Received Date:
10 January 2019 Available Online:
15 June 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21573031)
Abstract:
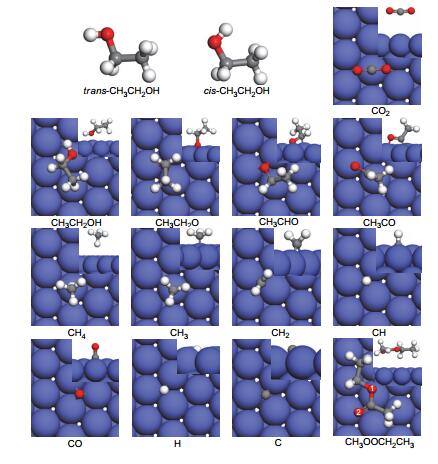
The detailed reaction mechanism of ethanol dehydrogenation on Co(111) surface was studied using the density functional theory (DFT) and slab periodic model. The structures and energies of the species involved in the reaction adsorbed on different adsorption sites (top, fcc, hcp and bridge sites) of the surface were calculated and compared. The calculated results show that ethanol adsorbs weakly on the Co(111) surface. CH3CH2O, CH and C prefer hcp sites with adsorption energies of -2.72, -6.85, and -6.92 eV, respectively. CH3CHO adsorbs weakly at the bridge-η1(O)-η1(Cα) site with adsorption energy of -0.47 eV. CH3CO and CH2 adsorb stably on Co(111) surface through their unsaturated C atoms with binding energies of -2.31 and -3.90 eV, respectively. CH3 and CH4 prefer to locate at top sites through the C atom with adsorption energies of -1.95 and -0.12 eV, respectively. CO and H are bind stably at fcc sites with binding energies of -1.62 and -2.77 eV, respectively. Due to the complexity of the decomposition of ethanol, the scissions of O-H, C-H, C-O and C-C bonds of CH3CH2OH were examined. The results show that ethanol decomposition on Co(111) surface starts with the scission of the O-H bond, and the dehydrogenation reaction of ethanol on Co(111) surface can be described as three reaction pathways:Path Ⅰ is the gradual dehydrogenation of CH3CH2OH via intermediate CH3CHO, which ultimately produces CH4 and CO; Path Ⅱ is the reaction of CH3CH2O and CH3CHO which were generated by dehydrogenation of ethanol, to form CH4 and CO2 via CH3COOH intermediate; Path Ⅲ is the process of CH3CH2O reacts with CH3CO to generate CH3COOC2H5. On the basis of our computational results, Path Ⅰ (CH3CH2OH→CH3CH2O→CH3CHO→CH3CO→CH3+CO→CH2→CH→CH4+CO+C+H) is more favorable than Paths Ⅱ and Ⅲ and the dehydrogenation of CH3CH2O to CH3CHO is the rate-determining step with a reaction energy barrier of 1.61 eV.
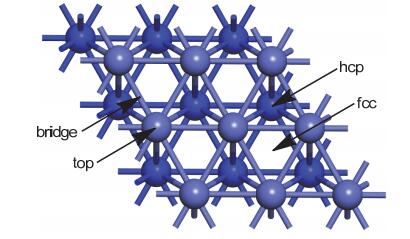
Figure 1.
Top views of Co(111) surface with marked adsorption sites (top site, bridge site, hexagonal-close-packed (hcp) site, and face-centered-cubic (fcc) sites.) For clarity, only two layers of Co atoms are illustrated
Table 1.
Stable adsorption sites, adsorption energies and structural parameters for intermediates of ethanol dehydrogenation on Co(111) (Zero point correction value is in parentheses)
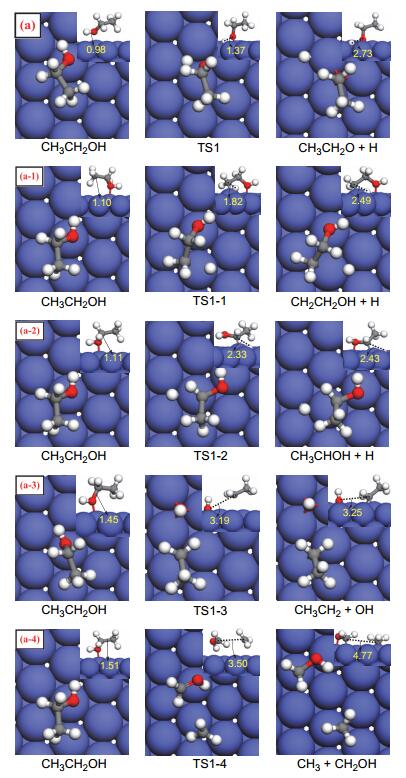
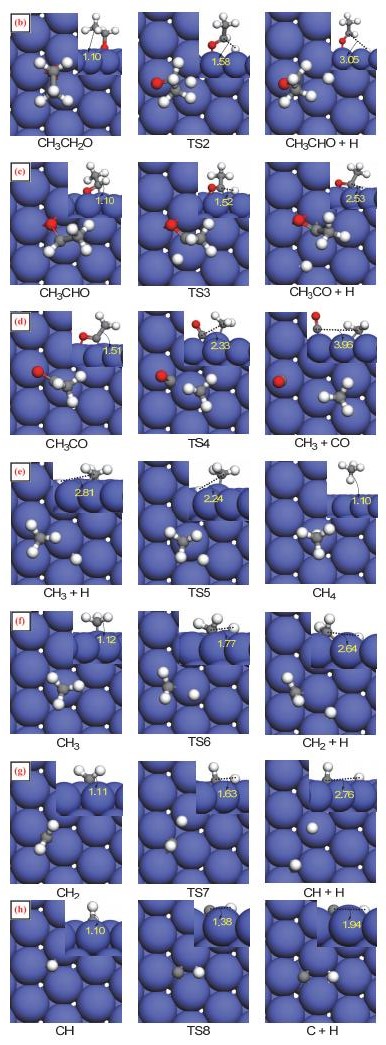
Figure 3.
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the O—H, C—H, C—O, and C—C bonds scission of ethanol on Co(111) (The value is the length of the broken bond)
Figure 5.
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in ethanol dehydrogenation on Co(111) (Path Ⅰ)
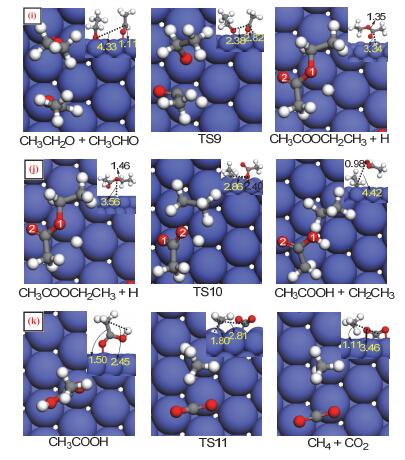
Figure 6.
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the reaction of CH3CH2O and CH3CHO on Co(111) (Path Ⅱ)
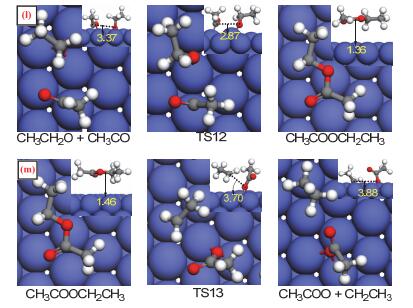
Figure 7.
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the reaction of CH3CH2O and CH3CO on Co(111) (Path Ⅲ)
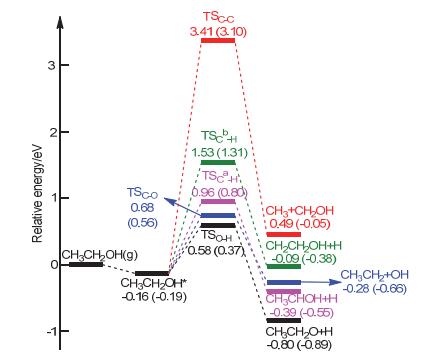
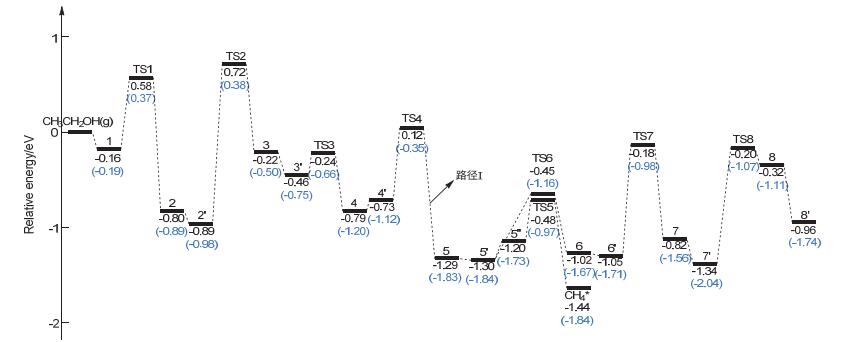
Figure 8.
Potential energy surface of ethanol dehydrogenation (Path Ⅰ); [A+B]* represents the co-adsorbed A and B, and A*+B* represents the respective adsorption of A and B on two separate catalysts. 1, CH3CH2OH*; 2, [CH3CH2O+H]*; 2', CH3CH2O*+H*; 3, [CH3CHO+H]*+H*; 3', CH3CHO*+2H*; 4, [CH3CO+H]*+2H*; 4', CH3CO*+3H*; 5, [CH3+CO]*+3H*; 5', CH3*+CO*+3H*; 5'', [CH3+H]*+CO*+2H*; 6, [CH2+H]*+CO*+3H*; 6', CH2*+CO*+4H*; 7, [CH+H]*+CO*+4H*; 7', CH*+CO*+5H*; 8, [C+H]*+CO*+5H*; 8', C*+CO*+6H*
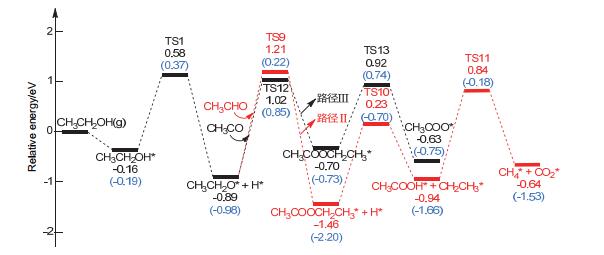
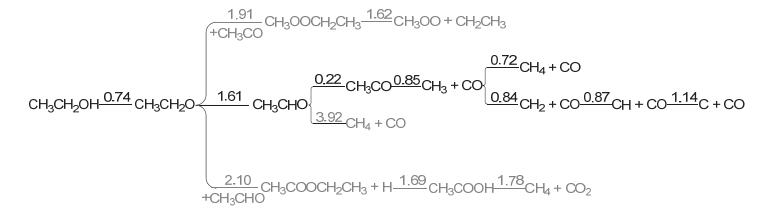
Figure 10.
Reaction network and energy barriers (eV) of ethanol dehydrogenation on Co(111) (The values on the lines are energy barriers for the relevant steps)
饶路, 姜艳霞, 张斌伟, 游乐星, 李崭红, 孙世刚, 化学进展, 2014, 26, 727.Rao, L.; Jiang, Y. X.; Zhang, B. W.; You, L. X.; Li, Z. H.; Sun, S. G. Prog. Chem. 2014, 26, 727(in Chinese).
[6]
Rodríguez, L. A.; Toro, M. E.; Vazquez, F.; Correa-Daneri, M. L.; Gouiric, S. C.; Vallejo, M. D. Int. J. Hydrogen Energy 2010, 35, 5914. doi: 10.1016/j.ijhydene.2009.12.112
[7]
Lamy, C.; Belgsir, E. M.; Léger, J. M. J. Appl. Electrochem. 2001, 31, 799. doi: 10.1023/A:1017587310150
[8]
Llorca, J.; Homs, N.; Sales, J.; de la Poscina, P. R. J. Catal. 2002, 209, 306. doi: 10.1006/jcat.2002.3643
吴匡衡, 周亚威, 马宪印, 丁辰, 蔡文斌, 化学学报, 2018, 76, 292. doi: 10.7503/cjcu20170465Wu, K. H.; Zhou, Y. W.; Ma, X. Y.; Ding, C.; Cai, W. B. Acta Chim. Sinica 2018, 76, 292(in Chinese). doi: 10.7503/cjcu20170465
[11]
Van Der Laan, G. P.; Beenackers, A. A. C. M. Catal. Rev. 1999, 41, 255. doi: 10.1081/CR-100101170
[12]
Shekhar, R.; Barteau, M. A. Catal. Lett. 1995, 31, 221. doi: 10.1007/BF00808835
[13]
Bowker, M.; Holroyd, R. P.; Sharpe, R. G.; Corneille, J. S.; Francis, S. M.; Goodman, D. W. Surf. Sci. 1997, 370, 113. doi: 10.1016/S0039-6028(96)00959-4
[14]
Lee, A. F.; Naughton, J. N.; Liu, Z.; Wilson, K. ACS Catal. 2012, 2, 2235. doi: 10.1021/cs300450y
[15]
Lee, A. F.; Gawthrope, D. E.; Hart, N. J.; Wilson, K. Surf. Sci. 2004, 548, 200. doi: 10.1016/j.susc.2003.11.004
[16]
Vigier, F.; Coutanceau, C.; Hahn, F.; Belgsir, E. M.; Lamy, C. J. Electroanal. Chem. 2004, 563, 81. doi: 10.1016/j.jelechem.2003.08.019
Tian, Z. J.; Wei, X. M.; Zhai, R. S.; Ren, S. Z.; Liang, D. B.; Lin, L. W. Chem. Res. Chin. Univ. 1997, 7, 1158.
[20]
Vicente, J.; Montero, C.; Ereña, J.; Azkoiti, M. J.; Bilbao, J.; Gayubo, A. G. Int. J. Hydrogen Energy 2014, 39, 12586. doi: 10.1016/j.ijhydene.2014.06.093
Balakrishnan, N.; Joseph, B.; Bhethanabotla, V. R. Surf. Sci. 2012, 606, 634. doi: 10.1016/j.susc.2011.11.033
[30]
van Helden, P.; van den Berg, J. A.; Weststrate, C. J. ACS Catal. 2012, 2, 1097. doi: 10.1021/cs2006586
[31]
Pour, A. N.; Keyvanloo, Z.; Izadyar, M.; Modaresi, S. M. Int. J. Hydrogen Energy 2015, 40, 7064. doi: 10.1016/j.ijhydene.2015.04.028
[32]
Swart, J. C. W.; Ciobîcä, L. M.; van Santen, R. A.; van Steen, E. J. Phys. Chem. C 2008, 112, 12899. doi: 10.1021/jp803305s
[33]
Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Phys. Rev. B 1997, 56, 13849. doi: 10.1103/PhysRevB.56.13849
[34]
Ashok, A.; Kumar, A.; Bhosale, R.; Saad, M. A. S.; AlMomani, F.; Tarlochan, F. Int. J. Hydrogen Energy 2017, 42, 23464. doi: 10.1016/j.ijhydene.2017.01.175
Figure 1
Top views of Co(111) surface with marked adsorption sites (top site, bridge site, hexagonal-close-packed (hcp) site, and face-centered-cubic (fcc) sites.) For clarity, only two layers of Co atoms are illustrated
Figure 3
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the O—H, C—H, C—O, and C—C bonds scission of ethanol on Co(111) (The value is the length of the broken bond)
Figure 5
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in ethanol dehydrogenation on Co(111) (Path Ⅰ)
Figure 6
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the reaction of CH3CH2O and CH3CHO on Co(111) (Path Ⅱ)
Figure 7
Calculated structures of the initial state (IS), transition state (TS), and final state (FS) involved in the reaction of CH3CH2O and CH3CO on Co(111) (Path Ⅲ)
Figure 8
Potential energy surface of ethanol dehydrogenation (Path Ⅰ); [A+B]* represents the co-adsorbed A and B, and A*+B* represents the respective adsorption of A and B on two separate catalysts. 1, CH3CH2OH*; 2, [CH3CH2O+H]*; 2', CH3CH2O*+H*; 3, [CH3CHO+H]*+H*; 3', CH3CHO*+2H*; 4, [CH3CO+H]*+2H*; 4', CH3CO*+3H*; 5, [CH3+CO]*+3H*; 5', CH3*+CO*+3H*; 5'', [CH3+H]*+CO*+2H*; 6, [CH2+H]*+CO*+3H*; 6', CH2*+CO*+4H*; 7, [CH+H]*+CO*+4H*; 7', CH*+CO*+5H*; 8, [C+H]*+CO*+5H*; 8', C*+CO*+6H*
Figure 10
Reaction network and energy barriers (eV) of ethanol dehydrogenation on Co(111) (The values on the lines are energy barriers for the relevant steps)
Table 1.
Stable adsorption sites, adsorption energies and structural parameters for intermediates of ethanol dehydrogenation on Co(111) (Zero point correction value is in parentheses)

 下载:
下载:

 下载:
下载:












 下载:
下载:
