron Lett. 1984, 25, 1379. doi: 10.1016/S0040-4039(01)80163-5
Mukaiyama, T.; Jona, H.; Takeuchi, K. Chem. Lett. 2000, 29, 696. doi: 10.1246/cl.2000.696
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900. doi: 10.1002/anie.v48:11
El-Badry, M. H.; Gervay-Hague, J. Tetrahedron Lett. 2005, 46, 6727. doi: 10.1016/j.tetlet.2005.07.129
(a) Lam, S. N.; Gervay-Hague, J. Carbohydr. Res. 2002, 337, 1953; (b) Lam, S. N.; Gervay-Hague, J. Org. Lett. 2002, 4, 2039; (c) Lam, S. N.; Gervay-Hague, J. J. Org. Chem. 2005, 70, 2387.
Sun, L.; Wu, X.; Xiong, D.-C.; Ye, X.-S. Angew. Chem. Int. Ed. 2016, 55, 8041. doi: 10.1002/anie.201600142
Park, Y.; Harper, K. C.; Kuhl, N.; Kwan, E. E.; Liu, R. Y.; Jacobsen, E. N. Science 2017, 355, 162. doi: 10.1126/science.aal1875
Schmidt, R. R.; Toepfer, A. Tetrahedron Lett. 1991, 32, 3353. doi: 10.1016/S0040-4039(00)92704-7
Yu, B.; Tao, H. Tetrahedron Lett. 2001, 42, 2405. doi: 10.1016/S0040-4039(01)00157-5
Ferrier, R. J.; Hay, R. W.; Vethaviyasar, N. Carbohydr. Res. 1973, 27, 55. doi: 10.1016/S0008-6215(00)82424-6
Veeneman, G. H.; Van Leeuwen, S. H.; Van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331. doi: 10.1016/S0040-4039(00)88799-7
Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313. doi: 10.1016/S0040-4039(00)97609-3
Andersson, F.; Fúgedi, P.; Garegg, P. J.; Nashed, M. Tetrahedron Lett. 1986, 27, 3919. doi: 10.1016/S0040-4039(00)83917-9
Martichonok, V.; Whitesides, G. M. J. Org. Chem. 1996, 61, 1702. doi: 10.1021/jo951711w
Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015. doi: 10.1021/ja0111481
Codée, J. D. C.; Litjens, R. E. J. N.; den Heeten, R.; Overkleeft, H. S.; van Boom, J. H.; van der Marel, G. A. Org. Lett. 2003, 5, 1519. doi: 10.1021/ol034312t
Wang, C.; Wang, H.; Huang, X.; Zhang, L.-H.; Ye, X.-S. Synlett 2006, 2846.
Marra, A.; Mallet, J. M.; Amatore, C.; Sinaÿ, P. Synlett 1990, 572. doi: 10.1055/s-1990-22045
(a) Mitsudo, K.; Kawaguchi, T.; Miyahara, S.; Matsuda, W.; Kuroboshi, M.; Tanaka, H. Org. Lett. 2005, 7, 4649; (b) Nokami, T.; Shibuya, A.; Tsuyama, H.; Suga, S.; Bowers, A. A.; Crich, D.; Yoshida, J. I. J. Am. Chem. Soc. 2007, 129, 10922.
(a) Nakanishi, M.; Takahashi, D.; Toshima, K. Org. Biomol. Chem. 2013, 11, 5079; (b) Wever, W. J.; Cinelli, M. A.; Bowers, A. A. Org. Lett. 2012, 15, 30; (c) Mao, R.-Z.; Guo, F.; Xiong, D.-C.; Li, Q.; Duan, J.; Ye, X.-S. Org. Lett. 2015, 17, 5606; (d) Mao, R.-Z.; Xiong, D.-C.; Guo, F.; Li, Q.; Duan, J.; Ye, X.-S. Org. Chem. Front. 2016, 3, 737; (e) Spell, M. L.; Deveaux, K.; Bresnahan, C. G.; Bernard, B. L.; Sheffield, W.; Kumar, R.; Ragains, J. R. Angew. Chem. Int. Ed. 2016, 55, 6515; (f) Yu, Y.; Xiong, D.-C.; Mao, R.-Z.; Ye, X.-S. J. Org. Chem. 2016, 81, 7134. (g) Wang, H.; Wu, P.; Zhao, X.; Zeng, J.; Wan, Q. Acta Chim. Sinica 2019, 77, 231. (王浩, 吴品儒, 赵祥, 曾静, 万谦, 化学学报, 2019, 77, 231.); (h) Ye, H.; Xiao, C.; Lu, L. Chin. J. Org. Chem. 2018, 38, 1897. (叶辉, 肖聪, 陆良秋, 有机化学, 2018, 38, 1897.)
Goswami, M.; Ellern, A.; Pohl, N. L. B. Angew. Chem. Int. Ed. 2013, 52, 8441. doi: 10.1002/anie.v52.32
Yamada, H.; Harada, T.; Miyazaki, H.; Takahashi, T. Tetrahedron Lett. 1994, 35, 3979. doi: 10.1016/S0040-4039(00)76718-9
Zhang, Z.; Ollmann, I. R.; Ye, X.-S.; Wischnat, R.; Baasov, T.; Wong, C.-H. J. Am. Chem. Soc. 1999, 121, 734. doi: 10.1021/ja982232s
Huang, X.; Huang, L.; Wang, H.; Ye, X.-S. Angew. Chem Int. Ed. 2004, 43, 5221. doi: 10.1002/(ISSN)1521-3773
Plante, O. J.; Palmacci, E. R.; Seeberger, P. H. Science 2001, 291, 1523. doi: 10.1126/science.1057324
Tanaka, H.; Adachi, M.; Tsukamoto, H.; Ikeda, T.; Yamada, H.; Takahashi, T. Org. Lett. 2002, 4, 4213. doi: 10.1021/ol020150+
Yu, B.; Yu, H.; Hui, Y.; Han, X. Tetrahedron Lett. 1999, 40, 8591. doi: 10.1016/S0040-4039(99)01839-0
Wang, P.; Lee, H.; Fukuda, M.; Seeberger, P. H. Chem. Commun. 2007, 1963.
Vohra, Y.; Buskas, T.; Boons, G.-J. J. Org. Chem. 2009, 74, 6064. doi: 10.1021/jo901135k
(a) Hsu, C.-H.; Chu, K. C.; Lin, Y. S.; Han, J. L.; Peng, Y. S.; Ren, C. T.; Wong, C.-H. Chem. Eur. J. 2010, 16, 1754; (b) Tanaka, H.; Tateno, Y.; Nishiura, Y.; Takahashi, T. Org. Lett. 2008, 10, 5597; (c) Tanaka, H.; Adachi, M.; Takahashi, T. Chem. Eur. J. 2005, 11, 849.
(a) Dinkelaar, J.; Gold, H.; Overkleeft, H. S.; Codée, J. D.; van der Marel, G. A. J. Org. Chem. 2009, 74, 4208; (b) Hu, Y. P.; Lin, S. Y.; Huang, C. Y.; Zulueta, M. M. L.; Liu, J. Y.; Chang, W.; Hung, S.-C. Nat. Chem. 2011, 3, 557.
Sarkar, S.; Dutta, S.; Das, G.; Sen, A. K. Tetrahedron 2011, 67, 4118. doi: 10.1016/j.tet.2011.03.109
Burkhart, F.; Zhang, Z.; Wacowich-Sgarbi, S.; Wong, C.-H. Angew. Chem. Int. Ed. 2001, 40, 1274. doi: 10.1002/(ISSN)1521-3773
Tsai, B. L.; Han, J. L.; Ren, C. T.; Wu, C.-Y.; Wong, C.-H. Tetrahedron Lett. 2011, 52, 2132. doi: 10.1016/j.tetlet.2010.11.055
Mong, K. K. T.; Wong, C.-H. Angew. Chem. Int. Ed. 2002, 41, 4087. doi: 10.1002/1521-3773(20021104)41:21<4087::AID-ANIE4087>3.0.CO;2-X
Lee, J. C.; Wu, C.-Y.; Apon, J. V.; Siuzdak, G.; Wong, C.-H. Angew. Chem. Int. Ed. 2006, 45, 2753. doi: 10.1002/(ISSN)1521-3773
Mong, T. K. K.; Lee, H. K.; Durón, S. G.; Wong, C.-H. PNAS 2003, 100, 797. doi: 10.1073/pnas.0337590100
Polat, T.; Wong, C.-H. J. Am. Chem. Soc. 2007, 129, 12795. doi: 10.1021/ja073098r
Hsu, Y.; Lu, X. A.; Zulueta, M. M. L.; Tsai, C. M.; Lin, K. I.; Hung, S.-C.; Wong, C.-H. J. Am. Chem. Soc. 2012, 134, 4549. doi: 10.1021/ja300284x
Wang, Z.; Zhou, L.; El-Boubbou, K.; Ye, X.-S.; Huang, X. J. Org. Chem. 2007, 72, 6409. doi: 10.1021/jo070585g
Li, Q.; Guo, Z. Org. Lett. 2017, 19, 6558. doi: 10.1021/acs.orglett.7b03275
Huang, L.; Huang, X. Chem. Eur. J. 2007, 13, 529. doi: 10.1002/(ISSN)1521-3765
Miermont, A.; Zeng, Y.; Jing, Y.; Ye, X.-S.; Huang, X. J. Org. Chem. 2007, 72, 8958. doi: 10.1021/jo701694k
Wang, Z.; Xu, Y.; Yang, B.; Tiruchinapally, G.; Sun, B.; Liu, R.; Huang, X. Chem. Eur. J. 2010, 16, 8365. doi: 10.1002/chem.v16:28
Sun, B.; Srinivasan, B.; Huang, X. Chem. Eur. J. 2008, 14, 7072. doi: 10.1002/chem.v14:23
Wang, Y.-S.; Wu, Y.; Xiong, D.-C.; Ye, X.-S. Chin. J. Chem. 2019, 37, 42. doi: 10.1002/cjoc.v37.1
(a) Gao, J.; Guo, Z. J. Org. Chem. 2013, 78, 12717; (b) Gao, J.; Liao, G.; Wang, L.; Guo, Z. Org. Lett. 2014, 16, 988; (c) Gao, J.; Guo, Z. Org. Lett. 2016, 18, 5552. (d) Wang, D.; Xiong, D.-C.; Ye, X.-S. Chin. Chem. Lett. 2018, 29, 1340; (e) Wu, Y.; Xiong, D.-C.; Chen, S.-C.; Wang, Y.-S.; Ye, X.-S. Nat. Commun. 2017, 8, 14851.
Werz, D. B.; Castagner, B.; Seeberger, P. H. J. Am. Chem. Soc. 2007, 129, 2770. doi: 10.1021/ja069218x
Routenberg, L. K.; Seeberger, P. H. Angew. Chem. Int. Ed. 2004, 43, 602. doi: 10.1002/(ISSN)1521-3773
Ratner, D. M.; Swanson, E. R.; Seeberger, P. H. Org. Lett. 2003, 5, 4717. doi: 10.1021/ol035887t
Codée, J. D. C.; Kröck, L.; Castagner, B.; Seeberger, P. H. Chem. Eur. J. 2008, 14, 3987. doi: 10.1002/chem.v14:13
(a) Walvoort, M. T. C.; Volbeda, A. G.; Reintjens, N. R. M.; van den Elst, H.; Plante, O. J.; Overkleeft, H. S.; Codée, J. D. Org. Lett. 2012, 14, 3776; (b) Hahm, H. S.; Broecker, F.; Kawasaki, F.; Mietzsch, M.; Heilbronn, R.; Fukuda, M.; Seeberger, P. H. Chem 2017, 2, 114.
Hewitt, M. C.; Snyder, D. A.; Seeberger, P. H. J. Am. Chem. Soc. 2002, 124, 13434. doi: 10.1021/ja027538k
Matsuzaki, Y.; Ito, Y.; Nakahara, Y.; Ogawa, T. Tetrahedron Lett. 1993, 34, 1061. doi: 10.1016/S0040-4039(00)77492-2
Hansen, S. U.; Miller, G. J.; Cliff, M. J.; Jayson, G. C.; Gardiner, J. M. Chem. Sci. 2015, 6, 6158. doi: 10.1039/C5SC02091C
Li, A.; Kong, F. Bioorg. Med. Chem. 2005, 13, 839. doi: 10.1016/j.bmc.2004.10.035
Pozsgay, V. Angew. Chem. Int. Ed. 1998, 37, 138. doi: 10.1002/(ISSN)1521-3773
Pozsgay, V.; Chu, C.; Pannell, L.; Wolfe, J.; Robbins, J. B.; Schneerson, R. PNAS 1999, 96, 5194. doi: 10.1073/pnas.96.9.5194
Pozsgay, V. Tetrahedron:Asymmetry 2000, 11, 151. doi: 10.1016/S0957-4166(99)00553-4
Joe, M.; Bai, Y.; Nacario, R. C.; Lowary, T. L. J. Am. Chem. Soc. 2007, 129, 9885. doi: 10.1021/ja072892+
Ishiwata, A.; Ito, Y. J. Am. Chem. Soc. 2011, 133, 2275. doi: 10.1021/ja109932t
Thadke, S. A.; Mishra, B.; Islam, M.; Pasari, S.; Manmode, S.; Rao, B. V.; Hotha, S. Nat. Commun. 2017, 8, 14019. doi: 10.1038/ncomms14019
Pasari, S.; Manmode, S.; Walke, G.; Hotha, S. Chem. Eur. J. 2018, 24, 1128. doi: 10.1002/chem.201704009
Mishra, B.; Neralkar, M.; Hotha, S. Angew. Chem. Int. Ed. 2016, 55, 7786. doi: 10.1002/anie.201511695
Fraser-Reid, B.; Lu, J.; Jayaprakash, K. N.; Lopez, J. C. Tetrahedron:Asymmetry 2006, 17, 2449. doi: 10.1016/j.tetasy.2006.09.008
Islam, M.; Shinde, G. P.; Hotha, S. Chem. Sci. 2017, 8, 2033. doi: 10.1039/C6SC04866H
Calin, O.; Eller, S.; Seeberger, P. H. Angew. Chem. Int. Ed. 2013, 52, 5862. doi: 10.1002/anie.201210176
Naresh, K.; Schumacher, F.; Hahm, H. S.; Seeberger, P. H. Chem. Commun. 2017, 53, 9085. doi: 10.1039/C7CC04380E
Yu, Y.; Kononov, A.; Delbianco, M.; Seeberger, P. H. Chem. Eur. J. 2018, 24, 6075. doi: 10.1002/chem.v24.23

图 3 糖基三氯乙酰亚胺酯的制备及其糖基化反应
Figure 3 Preparation of glycosyl trichloroacetimidates (TCAI) and their glycosylation reactions

图 6 正交选择性的液相一釜合成策略
Figure 6 Orthogonal selective one-pot glycosylation strategy in solution phase

图 7 基于糖基供体活性的程序化液相一釜合成策略
Figure 7 Reactivity-based one-pot glycosylation strategy in solution phase

图 8 预活化液相一釜合成策略
Figure 8 Preactivation-based one-pot glycosylation strategy in solution phase

图 10 Ogawa小组化学合成兔红细胞膜多糖7
Figure 10 Ogawa's chemical synthesis of polysaccharide 7 from rabbit erythrocyte membrane

图 11 Gardiner小组化学合成肝素类多糖工作
Figure 11 Gardiner's work on chemical syntheses of heparin-related polysaccharides

图 12 对全保护多糖21进行脱保护和硫酸化制备肝素类多糖22
Figure 12 Deprotection and sulfation of fully protected polysaccharide 21 to afford heparin-like polysaccharide 22

图 13 Kong小组化学合成植物来源多糖23的工作
Figure 13 Kong's work on the chemical synthesis of plant polysaccharide 23

图 14 Pozsgay小组化学合成痢疾志贺氏菌O-抗原多糖32
Figure 14 Pozsgay's chemical synthesis of the O-specific polysaccharide 32 of Shigella dysenteriae

图 15 Pozsgay小组化学合成痢疾志贺氏菌O-抗原多糖35
Figure 15 Pozsgay's chemical synthesis of the O-specific polysaccharide 35 of Shigella dysenteriae

图 16 Lowary小组化学合成结核分枝杆菌细胞壁多糖37
Figure 16 Lowary's chemical synthesis of polysaccharide 37 of Mycobacterium tuberculosis cell wall

图 17 Ito小组化学合成结核分枝杆菌细胞壁多糖38
Figure 17 Ito's chemical synthesis of polysaccharide 38 of Mycobacterium tuberculosis cell wall

图 18 Hotha小组化学合成结核分枝杆菌细胞壁多糖52
Figure 18 Hotha's chemical synthesis of polysaccharide 52 of Mycobacterium tuberculosis cell wall

图 19 结核分枝杆菌细胞壁多糖Arabinogalactan (57)的化学结构
Figure 19 Chemical structure of arabinogalactan polysaccharide (57) of Mycobacterium tuberculosis cell wall

图 20 结核分枝杆菌细胞壁多糖Arabinogalactan (57)的化学合成
Figure 20 Chemical synthesis of arabinogalactan polysaccharide (57) of Mycobacterium tuberculosis cell wall

图 21 Seeberger小组用自动化固相合成技术合成甘露聚糖30-mer 81
Figure 21 Seeberger's automated solid phase synthesis of a 30-mer mannoside 81

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:





 下载:
下载: