Received Date:
10 January 2019 Available Online:
15 June 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21773169)
Abstract:
Molecular-scale electronics studies the charge transport properties across molecules by constructing "elec-trode-molecule-electrode" junctions based on the molecular electrodes and single molecule or small amounts of molecular aggregates. It examines the structure-property relationship between the physical and chemical properties of the molecule and the charge transport by combining the intrinsic chemical properties of molecule with device architecture, reveals the micro-scale quantum transport mechanics principle, and explores molecular-based functional electronic devices. It is a research field that integrates chemistry, physics and microelectronics. In this review, we summarize some of the representative progress of molecular electronics in basic research (device preparation, transport mechanism) and applications in recent years.
分子电子学是一个新兴的充满活力的研究领域.近年来, 有关分子电子学领域发表的英文综述和专著很多[8~21].比较有代表性的英文综述主要如Venkataraman等[22]从物理有机化学原理的角度分析了锚定基团, 电极, 分子桥三种必备组分在结构及种类上对单分子器件性能的影响. Li等[23]针对分子电子学中纳米间隙电极的制备及单分子器件进行了详细的整理和总结. Guo和Xiang等[24]发表的综述则对该领域进行了最全面的总结和介绍. Scheer和Cuevas[25]合著的《Molecular Electronics: An introduction to theory and experiment》是介绍分子电子学领域的理论和实验最全面也最为详尽的专著.
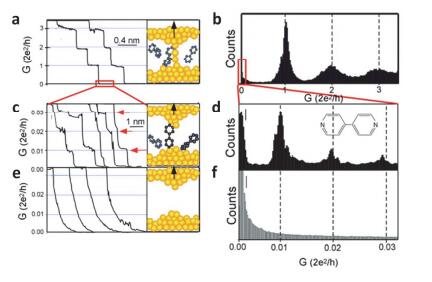
Figure 1.
(a) Conductance of a gold contact formed between a gold STM tip and a gold substrate decreases in quantum steps near multiples of G0 (2e2/h) as the tip is pulled away from the substrate. (b) A corresponding conductance histogram constructed as shown in (a) shows well-defined peaks near 1 G0, 2 G0, and 3 G0 due to conductance quantization. (c) A new series of conductance steps appears if molecules such as 4, 4-bipyridine are present in the solution. These steps are due to the formation of the stable molecular junction between the tip and the substrate electrodes. (d) (c) Corresponding conductance histogram, peaks near 1×, 2×, and 3×0.01 G0 that are ascribed to one, two, and three molecules, respectively. (e and f) In the absence of molecules, no such steps or peaks are observed within the same conductance range[29]
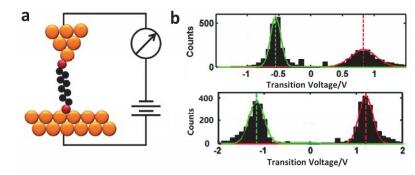
Figure 2.
(a) Schematic diagram of STM-BJ technology for alkyl dithiol molecules. (b) Statistical analysis of TVS of alkyldithiols and biphenyldithiols[30]
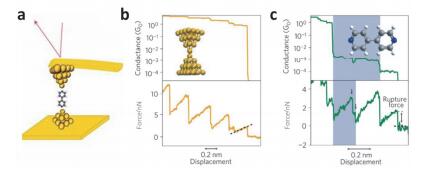
Figure 3.
(a) Schematic of a 4, 4′- bipyridine junction formed between a gold-coated AFM tip and a gold substrate by CP-AFM. (b) Typical current-voltage and conductance-voltage curves and changes in conductance and force during molecular junction formation. (c) The shaded areas represent high-conductance molecular regimes. The shaded areas in the low panel represent structural rearrangements within the high- conductance regime. The dashed lines indicate a complete rupture of the molecular junctions[37]
Figure 4.
(a) AFM-BJ technology diagram. (b) Diagram of the relationship between the conductance of dithiol molecules and the modulation force and amplitude of piezoelectric elements[39, 40]
Figure 5.
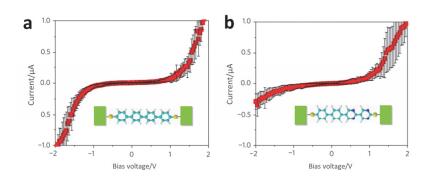
(a) MCBJ schematic diagram where a, b, c, d, e, f are curved beam, reverse support, notched gold wire, glue joint, piezoelectric element and glass tube. (b) Typical benzenedithiol single molecule current-voltage and conductance-voltage curves[5]
Figure 6.
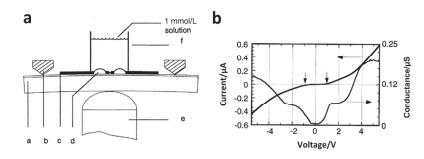
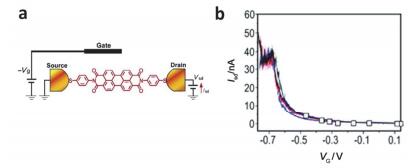
(a) Schematic diagram of a single molecule junction apply gate formed by MCBJ technology. (b) Diagram of the relationship between the current of 1, 4-benzenedithiol molecular knot and the gate voltage[44]
Figure 8.
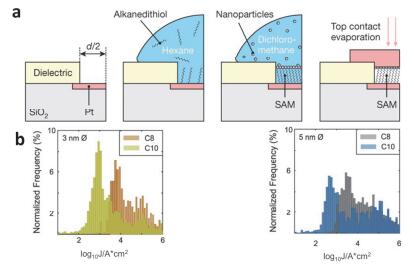
(a) Device preparation process using gold nanoparticle as a protective layer. (b) Effect of different sizes of gold nanoparticles on the electrical properties of molecular junctions[60]
Figure 9.
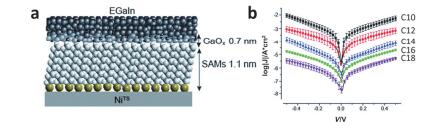
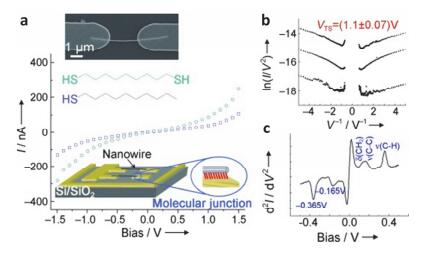
(a) Representation of a "suspended-wire" molecular junction and typical I-V curves of the molecules used in this study, inset: SEM image of a suspended nanowire. (b) Transition voltage spectroscopy of three different C9 junctions. (c) Representative IETS measurements of a C9 junction measured at 5 K, showing characteristic peaks of alkyl chains (C10 junctions have similar peaks)[61]
Figure 11.
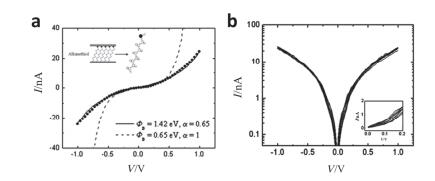
(a) The C12 I-V data curves that was measured (circular symbols) and calculated (solid curve). Dash line show the calculated I-V from a simple rectangular barrier (illustration is a schematic diagram of the device structure). (b) Temperature-dependent log I-V curves from 300 to 80 K with 20 K steps of C12[64]
Figure 12.
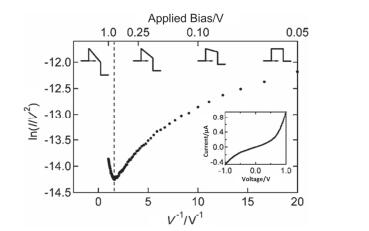
The average of 100 I-V curves of the Au-anthracenethiol-Au junction measured by CP-AFM. The illustration shows current-voltage data on a standard axis. the dotted line indicates the conversion voltage[65]
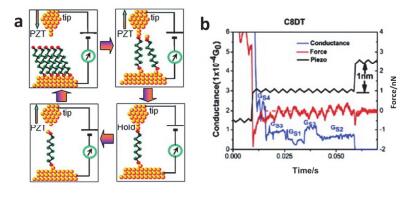
Figure 13.
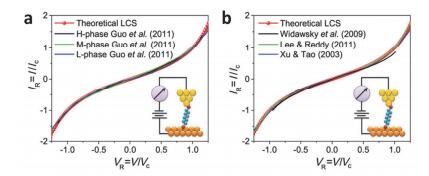
(a) averages of 2151, 1661, 1661 I-V traces for the high- (H); medium- (M); low- (L) conductance phases of octanedithiol (C8DT) junctions with gold electrodes and (b) In the measured STM-BJ technique, the gold electrode is connected to a single I-V curve connected to 4, 4'-diaminopurine, C8DT and 44bpy≡4, and the ohmic resistance R varies in three orders of magnitude [R≈1.3 MΩ to R≈1.2 GΩ][29, 30, 70~72]
Figure 15.
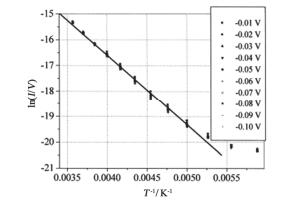
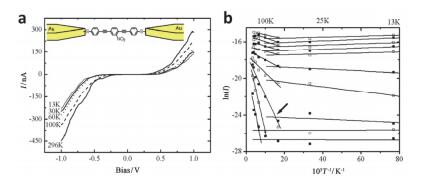
(a)
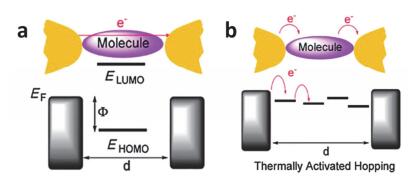
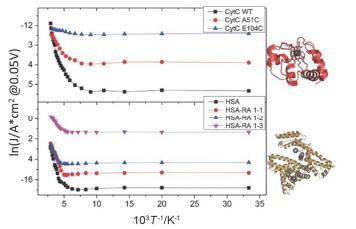
I-V curves measured on one of the junctions for several temperatures. Inset is the schematic of metal-molecule-metal junction. (b) Arrhenius plots of ln(I) vs 1/T at different bias voltages showing a transition in conductance from temperature independent at low temperature range and temperature dependent at high temperature. The arrow shows the turning point from temperature-independent to thermal activated process[74]
Figure 16.
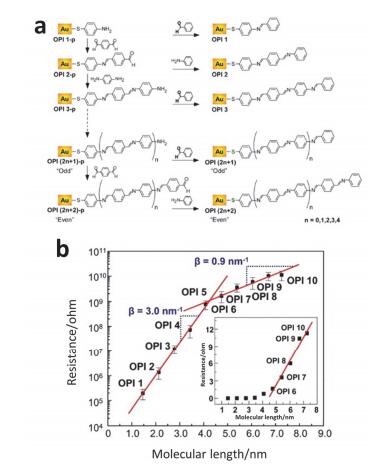
(a) The synthetic route of various length OPI wires. (b) Semilog plot of R versus L for the OPI junctions measured by CP-AFM. (Inset a linear plot of R versus L)[62]
Figure 17.
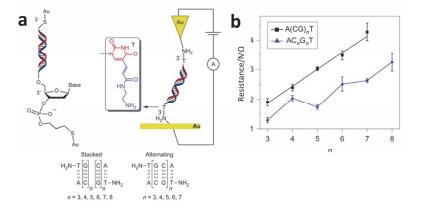
(a) DNA molecules connected to two electrodes via the sugar (left) and directly via the thymine base (T, right), A(CG)nT and ACnGnT denote alternating and stacked G sequences, respectively. (b) Resistance of alternating (black squares) and stacked (blue triangles) DNA sequences versus the number of CG in the sequences[80]
Figure 19.
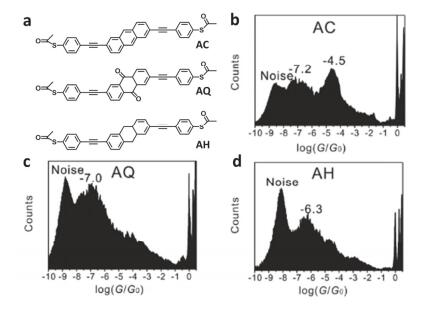
(a) Molecular structures of AC, AQ, and AH, (b) 1-D conductance histograms constructed from 500 individual traces of AC, (c) of AQ and (d) of AH [84]
Figure 21.
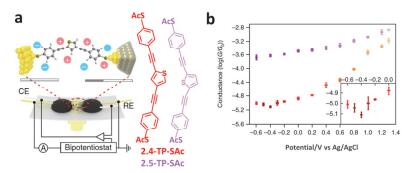
(a) Schematics of the electrochemically gated MCBJ technique and molecular structures of thiophene derivatives with anchoring groups of thioacetyl (–SAc). (b) Tendency of the molecular conductance of 2, 5-TP-SAc (purple) and 2, 4-TP-SAc (orange) versus electrode potentials from -0.6 V to 1.3 V. Inset, magnification from -0.6 V to 0 V [91]
Figure 23.
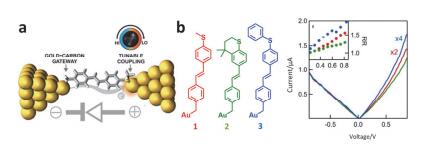
(a) Molecular junction device formed by STM-BJ. (b) The structure of the three molecules and the resulting rectification characteristics[99]
Figure 24.
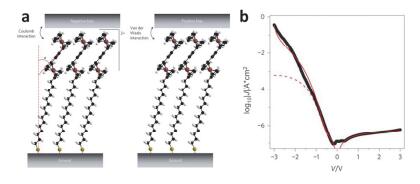
(a) Structure of HSC15Fc–C≡C–Fc molecular junction device. (b) log10|J|(V) curve of the molecular diode on Pt (black dots) with fits to with (solid red line) and without (dashed red line) the functional n(V)[102]
Figure 25.
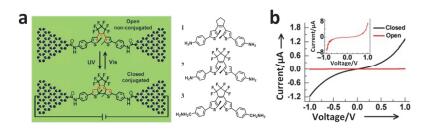
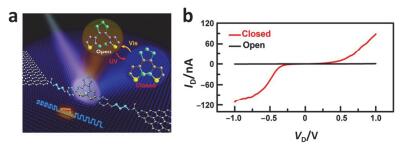
(a) Molecular bridge between the ends of a single SWNT electrode and switching between conjugated and non-conjugated molecular structures. (b) Schematic diagram of a separate open-state SWNT device and the device becomes off after UV illumination[105]
Figure 26.
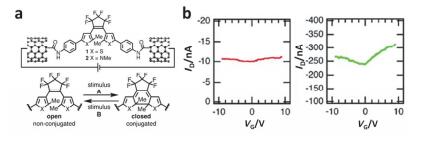
(a) Switching of graphene-diarylethene junctions and molecular structure. (b) I-V characteristics of open (red line) and closed (dark line) states at VG=0 V. The inset shows the enlarged I-V curve for the open state[106]
Figure 27.
(a) Schematic of a graphene-diarylethene-graphene junction that highlights the expansion of the molecular bridge by methylene groups. (b) I-V characteristics of individual diarylethenes in open (black line) and closed (red line) format gate voltage VG=0 V. VD is drain voltage; ID is drain current[107]
Figure 28.
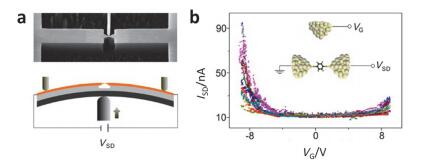
(a) Schematic of a single molecule transistor with an electrochemical gate. (b) Source-drain current (Isd) versus gate voltage (Vg) for a single PTCDI molecule transistor. The open squares were obtained from the peak position of the conductance histograms. The solid lines were obtained by directly recording the sourcedrain current while sweeping the gate voltage[111]
Figure 29.
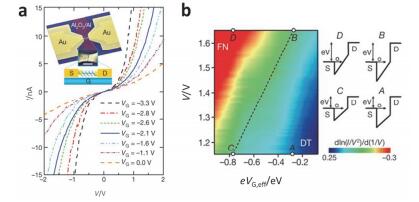
(a) Representative I-V curves measured at 4.2 K for different values of VG. Inset, the device structure and schematic. S, source; D, drain; G, gate. Scale bar, 100 nm. (b) Two-dimensional colour map of dln(I/V2)/d(1/V) (from Fowler-Nordheim plots). Energy-band diagrams corresponding to four different regions (points A~D) are also shown. F-N tunnelling; DT, direct tunneling[112]
Figure 30.
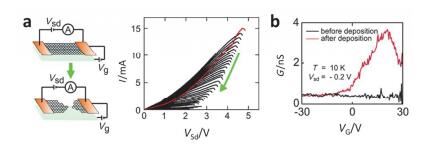
(a) Schematic of the feedback-controlled electroburning process, before (top) and after (bottom), the formation of nanometer sized gaps in few-layer graphite flakes. and Current-voltage (I-V) traces of the evolution (green arrow) of the feedback-controlled electroburning. The first I-V trace is displayed in red. (b) Conductance as a function of the applied back-gate voltage of the nanogapped electrodes bridged by 9Accm molecules at 10 K. While the empty nanogap electrodes show no dependence of the applied back-gate voltage, a clear conductance modulation as a function of Vg is observed after deposition of 9Accm molecules[113]
Figure 31.
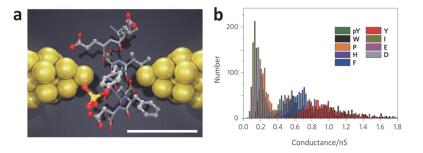
(a) Schematic diagram of preparation of molecular junction devices by MCBJ. (b) Conductance histogram of a certain number of different amino acids[114]
Figure 32.
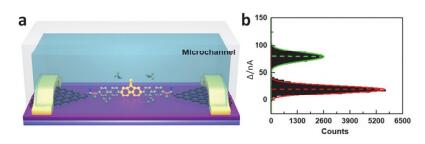
(a) Schematic diagram of real-time measurement setup with a home-made microchannel for single-molecule dynamics characterization. (b) Corresponding histogram of current values, showing a bimodal current distribution[115]
Figure 33.
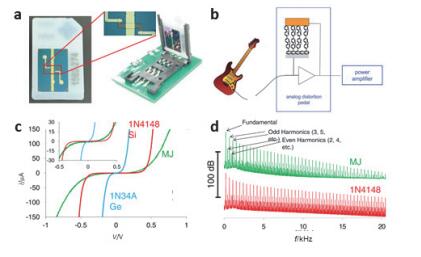
(a) Photograph of the molecular junctions fabricated using a SIM card pin out to simplify testing and integration into analog circuits, as shown at right. (b) Schematic diagram of applying a molecular junction device to an amplifier component of an electric guitar. (c) I-V characteristics of molecular junction devices and conventional diode devices. (d) Odd and even harmonic distribution of conventional diode and molecular junction limiting components at different frequencies[116, 117]
Guo, S.; Hihath, J.; Diez-Perez, I.; Tao, N. J. J. Am. Chem. Soc. 2011, 133, 19189. doi: 10.1021/ja2076857
[31]
Baldea, I. Phys. Rev. B 2012, 85, 9222.
[32]
Chen, F.; Li, X.; Hihath, J.; Huang, Z.; Tao, N. J. J. Am. Chem. Soc. 2006, 128, 15874. doi: 10.1021/ja065864k
[33]
Hines, T.; Diez-Perez, I.; Nakamura, H.; Shimazaki, T.; Asai, Y.; Tao, N. J. J. Am. Chem. Soc. 2013, 135, 3319. doi: 10.1021/ja3106434
[34]
Li, H.; Su, T. A.; Zhang, V.; Steigerwald, M. L.; Nuckolls, C.; Venkataraman, L. J. Am. Chem. Soc. 2015, 137, 5028. doi: 10.1021/ja512523r
[35]
Dell, E. J.; Capozzi, B.; DuBay, K. H.; Berkelbach, T. C.; Moreno, J. R.; Reichman, D. R.; Venkataraman, L.; Campos, L. M. J. Am. Chem. Soc. 2013, 135, 11724. doi: 10.1021/ja4055367
[36]
Wold, D. J.; Frisbie, C. D. J. Am. Chem. Soc. 2000, 122, 2970. doi: 10.1021/ja994468h
[37]
Aradhya, S. V.; Frei, M.; Hybertsen, M. S.; Venkataraman, L. Nat. Mater. 2012, 11, 872. doi: 10.1038/nmat3403
Thuo, M. M.; Reus, W. F.; Nijhuis, C. A.; Barber, J. R.; Kim, C.; Schulz, M. D.; Whitesides, G. M. J. Am. Chem. Soc. 2011, 133, 2962. doi: 10.1021/ja1090436
[47]
Nijhuis, C. A.; Reus, W. F.; Barber, J. R.; Dickey, M. D.; Whitesides, G. M. Nano Lett. 2010, 10, 3611. doi: 10.1021/nl101918m
[48]
Chiechi, R. C.; Weiss, E. A.; Dickey, M. D.; Whitesides, G. M. Angew. Chem., Int. Ed. Engl. 2008, 47, 142. doi: 10.1002/(ISSN)1521-3773
[49]
Senthil kumar, K.; Jiang, L.; Nijhuis, C. A. RSC Adv. 2017, 7, 14544. doi: 10.1039/C6RA27280K
[50]
Walker, A. V.; Tighe, T. B.; Haynie, B. C.; Uppili, S.; Winograd, N.; Allara, D. L. J. Phys. Chem. B 2005, 109, 11263. doi: 10.1021/jp0506484
[51]
Mahmoud, A. M.; Bergren, A. J.; Pekas, N.; McCreery, R. L. Adv. Funct. Mater. 2011, 21, 2273. doi: 10.1002/adfm.v21.12
[52]
Zhu, Z.; Daniel, T. A.; Maitani, M.; Cabarcos, O. M.; Allara, D. L.; Winograd, N. J. Am. Chem. Soc. 2006, 128, 13710. doi: 10.1021/ja060084x
[53]
Walker, A. V.; Tighe, T. B.; Cabarcos, O. M.; Reinard, M. D.; Haynie, B. C.; Uppili, S.; Winograd, N.; Allara, D. L. J. Am. Chem. Soc. 2004, 126, 3954. doi: 10.1021/ja0395792
[54]
DeIonno, E.; Tseng, H. R.; Harvey, D. D.; Stoddart, J. F.; Heath, J. R. J. Phys. Chem. B 2006, 110, 7609.
[55]
Bonifas, A. P.; McCreery, R. L. Nat. Nanotechnol. 2010, 5, 612. doi: 10.1038/nnano.2010.115
[56]
Honciuc, A.; Metzger, R. M.; Gong, A.; Spangler, C. W. J. Am. Chem. Soc. 2007, 129, 8310. doi: 10.1021/ja068729g
[57]
Bonifas, A. P.; McCreery, R. L. Nano Lett. 2011, 11, 4725. doi: 10.1021/nl202495k
[58]
Akkerman, H. B.; Blom, P. W. M.; de Leeuw, D. M.; de Boer, B. Nature 2006, 441, 69. doi: 10.1038/nature04699
[59]
Katsouras, I.; Piliego, C.; Blom, P. W. M.; Leeuwa, D. M. Nanoscale 2013, 5, 9882. doi: 10.1039/c3nr03183g
[60]
Puebla-Hellmann, G.; Venkatesan, K.; Mayor, M.; Lörtscher, E. Nature 2018, 559, 232. doi: 10.1038/s41586-018-0275-z
Baldea, I.; Xie, Z.; Frisbie, C. D. Nanoscale 2015, 7, 10465. doi: 10.1039/C5NR02225H
[70]
Widawsky, J. R.; Kamenetska, M.; Klare, J.; Nuckolls, C.; Stei-gerwald, M. L.; Hybertsen, M. S.; Venkataraman, L. Nanotechnology 2009, 20, 434009. doi: 10.1088/0957-4484/20/43/434009
Chen, J.; Calvet, L. C.; Reed, M. A.; Carr, D. W.; Grubisha, D. S.; Bennett, D. W. Chem. Phys. Lett. 1999, 313, 741. doi: 10.1016/S0009-2614(99)01060-X
[74]
Selzer, Y.; Cabassi, M. A.; Mayer, T. S.; Allara, D. L. J. Am. Chem. Soc. 2004, 126, 4052. doi: 10.1021/ja039015y
[75]
Choi, S. H.; Risko, C.; Delgado, M. C. R.; Kim, B.; Bredas, J. L.; Frisbie, C.D. J. Am. Chem. Soc. 2010, 132, 4358. doi: 10.1021/ja910547c
Eley, D. D.; Spivey, D. I. Trans. Faraday Soc. 1962, 58, 411. doi: 10.1039/TF9625800411
[79]
Genereux, J. C.; Barton, J. K. Chem. Rev. 2010, 110, 1642. doi: 10.1021/cr900228f
[80]
Xiang, L.; Palma, J. L.; Bruot, C.; Mujica, V.; Ratner, M. A.; Tao, N. J. Nat. Chem. 2015, 7, 221. doi: 10.1038/nchem.2183
[81]
Bostick, C. D.; Mukhopadhyay, S.; Pecht, I.; Sheves, M.; Cahen, D.; Lederman, D. Rep. Prog. Phys. 2018, 81, 026601. doi: 10.1088/1361-6633/aa85f2
[82]
Amdursky, N.; Marchak, D.; Sepunaru, L.; Pecht, I.; Sheves, M.; Cahen, D. Adv. Mater. 2014, 26, 7142. doi: 10.1002/adma.v26.42
[83]
Andrews. D. Q.; Solomon, G. C.; Goldsmith, R. H.; Hansen, T.; Wasielewski, M. R.; Duyne, R. P.; Ratner, M. A. J. Am. Chem. Soc. 2008, 130, 17301. doi: 10.1021/ja8044053
[84]
Hong, W. J.; Valkenier, H.; Meszaros, G.; Manrique, D. Z.; Mishchenko, A.; Putz, A.; Garcia, P. M.; Lambert, C. J.; Hummelen, J. C.; Wandlowski, T. Beilstein J. Nanotechnol. 2011, 2, 699. doi: 10.3762/bjnano.2.76
[85]
Fracasso, D.; Valkenier, H.; Hummelen, J. C.; Solomon, G. C.; Chiechi, R. C. J. Am. Chem. Soc. 2011, 133, 9556 doi: 10.1021/ja202471m
[86]
Guedon, C. M.; Valkenier, H.; Markussen, T.; Thygesen, K. S.; Hummelen, J. C.; Molen, S. J. Nat. Nanotechnol. 2012, 7, 304..
[87]
Rabache, V.; Chaste, J.; Petit, P.; Della Rocca, M. L.; Martin, P.; Lacroix, J. C.; McCreery, R. L.; Lafarge, P. J. Am. Chem. Soc. 2013, 135, 10218. doi: 10.1021/ja403577u
[88]
Manrique, D. Z.; Huang, C.; Baghernejad, M.; Zhao, X.; AlOwaedi, O. A.; Sadeghi, H.; Kaliginedi, V.; Hong, W. J.; Wandlowski, M.; Gulcur, T.; Bryce, M. R.; Lambert, C. J. Nat. Commun. 2015, 6, 6389. doi: 10.1038/ncomms7389
Zhang, P.; Chen, L. C.; Zhang, Z. Q.; Cao, J. J.; Tang, C.; Liu, J.; Duan, L. L.; Huo, Y.; Shao, X.; Hong, W. J.; Zhang, H. L. J. Am. Chem. Soc. 2018, 140, 6531. doi: 10.1021/jacs.8b02825
Migliore, A.; Jia, C. C.; Xin, N.; Huang, S. Y.; Wang, J. Y.; Yang, Q.; Wang, S. P.; Chen, H. L.; Wang, D. M.; Feng, B. Y.; Liu, Z. R.; Zhang, G. Y.; Qu, D. H.; Tian, H.; Ratner, M. A.; Xu, H. Q.; Nitzan, A.; Guo, X. F. Science 2016, 352, 1443. doi: 10.1126/science.aaf6298
[108]
Javey, A.; Guo, J.; Wang, Q.; Lundstrom, M.; Dai, H. Nature 2003, 424, 654. doi: 10.1038/nature01797
Damle, P.; Rakshit, T.; Paulsson, M.; Datta, S. IEEE T. Nanotechnol. 2002, 1, 145. doi: 10.1109/TNANO.2002.806825
[111]
Xu, B. Q.; Xiao, X. Y.; Yang, X. M.; Zang, L.; Tao, N. J. J. Am. Chem. Soc. 2005, 127, 2386. doi: 10.1021/ja042385h
[112]
Song, H.; Kim, Y.; Jang, Y. H.; Jeong, H.; Reed, M. A.; Lee, T. Nature 2009, 462, 1039. doi: 10.1038/nature08639
[113]
Prins, F.; Barreiro, A.; Ruitenberg, J. W.; Seldenthuis, J. S.; Ali-aga-Alcalde, N.; Vandersypen, L. M.; van der Zant, H. S. Nano Lett. 2011, 11, 4607. doi: 10.1021/nl202065x
[114]
Ohshiro, T.; Tsutsui, M.; Yokota, K.; Furuhashi, M.; Taniguchi, M.; Kawai, T. Nat. Nanotechnol. 2014, 9, 835. doi: 10.1038/nnano.2014.193
Bergren, A. J.; Zeer-Wanklyn, L.; Semple, M.; Pekas, N.; Szeto, B.; McCreery, R. L. J. Phys. Condens. Matter. 2016, 28, 094011. doi: 10.1088/0953-8984/28/9/094011
[117]
McCreery, R. L.; Bergren, A.; Morteza-Najarian, A.; Sayed, S. Y.; Yan, H. Faraday Discuss 2014, 172, 9. doi: 10.1039/C4FD00172A
Xiang, D.; Sydoruk, V.; Vitusevich, S.; Petrychuk, M. V.; Offenhaeusser, A.; Kochelap, V. A.; Belyaev, A. E.; Mayer, D. Appl. Phys. Lett. 2015, 106, 063702-1.
Figure 1
(a) Conductance of a gold contact formed between a gold STM tip and a gold substrate decreases in quantum steps near multiples of G0 (2e2/h) as the tip is pulled away from the substrate. (b) A corresponding conductance histogram constructed as shown in (a) shows well-defined peaks near 1 G0, 2 G0, and 3 G0 due to conductance quantization. (c) A new series of conductance steps appears if molecules such as 4, 4-bipyridine are present in the solution. These steps are due to the formation of the stable molecular junction between the tip and the substrate electrodes. (d) (c) Corresponding conductance histogram, peaks near 1×, 2×, and 3×0.01 G0 that are ascribed to one, two, and three molecules, respectively. (e and f) In the absence of molecules, no such steps or peaks are observed within the same conductance range[29]
Figure 2
(a) Schematic diagram of STM-BJ technology for alkyl dithiol molecules. (b) Statistical analysis of TVS of alkyldithiols and biphenyldithiols[30]
Figure 3
(a) Schematic of a 4, 4′- bipyridine junction formed between a gold-coated AFM tip and a gold substrate by CP-AFM. (b) Typical current-voltage and conductance-voltage curves and changes in conductance and force during molecular junction formation. (c) The shaded areas represent high-conductance molecular regimes. The shaded areas in the low panel represent structural rearrangements within the high- conductance regime. The dashed lines indicate a complete rupture of the molecular junctions[37]
Figure 4
(a) AFM-BJ technology diagram. (b) Diagram of the relationship between the conductance of dithiol molecules and the modulation force and amplitude of piezoelectric elements[39, 40]
Figure 5
(a) MCBJ schematic diagram where a, b, c, d, e, f are curved beam, reverse support, notched gold wire, glue joint, piezoelectric element and glass tube. (b) Typical benzenedithiol single molecule current-voltage and conductance-voltage curves[5]
Figure 6
(a) Schematic diagram of a single molecule junction apply gate formed by MCBJ technology. (b) Diagram of the relationship between the current of 1, 4-benzenedithiol molecular knot and the gate voltage[44]
Figure 8
(a) Device preparation process using gold nanoparticle as a protective layer. (b) Effect of different sizes of gold nanoparticles on the electrical properties of molecular junctions[60]
Figure 9
(a) Representation of a "suspended-wire" molecular junction and typical I-V curves of the molecules used in this study, inset: SEM image of a suspended nanowire. (b) Transition voltage spectroscopy of three different C9 junctions. (c) Representative IETS measurements of a C9 junction measured at 5 K, showing characteristic peaks of alkyl chains (C10 junctions have similar peaks)[61]
Figure 11
(a) The C12 I-V data curves that was measured (circular symbols) and calculated (solid curve). Dash line show the calculated I-V from a simple rectangular barrier (illustration is a schematic diagram of the device structure). (b) Temperature-dependent log I-V curves from 300 to 80 K with 20 K steps of C12[64]
Figure 12
The average of 100 I-V curves of the Au-anthracenethiol-Au junction measured by CP-AFM. The illustration shows current-voltage data on a standard axis. the dotted line indicates the conversion voltage[65]
Figure 13
(a) averages of 2151, 1661, 1661 I-V traces for the high- (H); medium- (M); low- (L) conductance phases of octanedithiol (C8DT) junctions with gold electrodes and (b) In the measured STM-BJ technique, the gold electrode is connected to a single I-V curve connected to 4, 4'-diaminopurine, C8DT and 44bpy≡4, and the ohmic resistance R varies in three orders of magnitude [R≈1.3 MΩ to R≈1.2 GΩ][29, 30, 70~72]
Figure 15
(a)
I-V curves measured on one of the junctions for several temperatures. Inset is the schematic of metal-molecule-metal junction. (b) Arrhenius plots of ln(I) vs 1/T at different bias voltages showing a transition in conductance from temperature independent at low temperature range and temperature dependent at high temperature. The arrow shows the turning point from temperature-independent to thermal activated process[74]
Figure 16
(a) The synthetic route of various length OPI wires. (b) Semilog plot of R versus L for the OPI junctions measured by CP-AFM. (Inset a linear plot of R versus L)[62]
Figure 17
(a) DNA molecules connected to two electrodes via the sugar (left) and directly via the thymine base (T, right), A(CG)nT and ACnGnT denote alternating and stacked G sequences, respectively. (b) Resistance of alternating (black squares) and stacked (blue triangles) DNA sequences versus the number of CG in the sequences[80]
Figure 19
(a) Molecular structures of AC, AQ, and AH, (b) 1-D conductance histograms constructed from 500 individual traces of AC, (c) of AQ and (d) of AH [84]
Figure 21
(a) Schematics of the electrochemically gated MCBJ technique and molecular structures of thiophene derivatives with anchoring groups of thioacetyl (–SAc). (b) Tendency of the molecular conductance of 2, 5-TP-SAc (purple) and 2, 4-TP-SAc (orange) versus electrode potentials from -0.6 V to 1.3 V. Inset, magnification from -0.6 V to 0 V [91]
Figure 24
(a) Structure of HSC15Fc–C≡C–Fc molecular junction device. (b) log10|J|(V) curve of the molecular diode on Pt (black dots) with fits to with (solid red line) and without (dashed red line) the functional n(V)[102]
Figure 25
(a) Molecular bridge between the ends of a single SWNT electrode and switching between conjugated and non-conjugated molecular structures. (b) Schematic diagram of a separate open-state SWNT device and the device becomes off after UV illumination[105]
Figure 26
(a) Switching of graphene-diarylethene junctions and molecular structure. (b) I-V characteristics of open (red line) and closed (dark line) states at VG=0 V. The inset shows the enlarged I-V curve for the open state[106]
Figure 27
(a) Schematic of a graphene-diarylethene-graphene junction that highlights the expansion of the molecular bridge by methylene groups. (b) I-V characteristics of individual diarylethenes in open (black line) and closed (red line) format gate voltage VG=0 V. VD is drain voltage; ID is drain current[107]
Figure 28
(a) Schematic of a single molecule transistor with an electrochemical gate. (b) Source-drain current (Isd) versus gate voltage (Vg) for a single PTCDI molecule transistor. The open squares were obtained from the peak position of the conductance histograms. The solid lines were obtained by directly recording the sourcedrain current while sweeping the gate voltage[111]
Figure 29
(a) Representative I-V curves measured at 4.2 K for different values of VG. Inset, the device structure and schematic. S, source; D, drain; G, gate. Scale bar, 100 nm. (b) Two-dimensional colour map of dln(I/V2)/d(1/V) (from Fowler-Nordheim plots). Energy-band diagrams corresponding to four different regions (points A~D) are also shown. F-N tunnelling; DT, direct tunneling[112]
Figure 30
(a) Schematic of the feedback-controlled electroburning process, before (top) and after (bottom), the formation of nanometer sized gaps in few-layer graphite flakes. and Current-voltage (I-V) traces of the evolution (green arrow) of the feedback-controlled electroburning. The first I-V trace is displayed in red. (b) Conductance as a function of the applied back-gate voltage of the nanogapped electrodes bridged by 9Accm molecules at 10 K. While the empty nanogap electrodes show no dependence of the applied back-gate voltage, a clear conductance modulation as a function of Vg is observed after deposition of 9Accm molecules[113]
Figure 31
(a) Schematic diagram of preparation of molecular junction devices by MCBJ. (b) Conductance histogram of a certain number of different amino acids[114]
Figure 32
(a) Schematic diagram of real-time measurement setup with a home-made microchannel for single-molecule dynamics characterization. (b) Corresponding histogram of current values, showing a bimodal current distribution[115]
Figure 33
(a) Photograph of the molecular junctions fabricated using a SIM card pin out to simplify testing and integration into analog circuits, as shown at right. (b) Schematic diagram of applying a molecular junction device to an amplifier component of an electric guitar. (c) I-V characteristics of molecular junction devices and conventional diode devices. (d) Odd and even harmonic distribution of conventional diode and molecular junction limiting components at different frequencies[116, 117]

 下载:
下载:

 下载:
下载:


































 下载:
下载:
