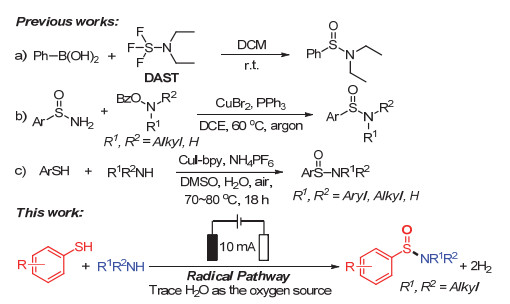
图 1.
亚磺酰胺化合物的制备方法
Figure 1.
Synthetic methods of sulfinamides

亚磺酰胺及其衍生物是一类含有N—S键的有机分子, 在有机合成[1]、生物化学[2]及材料科学[3]等领域有着重要的应用价值.传统上合成亚磺酰胺化合物是通过亚磺酰氯与胺类化合物的亲核加成[4].但是, 亚磺酰卤化合物活性较高, 合成效果并不理想.经过多年的探究, 化学家们遴选出亚磺酸酯[5]或者二硫化物[6]等作为亚磺酰卤的替代品经两步或者多步反应制备亚磺酰胺化合物.但繁琐的反应步骤、苛刻的反应条件及过氧化现象等依然是亟待解决的问题. 2016年, Shi小组[7]使用芳基硼酸及其衍生物与二乙胺基三氟化硫(DAST)于室温条件下制备了亚磺酰胺化合物(图 1, a). 2018年, Bolm小组[8]借助过渡金属催化的方式实现了一级亚磺酰胺化合物同苯甲酰羟胺化合物的亚磺酰胺化反应, 制备了二级和三级的亚磺酰胺化合物(图 1, b).同年, Zhang小组[9]利用CuI为催化剂, 实现了β-亚磺酰酯同苯甲酰羟胺化合物间的偶联反应, 合成了亚磺酰胺化合物.值得注意的是, 2016年Taniguchi小组[10]使用未经预官能团化的巯基化合物和胺类化合物在CuI催化作用下, 实现了亚磺酰胺化合物的制备(图 1, c).显然, 直接活化S—H键和N—H键实现偶联是合成亚磺酰胺化合物最为有效的方法, 此方法无需反应底物的预官能团化即可实现N—S键的构筑.
本文我们借助电化学活化S—H键和N—H键交叉偶联放氢的策略[11], 使用未经预官能团化的苯硫酚及胺类化合物为反应底物, 水为氧源, 实现了亚磺酰胺化合物的制备(图 1).此反应简单温和、绿色友好, 有利于工业化的应用.电化学催化的巯基化合物和胺类化合物间的氧化交叉偶联放氢在此展示了较好的官能团耐受性.实验结果证实此反应通过自由基路径实现N—S键的构筑, 生成次磺酰胺化合物中间产物, 在电化学进一步作用下, 利用溶剂中的H2O作为氧源, 实现了亚磺酰胺化合物的合成.
我们使用对甲氧基苯硫酚1a, 吗啉2a作为反应底物, 四正丁基碘化铵(TBAI)作为电解质, 六氟异丙醇(HFIP)作为反应溶剂, 使用碳泡沫电极作为阳极, 铂片电极作为阴极, 对亚磺酰胺化合物的合成反应进行了探索.实验结果如表 1所示, 当以0.2 mmol对甲氧基苯硫酚1a和0.6 mmol的吗啉2a作为反应底物, 0.1 mol/L的四正丁基碘化铵作为电解质溶解于5 mL六氟异丙醇溶剂中, 使用10 mA恒定电流于一体池中进行电解时, 6小时后可得到99%分离产率的目标产物3a (表 1, Entry 1).对不同电解质进行筛选的结果显示, 电解质的类型对反应有较大的影响(表 1, Entries 2~5).四正丁基四氟硼酸铵(nBu4NBF4)或四正丁基六氟磷酸铵(nBu4NPF6)为电解质时, 可得到中等到较好的反应收率(48%~70%); 四正丁基卤化铵为电解质时, 获得很好的反应收率(73%~99%), 尤其是四正丁基溴化铵作为电解质时, 得到了99%收率的预期产物.由此可见, 多种电解质适用于此反应体系.对反应溶剂进行筛选的结果发现, 六氟异丙醇为反应溶剂时, 可以很好地适用于此反应体系, 乙腈和甲醇为反应溶剂时, 没有得到预期产物(表 1, Entries 1, 6~7).对电解质及反应底物的用量进行筛选的结果表明, 即使电解质浓度降低为0.05 mol/L时, 仍然可以得到99%分离产率的预期产物(表 1, Entries 8~9).降低吗啉的量为0.3 mmol时, 反应效率依然很好(表 1, Entries 10~12).然而, 当恒定电流降低为5 mA时, 反应效率大大降低(表 1, Entry 13).控制实验表明在没有恒定电流通过的条件下, 没有预期反应产物生成, 说明电化学催化对此反应是不可或缺的.
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entrya | 2a (mmol) | Electrolyte | Solvent | Yieldb/% |
| 1 | 0.6 | TBAI | HFIP | 99 |
| 2 | 0.6 | nBu4NBF4 | HFIP | 48 |
| 3 | 0.6 | nBu4NPF6 | HFIP | 70 |
| 4 | 0.6 | nBu4NCl | HFIP | 73 |
| 5 | 0.6 | nBu4NBr | HFIP | 99 |
| 6 | 0.6 | TBAI | MeCN | trace |
| 7 | 0.6 | TBAI | MeOH | trace |
| 8c | 0.6 | TBAI | HFIP | 70 |
| 9d | 0.6 | TBAI | HFIP | 99 |
| 10 | 0.4 | TBAI | HFIP | 99 |
| 11 | 0.3 | TBAI | HFIP | 99 |
| 12 | 0.2 | TBAI | HFIP | 89 |
| 13e | 0.3 | TBAI | HFIP | 47 |
| 14f | 0.6 | TBAI | HFIP | 0 |
| a Standard reaction conditions: RVC (10 mm×10 mm×5 mm) anode, Pt plate (10 mm×5 mm×0.3 mm) cathode, constant current I=10 mA, 1a (0.2 mmol), 2a (0.6 mmol), TBAI (0.1 mol/L), HFIP (5 mL), argon, room temperature, 6 h, undivided cell. b Yields of isolated products. c TBAI (0.01 mol/L). d TBAI (0.05 mol/L). e Constant current I=5 mA. f Constant current I=0 mA. | ||||
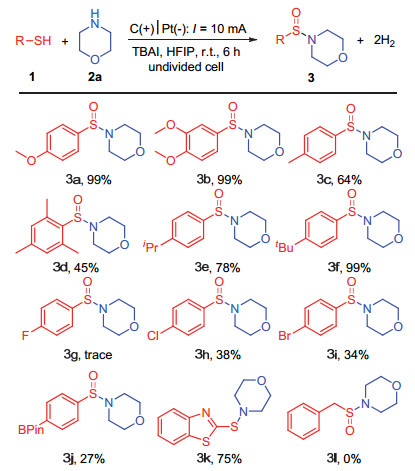
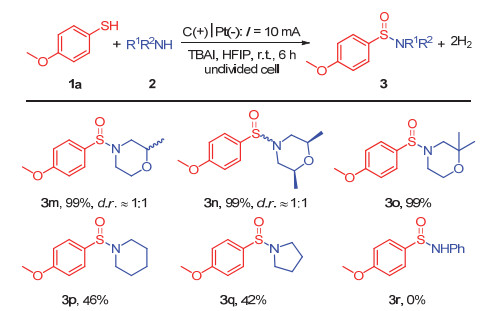
通过对反应条件的优化, 我们确定了最优的反应条件:以巯基化合物(0.2 mmol)和胺类化合物(0.3 mmol)为反应底物, 四正丁基碘化铵(0.05 mol/L)为电解质, 六氟异丙醇(5 mL)为反应溶剂, 阳极为碳泡沫电极, 阴极为铂电极, 氩气保护, 室温条件下, 于一体池中通以10 mA恒定电流电解催化反应6 h (Table 1, Entry 11).在此最优反应条件下, 我们对底物的普适性进行了探究.
首先, 我们对巯基化合物进行了普适性考察, 如图 2所示.含有给电子或吸电子取代基的苯硫酚类化合物均可以很好地适用于此反应体系. 3, 4-二甲氧基苯硫酚作为反应底物时, 能够以99%的高分离产率得到预期的亚磺酰胺化合物3b.当使用对甲基苯硫酚作为反应底物时, 反应产率有所降低, 得到了中等产率(64%)的预期产物3c.使用2, 4, 6-三甲基苯硫酚作为反应底物时, 反应产率(3d, 45%)的降低可能与其位阻效应有关.进一步探究发现, 同样是烷基官能团, 对位是异丙基或者叔丁基的苯硫酚化合物显然可以更好地适用于此反应体系, 分别得到了78%和99%的收率(3e~3f).根据上述反应结果可以发现, 官能团的给电子特性越好则越有利于此反应的进行.对于含有吸电子官能团的卤代苯硫酚, 则只得到了中等及以下(34%~38%)的收率(3g~3i), 较低的反应收率可能与其吸电子效应有关. 4-巯基苯硼酸频那醇酯同样可以适用于此反应体系, 但产率较低(3j, 27%).然而, 杂环的2-巯基苯并噻唑作为反应底物时, 并没有得到预期的亚磺酰胺化合物, 所得产物为次磺酰胺化合物3k[11].反应结果说明, 2-巯基苯并噻唑结构的缺电子特性抑制了其进一步被氧化为亚磺酰胺化合物.烷基硫醇类化合物并不能很好地适用于此反应体系(3l).上述的反应结果说明, 反应体系对于苯硫酚类化合物具有很广的底物适用范围.
我们对胺类化合物的底物适用性进行了探究(图 3).实验结果发现, 甲基取代的吗啉的衍生物可以很好地适用于此反应体系, 均以99%的分离产率得到预期的反应产物(3m~3o).六氢哌啶以及四氢吡咯作为反应底物时, 也可以很好地适用于此反应体系, 得到中等收率(42%~46%)的预期产物(3p~3q).在该反应条件下, 原料2p和2q可能会经历脱氢芳构化过程, 因而降低了它们的反应浓度, 从而导致反应产率的降低.另外, 苯胺类化合物并不能很好地适用于此反应体系.
通过循环伏安法, 使用四正丁基六氟磷酸铵作为电解质, 六氟异丙醇作为溶剂, 对对甲氧基苯硫酚1a以及吗啉2a的氧化电位进行了检测.结果显示, 对甲氧基苯硫酚的氧化电位为Eox=1.07 V vs. SCE, 吗啉的氧化电位为Eox=2.07 V vs. SCE, 而在恒定电流10 mA的反应条件下, 阳极电位稳定在2.5~4.5 V vs. SCE之间, 可见其有足够的氧化能力促使反应的正常进行, 进而确保了此反应发生的可行性.
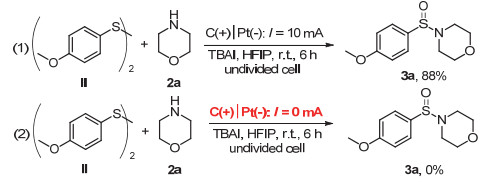
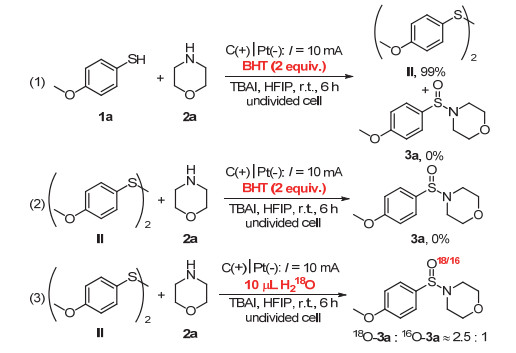
在反应过程中, 我们检测到有对甲氧基苯硫酚二聚产物生成, 推测其有可能作为反应的中间体参与N—S键的构筑反应.为此, 我们使用双(4-甲氧基苯基)二硫化物Ⅱ和吗啉2a作为反应底物, 于标准反应条件下进行此反应, 结果可得88%收率的产物3a(图 4, 1).然而, 在同样的反应条件下, 不施加恒定电流进行此反应, 则没有预期产物的生成(图 4, 2).上述实验结果说明, 在反应体系中, 双(4-甲氧基苯基)二硫化物可作为反应中间体参与反应过程, 而且此反应必须在电化学催化的条件下方可进行, 进而排除了吗啉对二硫化物亲核加成反应的可能性.
为了进一步验证反应历程, 我们进行了自由基抑制实验(图 5, 1~2).在标准的反应条件下, 加入2 equiv.的2, 6-二叔丁基-4-甲基苯酚(BHT)作为自由基抑制剂作用于此反应体系时, 我们只得到了当量的二硫化物中间产物Ⅱ, 并没有得到预期的亚磺酰胺化合物3a (图 5, 1).说明反应过程中有自由基中间体生成, 且BHT对此反应过程有很好的抑制作用.进一步我们使用二硫化物作为反应底物与吗啉进行反应时, BHT可以起到很好的抑制作用(图 5, 2).此结果说明, BHT作为自由基抑制剂作用于吗啉与二硫化物中间体N—S键的构筑过程.
相对于其他电解质, TBAI等四正丁基卤代铵化合物作为电解质时, 具有更好的反应效果(表 1, Entries 1~5).为此, 我们推测TBAI作为电解质的同时, 亦可作为中间体参与此反应过程, 进而提高了反应效率.反应结束后肉眼可见的碘单质的生成说明上述推论的合理性.高分辨质谱检测结果说明, 亚磺酰胺化合物可能通过次磺酰胺化合物的进一步氧化转化生成.重氧水(H218O)标记实验证实亚磺酰胺化合物3的氧来源于反应溶剂中的水(图 5, 3).
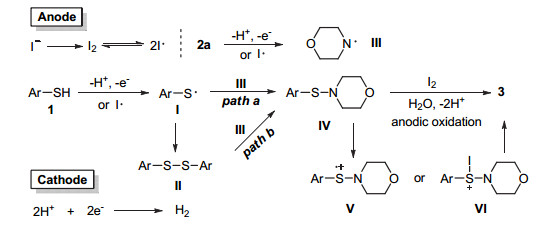
基于上述实验结果和相关的文献报道[12, 13], 我们提出了如下反应机理(图 6).反应体系中, 电解质解离的碘负离子在阳极的氧化作用下转化为碘单质, 亦即碘自由基的可逆形态.对甲氧基苯硫酚在阳极或者碘自由基的作用下, 转化为巯基自由基Ⅰ, 其可通过二聚的方式转化为二硫化物Ⅱ.此时, 经阳极电催化氧化或者碘自由基氢提取作用形成的胺基自由基Ⅲ可以通过自由基加成的方式作用于巯基自由基或者二硫化物中间体, 进而形成次磺酰胺化合物中间体Ⅳ.由于反应体系中有碘单质的生成, 次磺酰胺中间产物也可通过巯基化合物或胺类化合物亲核加成于反应过程中形成的N—I化合物中间体或S—I化合物中间体产生.次磺酰胺化合物中间体在阳极的氧化或碘的作用下, 转化为硫的自由基正离子中间体Ⅴ或S—I化合物中间体Ⅵ, 反应溶剂中的水则作为亲核试剂加成于上述中间体Ⅴ和Ⅵ, 经进一步电化学氧化生成亚磺酰胺3[14].反应体系中的质子在阴极被还原放出氢气.
本文以苯硫酚类化合物和胺类化合物为反应底物, 通过交叉偶联放氢反应于电化学一体池中合成了亚磺酰胺化合物.在反应体系中, TBAI起到电解质和中间体的双重作用, 可加速反应的进行, 提高反应效率.机理探究结果说明, 苯硫酚类化合物和胺类化合物在此反应条件下, 通过自由基反应的方式生成次磺酰胺化合物.在电化学氧化或碘的作用下, 次磺酰胺化合物转化为硫的自由基正离子中间体或S-I中间体, 与反应溶剂中的水作用后, 经进一步氧化形成相应的亚磺酰胺化合物.该转化构建了一系列的亚磺酰胺化合物, 反应的唯一副产物是氢气, 具有简单易操作、反应条件温和、官能团兼容性好等优点.
(a) Sola, J.; Reves, M.; Riera, A.; Verdaguer, X. Angew. Chem. Int. Ed. 2007, 46, 5020. (b) Beck, E. M.; Hyde, A. M.; Jacobsen, E. N. Org. Lett. 2011, 13, 4260. (c) Viso, A.; de la Pradilla, R. F.; Urena, M.; Bates, R. H.; del Aguila, M. A.; Colomer, I. J. Org. Chem. 2012, 77, 525. (d) Zhang, Z. M.; Chen, P.; Li, W. B.; Niu, Y. F.; Zhao, X. L.; Zhang, J. L. Angew. Chem. Int. Ed. 2014, 53, 4350. (e) Fjelbye, K.; Svenstrup, N.; Puschl, A. Synthesis-Stuttgart 2015, 47, 3231. (f) Su, X.; Zhou, W.; Li, Y. Y.; Zhang, J. L. Angew. Chem. Int. Ed. 2015, 54, 6874. (g) Zhou, W.; Su, X.; Tao, M. N.; Zhu, C. Z.; Zhao, Q. J.; Zhang, J. L. Angew. Chem. Int. Ed. 2015, 54, 14853. (h) Chelouan, A.; Recio, R.; Borrego, L. G.; Alvarez, E.; Khiar, N.; Fernandez, I. Org. Lett. 2016, 18, 3258.
(a) Moree, W. J.; Vandermarel, G. A.; Liskamp, R. M. J. Tetrahedron Lett. 1991, 32, 409. (b) Viswanadhan, V. N.; Ghose, A. K.; Hanna, N. B.; Matsumoto, S. S.; Avery, T. L.; Revankar, G. R.; Robins, R. K. J. Med. Chem. 1991, 34, 526. (c) Carreno, M. C. Chem. Rev. 1995, 95, 1717. (d) Khiar, N.; Werner, S.; Mallouk, S.; Lieder, F.; Alcudia, A.; Fernández, I. J. Org. Chem. 2009, 74, 6002. (e) Chelouan, A.; Recio, R.; Borrego, L. G.; Alvarez, E.; Khiar, N.; Fernandez, I. Org. Lett. 2016, 18, 3258.
(a) Andreassen, T.; Lorentzen, M.; Hansen, L.-K.; Gautun, O. R. Tetrahedron 2009, 65, 2806. (b) Chen, D.; Xu, M.-H. J. Org. Chem. 2014, 79, 7746.
Uchino, M.; Sekiya, M. Chem. Pharm. Bull. 1980, 28, 126. doi: 10.1248/cpb.28.126
(a) Billard, T.; Greiner, A.; Langlois, B. R. Tetrahedron 1999, 55, 7243. (b) Davis, F. A.; Zhang, Y.; Andemichael, Y.; Fang, T.; Fanelli, D. L.; Zhang, H. J. Org. Chem. 1999, 64, 1403. (c) Zhou, P.; Chen, B.-C.; Davis, F. A. Tetrahedron 2004, 60, 8003.
Cogan, D. A.; Liu, G.; Kim, K.; Backes, B. J.; Ellman, J. A. J. Am. Chem. Soc. 1998, 120, 8011. doi: 10.1021/ja9809206
Wang, Q.; Tang, X.-Y.; Shi, M. Angew. Chem. Int. Ed. 2016, 55, 10811. doi: 10.1002/anie.201605066
Yu, H.; Li, Z.; Bolm, C. Angew. Chem. Int. Ed. 2018, 57, 15602. doi: 10.1002/anie.201810548
Dai, Q.; Zhang, J. Adv. Synth. Catal. 2018, 360, 1123. doi: 10.1002/adsc.201701510
Taniguchi, N. Eur. J. Org. Chem. 2016, 2016, 2157. doi: 10.1002/ejoc.201600091
钟建基, 孟庆元, 陈彬, 佟振合, 吴骊珠, 化学学报, 2017, 75, 34. doi: 10.3969/j.issn.0253-2409.2017.01.006Zhong, J.; Meng, Q.; Chen, B.; Tung, C.; Wu, L. Acta Chim. Sinica 2017, 75, 34(in Chinese). doi: 10.3969/j.issn.0253-2409.2017.01.006
(a) Tang, S.; Liu, Y.; Li, L.; Ren, X.; Li, J.; Yang, G.; Li, H.; Yuan, B. Org. Biomol. Chem. 2019, 17, 1370. (b) Yan, Y.; Cui, C.; Li, Z. Chin. J. Org. Chem., 2018, 38, 2501(in Chinese). (闫溢哲, 崔畅, 李政, 有机化学, 2018, 38, 2501.) (c) Yuan, Y.; Chen, Y.; Tang, S.; Huang, Z.; Lei, A. Sci. Adv. 2018, 4, eaat5312. (d) Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem. Int. Ed. 2017, 56, 3009.
(a) Wang, Y.; Qian, P.; Su, J.-H.; Li, Y.; Bi, M.; Zha, Z.; Wang, Z. Green Chem. 2017, 19, 4769. (b) Huang, P.; Wang, P.; Tang, S.; Fu, Z.; Lei, A. Angew. Chem. Int. Ed. 2018, 57, 8115; (c) Liu, K.; Song, C.; Lei, A. Org. Biomol. Chem. 2018, 16, 2375.
Gao, X.; Yuan, G.; Chen, H.; Jiang, H.; Li, Y.; Qi, C. Electrochem. Commun. 2013, 34, 242. doi: 10.1016/j.elecom.2013.06.022
表 1 反应条件的优化
Table 1. Reaction conditions optimization
|
|
||||
| Entrya | 2a (mmol) | Electrolyte | Solvent | Yieldb/% |
| 1 | 0.6 | TBAI | HFIP | 99 |
| 2 | 0.6 | nBu4NBF4 | HFIP | 48 |
| 3 | 0.6 | nBu4NPF6 | HFIP | 70 |
| 4 | 0.6 | nBu4NCl | HFIP | 73 |
| 5 | 0.6 | nBu4NBr | HFIP | 99 |
| 6 | 0.6 | TBAI | MeCN | trace |
| 7 | 0.6 | TBAI | MeOH | trace |
| 8c | 0.6 | TBAI | HFIP | 70 |
| 9d | 0.6 | TBAI | HFIP | 99 |
| 10 | 0.4 | TBAI | HFIP | 99 |
| 11 | 0.3 | TBAI | HFIP | 99 |
| 12 | 0.2 | TBAI | HFIP | 89 |
| 13e | 0.3 | TBAI | HFIP | 47 |
| 14f | 0.6 | TBAI | HFIP | 0 |
| a Standard reaction conditions: RVC (10 mm×10 mm×5 mm) anode, Pt plate (10 mm×5 mm×0.3 mm) cathode, constant current I=10 mA, 1a (0.2 mmol), 2a (0.6 mmol), TBAI (0.1 mol/L), HFIP (5 mL), argon, room temperature, 6 h, undivided cell. b Yields of isolated products. c TBAI (0.01 mol/L). d TBAI (0.05 mol/L). e Constant current I=5 mA. f Constant current I=0 mA. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们






 下载:
下载: