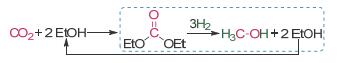
图式 1.
CO2经由碳酸二乙酯制甲醇转化路径
Scheme 1.
Strategy of CO2 to methanol based on diethyl carbonate

石油、煤炭和天然气等化石能源的大量消耗导致空气中二氧化碳的浓度逐年攀高, 引发全球变暖和气候变化等环境问题, 打破了全球的生态平衡.将二氧化碳转化为具有附加值的化学品(甲醇、甲酸、乙醇和烯烃等)[1], 不仅可以有效缓解因二氧化碳浓度偏高引发的温室效应, 同时也为碳一化学品的转化利用提供新的路径.甲醇作为重要的基础化学品和能源载体, 被广泛应用于化工、医药等领域.目前工业生产中主要采用Cu-ZnO基催化剂催化合成气反应制备甲醇, 由于热力学和动力学限制, 该反应需在较高的温度和压力下进行, 且存在反应活性较低等问题[2].基于二氧化碳直接加氢体系存在能耗和反应性能等方面的不足, Milstein等[3]提出了二氧化碳间接转化合成甲醇的新思路.通过温和条件下化学固定二氧化碳形成碳酸酯类衍生物, 利用Ru基均相加氢催化剂在较为温和的条件下实现了二氧化碳至醇类的高活性和高选择性转化.该工艺路线简单且原子经济性高, 但是均相催化剂与产物难于分离的缺点阻碍了其进一步规模化应用.为克服这一难题, 很多学者报道了Cu/HMS、CuCr2O4及Cu/CeO2等非均相体系用于催化碳酸酯加氢反应[4].
早期研究表明, Cu/SiO2催化剂对酯类选择性加氢反应具有较好的活性, 但是高分散的活性铜物种在高温、还原性气氛及较大空速条件下长时间运行时, SiO2载体无法有效稳定铜活性组分, 导致催化剂易失活.如何提高反应过程中活性物种的稳定性, 进而获得具有高稳定性的催化剂一直是相关领域亟待解决的科学问题[5].向催化剂中引入结构助剂可有效调变催化体系的织构特征和电子结构, 稳定催化剂中的活性金属物种, 显著提高其高温抗烧结能力, 进而改善其反应性能和稳定性[6].国内外学者开展了大量有关铜基催化剂助剂/修饰剂的研究工作.本课题组[7]曾报道了镍作为电子助剂修饰Cu/HMS催化剂, 在稳定活性铜物种的同时形成了部分合金相结构, 显著改变了草酸二甲酯加氢制乙二醇的选择性, 同时催化剂稳定性也得以提升.文献[8]报道Cu-Cr催化剂在催化甘油氢解反应体系中, 大量高度分散的CuCrxOy和Cr2O3晶相也可有效抑制铜物种的团聚, 并且铜和铬元素间的电子转移对反应活性也有一定的促进作用[9].此外, 双金属组分催化剂CuCr/SBA-15被用于催化反式肉桂醛加氢反应时, 铬作为助剂引入铜基催化剂对其反应性能也有一定的促进作用[10].
在本研究中, 我们采用蒸氨法引入助剂铬至Cu/ SiO2催化剂体系中, 系统考察了助剂铬的修饰对催化剂的物理化学特性及碳酸二乙酯选择加氢制甲醇反应性能的影响, 并对铬修饰所得的Crx-Cu/SiO2催化剂的反应活性中心进行了探讨.
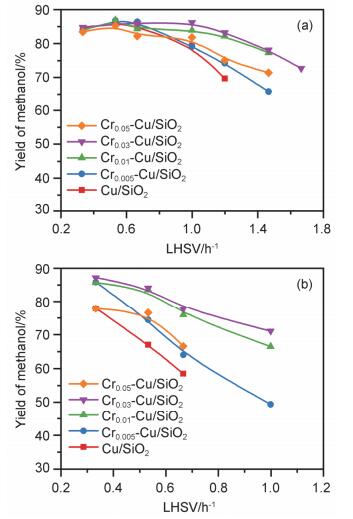
以碳酸二乙酯连续加氢制甲醇为目标反应, 通过调变铬的修饰量探究了助剂对Cu/SiO2催化体系的结构及其催化性能的影响, 所得催化活性结果如图 1所示. 图 1a为温度在503 K条件下不同铬含量修饰的Crx-Cu/ SiO2催化剂表现出的甲醇收率随液时空速(LHSV)的变化曲线, 在液时空速低于1.4 h-1的情况下, 催化剂的性能较佳, 碳酸二乙酯均可完全转化.加氢产物以甲醇、乙醇为主, 同时伴有少量脱羰和甲烷化副产物一氧化碳、二氧化碳和甲烷生成[4c].其中, 液时空速为1.0 h-1, 目标产物甲醇的收率可达86.2%, 其时空得率(STY)为5.6 mmolMeOH•gcat-1•h-1. 图 1b为该催化体系在反应温度为483 K时甲醇收率随碳酸二乙酯液时空速的变化趋势, 在较低空速条件下, 甲醇收率仍可达85%以上, 表明低空速条件下适当降低反应温度仍可获得较高的目标产物收率, 气相副产物的生成也会被抑制.结合503和483 K条件下催化剂的反应性能可知, 适量铬修饰后Crx-Cu/SiO2催化剂的活性均有不同程度的提升, 其中低温条件下效果更为显著, 并且碳酸二乙酯液时空速的可操作窗口也得以拓展.当铬修饰量大于3 wt%时, 催化剂在目标反应中甲醇的收率开始降低, 然而对比未引入铬的Cu/SiO2催化剂仍表现出一定的性能优势.在该反应中Cu/SiO2催化剂的失活机理研究表明[10], 当底物的液时空速较大且催化剂还原活化不足时, 催化剂的表面会被大量底物分子及中间物种吸附进而导致催化剂不可逆失活[11].对催化剂的稳定性测试也可以看出, 未经修饰的Cu/SiO2催化体系在100 h连续反应后, 碳酸二乙酯的转化率及目标产物甲醇收率开始迅速下降, 分别降低至35.7%和20.3%.而从铬含量为3 wt%的Cr0.03-Cu/ SiO2催化体系的稳定性测试结果可知, 铬的修饰使得催化剂的稳定性明显改善, 在经历150 h连续反应后, 碳酸二乙酯转化率及甲醇收率未见明显下降(图S1).综上所述, 少量铬修饰不仅可以有效提升碳酸二甲酯催化加氢制甲醇的反应性能, 同时也可显著改善其长周期反应稳定性, 这对其今后的工业化应用十分关键.
为了探究Crx-Cu/SiO2催化剂的反应性能及稳定性提升的原因, 对催化剂的物化结构进行了考察.铬的引入使得催化剂的物化结构发生了显著改变, 一系列Crx-Cu/SiO2催化剂的N2吸脱附等温线(图S2a)和孔径分布(图S2b)如附图所示. Crx-Cu/SiO2催化剂的孔道结构和滞后环形状相近, 均为Langmuir IV型等温线和H1型滞后环, 然而吸附等温线的滞后环面积有所减小.由孔径分布图可知, 少量铬的加入使得催化剂孔径略有减小, 而持续增加铬的量则会使得催化剂的平均孔径明显增大.其中Cu/SiO2催化剂的比表面积(SBET)为368 m2• g-1, 而随着铬含量的增加其比表面积呈现逐步降低的趋势, 当铬的引入量为5 wt%时, 达到最小值(185 m2• g-1).平均孔体积(Vpore)也有不同程度的减小, 由1.03 cm3•g-1减少至0.77 cm3•g-1.而其平均孔径(Dpore)则由11.8 nm增大至21.1 nm, 而后又减小至19.4 nm.以上结果表明少量铬的修饰显著改变了催化剂的织构特征.
焙烧后及还原后Cux-Cr/SiO2催化剂的X射线粉末衍射(XRD)如图 2所示.焙烧后铜和铬物种高度分散在催化剂的表面, 两种金属相关物相的衍射峰均未明显检出, 其中衍射角2θ位于22°处出现的衍射峰归属于无定型的SiO2物种(图 2a)[12].由还原后催化剂的衍射图可以看出, Cu/SiO2催化剂中出现了Cu和Cu2O的衍射峰(图 2b), 且Cux-Cr/SiO2催化剂中位于43.3°对应于金属铜(JCPDS: 04-0836)的{111}晶面衍射峰强度减弱并且出现宽化现象, 由Scherrer公式计算可发现金属铜的平均粒径由10.7 nm减小至7.5 nm (表 1), 表明铜物种的分散情况得到了有效改善[13].当铬含量为3 wt%时, 衍射角位于36.5°处对应于Cu2O相(JCPDS:05-0667)的衍射峰相对较强, 表明铬的引入改变了铜物种的还原程度并且有效调控了Cu(Ⅰ)物种在催化剂表面的分布.在酯加氢反应中, Cu(Ⅰ)物种常被认为是重要的羰基吸附位点, 对于反应有一定的促进作用[14].此外, 系列催化剂的XRD图中均未观察到与铬物种相关的衍射峰, 表明其颗粒较小且高度分散在催化剂中[15].

 下载:
导出CSV
下载:
导出CSV
| Catalyst | SBET/(m2•g-1) | Vpore/(cm3•g-1) | Dpore/nm | dCua/nm | DCub/% | SCub/(m2•gcat-1) |
| Cu/SiO2 | 368 | 1.03 | 11.8 | 10.7 | 31.5 | 40.9 |
| Cr0.005-Cu/SiO2 | 350 | 0.75 | 9.5 | 8.3 | 51.0 | 66.2 |
| Cr0.01-Cu/SiO2 | 196 | 0.84 | 19.5 | 7.6 | 52.8 | 68.5 |
| Cr0.03-Cu/SiO2 | 185 | 0.87 | 21.1 | 7.5 | 46.3 | 60.1 |
| Cr0.05-Cu /SiO2 | 183 | 0.77 | 19.4 | 8.9 | 32.5 | 42.2 |
| aCu average diameter of particle size calculated from XRD; bDispersion and surface area of Cu determined by N2O titration. | ||||||
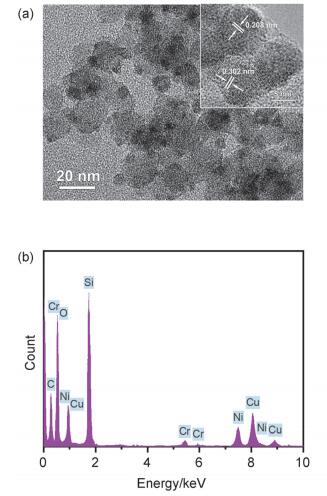
图 3a为还原后Cr0.03-Cu/SiO2催化剂的TEM图像, 可以发现金属Cu在SiO2载体上分散良好, 其平均粒径为6.3 nm.催化剂的EDX能谱分析结果如图 3b所示, 可以发现Cu和Cr元素共同存在于Cr0.03-Cu/SiO2催化剂中.同时, Cr0.03-Cu/SiO2催化剂的高分辨电镜图像(图 3a内插图)中分别观察到金属铜{111}面的晶格条纹(0.208 nm)和亚铬酸铜{200}面的晶格条纹(0.302 nm), 该结果表明铜和铬相互作用形成了CuCr2O4物相, 而CuCr2O4相可有效促进活性铜物种的分散[8b].
进一步研究了不同铬含量Crx-Cu/SiO2催化剂的还原特性, 通过催化剂的氢气程序升温还原峰的位置及峰面积变化判断催化剂中铜铬金属间以及金属与载体间的相互作用[15].对比图 4中不同催化剂的耗氢峰不难看出, Cu/SiO2催化剂的H2-TPR图谱中仅出现了位于539 K的对称还原峰, 而Cr修饰量为0.5 wt%时, 催化剂的耗氢峰相较于Cu/SiO2向低温方向位移, 由539 K降至535 K, 且耗氢峰面积微弱增大, 表明少量铬即可改变Crx-Cu/SiO2催化剂中铜物种的表面分布状况, 其还原峰的半峰宽及还原温度范围均呈现增大的趋势, 表明催化剂中出现了新的耗氢物种.据文献报道, 程序升温还原过程中位于503和533 K左右的两处耗氢峰, 分别对应于CuO至Cu(0)的还原及CuCr2O4的部分还原[16].结合还原后样品的HRTEM图像(图 3a), 我们推测本文报道的催化体系中, 随着Cr含量增加形成的CuCr2O4相导致了还原峰的宽化且还原峰逐步向高温区域发生偏移, 其低温区约530 K仍对应于小颗粒的CuO的还原; 对于Cr0.01-Cu/SiO2催化剂, 在550 K出现肩峰, 随着铬含量增大, 肩峰强度逐渐增强, 且还原温度向高温区偏移.进一步表明因铜铬较强的相互作用而产生的CuCr2O4相随着铬含量的增多而增多, 且被还原性能减弱.
为了进一步了解催化剂的活性物种的变化情况, 采用笑气(N2O)对催化剂的活性比表面(SCu(0))进行滴定, 结果如表 1中所示.不难看出, 铬的引入使得表面活性铜物种明显增多, 尤其在铬含量较低的情况下, 铜铬相互作用形成CuCr2O4物相能够较好的分散于载体表面, 阻滞了铜物种的团聚生长, 进而获得了较好的分散性.而铬修饰量为5 wt%时, 过量高分散的CuCr2O4又会覆盖铜纳米颗粒的表面, 减少了活性铜的可接触性, 导致表观活性铜比表面积的减小.因此, 适量助剂铬修饰对于活性铜比表面的提升起到积极作用, 为催化加氢过程提供更多活性位点, 有利于反应性能的提升[17].
为了探究助剂铬修饰对催化剂表面元素组成和价态的影响, 对还原后催化剂进行了X射线光电子能谱(XPS)及X射线激发俄歇电子能谱(XAES)表征. 图 5a为还原后催化剂的Cu 2p谱图, 在结合能位于约942.5 eV处并未发现轨道电子由2p跃迁到3d轨道所形成的卫星峰, 表明还原后催化剂中的铜元素已全部转变为Cu(0)和Cu(Ⅰ), 无高价态铜存在[12].随着Cr引入量的逐步增大, Cr 2p信号峰逐渐增强, 证明表面铬元素的含量逐渐增加(图 5b).其中Cr 2p3/2结合能位于577.8 eV处, 高于文献中报道的纯相Cr2O3的Cr 2p3/2结合能, 更接近于CuCr2O4和Cr(OH)3中Cr物种相应的结合能位置(分别为577.2和577.5 eV)[18], 然而由于CuCr2O4不易被还原, 因此Cr的相对含量较高. Yurieva等[19]曾报道在还原后的CuCr2O4催化剂中含有Cr2O3物种, 其表面可吸附大量H, 进一步形成-OH, 进而起到稳定质子氢的作用. Khassin等[20]同样证实了氢分子可以吸附在还原后的亚铬酸盐上并与晶格氧形成共价O…H键以增强氢的活化.综上可知, 结合H2-TPR表征结果, 我们推测Crx-Cu/SiO2催化剂中铬主要以CuCr2O4和类Cr2O3两种物态并存.
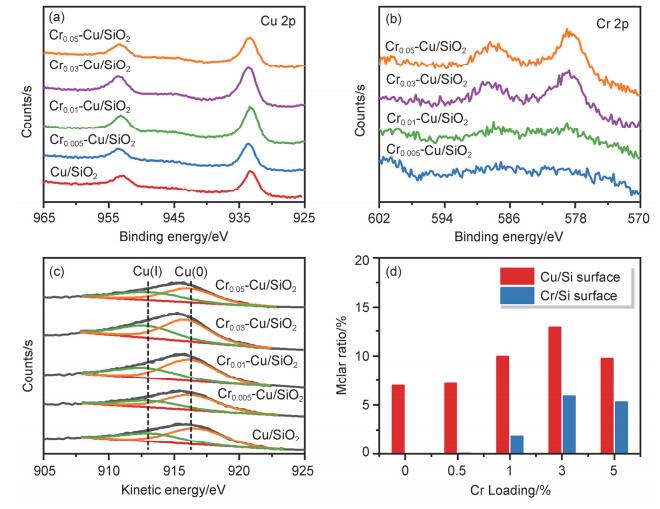
此外, 对XPS结果的进一步分析表明, 还原后Crx- Cu/SiO2催化剂表面Cu/Si与Cr/Si的物质的量比均随着铬引入量的增多呈现相似的火山型变化趋势(图 5d), Cu/Si物质的量比增大进一步表明铬的加入有利于铜物种的分散和表面富集.当铬引入量为5 wt%时, Cu/Si物质的量比的降低则说明, 过量的铬添加又会覆盖部分表面铜, 同时Cu/Si物质的量比的降低较Cr/Si物质的量比降低更显著, 这一发现证实了铬在催化剂表面的富集要强于铜. CuCr2O4•CuO催化剂中铬元素在还原过程中在还原性气氛驱动下同样会发生向铜表面迁移的现象[21].俄歇电子能谱表征结果进一步阐明了还原后Crx-Cu/ SiO2催化剂表面Cu(0)与Cu(Ⅰ)物种的分布状况, 所得结果如图 5c所示. Cu(0)与Cu(Ⅰ)含量的具体数据列于表 2中, 可观察到还原后的Crx-Cu/SiO2催化剂样品中均含有两种不同的还原态铜物种, 且随着铬含量的变化, 不同价态铜物种的相对含量存在一定差异.随着铬含量的增加, Cu(Ⅰ)/(Cu(0)+Cu(Ⅰ))的数值呈现先增大后减弱的趋势.且Cu(Ⅰ)的表面分布状况与XRD结果中Cu2O衍射峰相对强度的变化趋势相吻合.
下载:
导出CSV
| Catalyst | B. E.a/eV Cu 2p | K. E.b/eV | Cu(Ⅰ)/(Cu(0)+Cu(Ⅰ))c | |
| Cu(Ⅰ) | Cu(0) | |||
| Cu/SiO2 | 933.5 | 912.7 | 916.6 | 0.358 |
| Cr0.005-Cu/SiO2 | 933.6 | 912.8 | 916.4 | 0.291 |
| Cr0.01-Cu/SiO2 | 933.4 | 912.8 | 916.6 | 0.363 |
| Cr0.03-Cu/SiO2 | 933.5 | 912.8 | 916.5 | 0.387 |
| Cr0.05-Cu /SiO2 | 933.4 | 912.6 | 916.7 | 0.383 |
| a Binding energy; bKinetic energy; c Calculated from XAES of the reduced Crx-Cu/SiO2 catalysts. | ||||
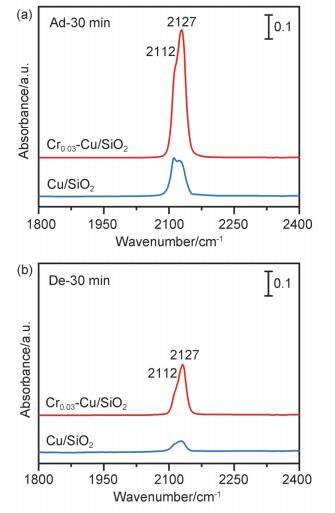
进一步采用原位漫反射傅里叶变换红外光谱(In-situ DRIFTS)对催化剂表面的CO吸附性能进行了表征, 进而阐明铬引入前后催化剂表面不同Cu(0)与Cu(Ⅰ)的分布状况对于羰基吸附构型的影响.铬修饰前后样品分别在2112和2127 cm-1出现吸收峰, 氩气吹扫一定时间后吸收峰强度均出现了不同程度的减弱(图S3), 尤其以低波数吸收峰减弱最为显著, 表明样品表面物理吸附的CO逐渐脱去(图 6).值得注意的是, Cr0.03-Cu/SiO2催化剂的CO红外吸收峰在吹扫前后强度均大于Cu/SiO2.通常情况下, CO吸附在Cu(0), Cu(Ⅰ)和Cu(Ⅱ)物种上伸缩振动频率存在明显的差异[22], 相应波数由大及小的顺序为: Cu(Ⅱ)-CO>Cu(Ⅰ)-CO>Cu(0)-CO, 其中CO在Cu(Ⅱ)和Cu(0)物种上的吸附较弱且在常温条件下可逆, 而在Cu(Ⅰ)物种上的吸附则较为稳定[23].即出现在2112和2127 cm-1的两个羰基峰分别归属于Cu(0)和Cu(Ⅰ)上羰基的伸缩振动[24].该现象进一步佐证了催化剂中确实存在两种还原态的铜物种, 其中Cr0.03-Cu/SiO2催化剂表面的Cu(Ⅰ)物种更为丰富, 对羰基有较强的吸附和锚定能力, 即Cu(Ⅰ)有利于碳酸酯底物中羰基在催化剂表面的吸附.同时该结果也进一步佐证Cu LMM结果中Crx-Cu/SiO2催化剂表面Cu(Ⅰ)物种含量出现微弱升高的现象[25].
助剂修饰作为提高催化活性的有效方式被广泛应用于催化加氢和氢解体系中.例如, 在Co/γ-Al2O3催化剂中加入修饰剂Zr可以促进Co-Al尖晶石相的形成, 改变活性组分Co的还原特性, 同时Zr引入的部分酸性位点有助于提高费托合成反应中长碳链产物的选择性[26].本文采用蒸氨法制备的铬修饰的Crx-Cu/SiO2催化剂与未经修饰的Cu/SiO2催化剂相比, 其还原特性、活性铜的分散度以及不同价态铜的表面分布状况均发生了不同程度的变化, 且碳酸二乙酯加氢反应的活性有不同程度的提升.随着铬修饰量的增加, 产物甲醇的选择性呈现先增大后减小的趋势, Cr0.03-Cu/SiO2催化剂在较高空速条件下仍能显示出较优催化性能和反应稳定性, 产物甲醇的收率最高可达86.2%, 其时空得率可达5.6 mmolMeOH•gcat-1•h-1.早期研究表明, Cu(0)和Cu(Ⅰ)在酯加氢反应中起到协同催化的作用, 其中Cu(0)主要为解离氢的作用, Cu(Ⅰ)则对酯类和酰类化合物中羰基的吸附和活化有着重要的影响, 因此通过不同方式调变Cu(0)和Cu(Ⅰ)的分布利于我们获得更优的反应性能.本文中XAES和In-situ DRIFTS表征均证明Crx-Cu/SiO2催化剂中Cu(Ⅰ)物种的表面分布得以改善, 该结果表明活性铜的分散度和Cu(Ⅰ)的分布状况对于碳酸二乙酯加氢反应性能起到重要作用.铬作为助剂修饰铜基催化剂时在催化剂中易形成尖晶石相的CuCr2O4结构, 在氢气活化过程中CuCr2O4物种部分还原促进活性铜的析出, 同时CuCr2O4由四方尖晶石结构转变为立方尖晶石结构并储存一定量的氢原子[8a].反应过程中, 体相结构储存的氢原子又可以释放至催化剂表面, 促进加氢活化过程, 因此部分还原的CuCr2O4结构为活泼氢的储存和释放搭建了一座桥梁[27].同时, CuCr2O4还可作为电荷转移载体降低活性铜物种周围电子密度, 利于稳定偏正性的铜物种, 间接调控Cu(Ⅰ)物种的表面分布, 为羰基的吸附和稳定提供了更多位点, 该结果也从原位红外表征中得到了进一步的佐证[28].
在铜基催化剂用于酯类气固相加氢反应时, 铜颗粒的聚集生长、分散度的降低及活性物种表面分布的改变通常是不可逆失活的主要原因[5a, 29].在寿命实验中, 相同反应条件下Cr0.03-Cu/SiO2催化剂的稳定性较不含铬的Cu/SiO2催化剂有明显改善, 经150 h持续运行, 催化活性未见明显降低.对反应后的样品进行电镜观察发现, Cr0.03-Cu/SiO2催化剂中铜的平均粒径由6.3 nm增大至7.2 nm (图S4、S5), 而Cu/SiO2催化剂中铜的平均粒径由6.7 nm增大至11.6 nm (图S6).该结果进一步表明铬物种(Cr2O3和CuCr2O4)有效阻滞了铜颗粒在高温下的熟化生长过程, 增强了活性组分与载体间的相互作用, 进而使得反应催化剂获得较好的稳定性.
基于以上讨论, 我们推断本研究中铬修饰在促进铜物种的分散状态的同时可进一步改变其电子状态, 促进对氢的解离活化和中间产物的稳定, 因此即使在较大碳酸二乙酯空速的情况下, 铬助剂修饰的Crx-Cu/SiO2催化剂在提高反应稳定性的前提下仍可提供足量的活性氢物种, 进而提高反应体系的加氢转化效率.
采用蒸氨法合成了一系列不同含量铬修饰的Crx- Cu/SiO2催化剂, 考察了其催化碳酸二乙酯气固相加氢制甲醇的反应性能.研究表明, Cr0.03-Cu/SiO2催化剂上甲醇收率和时空得率分别可达86.2%和5.6 mmolMeOH• gcat-1•h-1, 经150 h长时间运行未见催化剂活性明显降低.少量铬的加入提高了催化剂表面铜物种的分散度和活性比表面积, 优化了不同价态铜物种的分布状况.推测反应中生成的亚铬酸铜相增强了金属与载体间的相互作用及其活化氢的能力, 调控了催化剂的表面电子结构及对反应物的吸附构型, 进而显著提升了催化反应活性和长周期运行稳定性.
Cu/SiO2催化剂的制备:在室温及搅拌条件下, 称取2.853 g Cu(NO3)2•3H2O于200 mL去离子水中充分溶解, 逐滴加入氨水(25 wt%)调节溶液pH为11.0, 得到深蓝色铜氨溶液.称取10.0 g硅溶胶(30 wt%)加入至以上铜氨溶液中持续搅拌4 h, 所得溶胶体系于363 K水浴中蒸氨至中性.将所得悬浊液过滤, 以去离子水洗涤后于373 K干燥12 h, 干燥所得样品研磨后于723 K下焙烧4 h, 升温速率5 K•min-1.所得粉末记为Cu/SiO2催化剂, Cu的理论含量为20 wt%.
Crx-Cu/SiO2催化剂的制备:该系列催化剂制备方法与Cu/SiO2催化剂的制备过程相近, 保持铜的负载量为20 wt%, 引入不同量的Cr(NO3)3•9H2O至铜氨溶液, 其余步骤同上.所得粉末记为Crx-Cu/SiO2催化剂, 其中x代表助剂铬的负载量.
采用Tristar 3020型吸附仪对样品的比表面积和孔径分布进行表征; 采用D8 Advance型X-射线粉末衍射仪对样品的物相进行分析; 采用JEM-2010型透射电镜对样品的形貌结构及晶格参数进行观察; 采用TP5080型全自动多用吸附仪对催化剂的还原特性和活性比表面进行测定; 采用PHI 5000C ESCA型X-射线光电子能谱仪分析样品的表面元素组成及价态分布; 采用Nicolet IR-200型红外光谱仪测定催化剂的CO吸附特性.详细操作步骤见Supporting Information.
催化剂的性能评价在成套高压固定床微反应装置上进行, 反应管规格为500×20×10 mm(管长×外径×内径).对样品进行造粒过筛后, 称取40~60目催化剂0.8 g加入至反应管中, 床层两端用惰性石英砂进行填充.在装置气密性良好情况下, 调节H2压力为0.5 MPa, 流量50 mL•min-1, 以2 K/min速率升温至573 K对催化剂还原4 h.待催化剂床层温度降至反应温度后, 调节H2压力为2.5 MPa, 流量150 mL•min-1, 通过高压恒流泵进料.预反应6 h后收集产物, 采用岛津GC-2010 Plus气相色谱(毛细管柱, RTx-Wax 12442)对产物组成进行定量分析.
(a) Wang, J. J.; Li, G. N.; Li, Z. L.; Tang, C. Z.; Feng, Z. C.; An, H. Y.; Liu, H. L.; Liu, T. F.; Li, C. Sci. Adv. 2017, 3, e1701290;(b) Gao, P.; Li, S. G.; Bu, X. N.; Dang, S. S.; Liu, Z. Y.; Wang, H.; Zhong, L. S.; Qiu, M. H.; Yang, C. G.; Cai, J.; Wei, W.; Sun, Y. H. Nat. Chem. 2017, 9, 1019;(c) Bai, S. X.; Shao, Q.; Wang, P. T.; Dai, Q. G.; Wang, X. Y.; Huang, X. Q. J. Am. Chem. Soc. 2017, 139, 6827;(d) Gao, Y. N.; Liu, S. Z.; Zhao, Z. Q.; Tao, H. C.; Sun, Z. Y. Acta Phys.-Chim. Sin. 2018, 34, 858. (高云楠, 刘世桢, 赵振清, 陶亨聪, 孙振宇, 物理化学学报, 2018, 34, 858.)
Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; Tovar, M.; Fischer, R. W.; Norskov, J. K.; Schlogl, R. Science 2012, 336, 893. doi: 10.1126/science.1219831
(a) Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.; Milstein, D. Nat. Chem. 2011, 3, 609;(b) Balaraman, E.; Ben-David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2011, 50, 11702;(c) Han, Z. B.; Rong, L. C.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. L. Angew. Chem., Int. Ed. 2012, 51, 13041.
(a) Chen, X.; Cui, Y. Y.; Wen, C.; Wang, B.; Dai, W.-L. Chem. Commun. 2015, 51, 13776;(b) Lian, C.; Ren, F. M.; Liu, Y. X.; Zhao, G. F.; Ji, Y. J.; Rong, H. P.; Jia, W.; Ma, L.; Lu, H. Y.; Wang, D. S.; Li, Y. D. Chem. Commun. 2015, 51, 1252;(c) Tamura, M.; Kitanaka, T.; Nakagawa, Y.; Tomishige, K. ACS Catal. 2016, 6, 376;(d) Cui, Y. Y.; Dai, W.-L. Catal. Sci. Technol. 2016, 6, 7752.
(a) Wen, C.; Cui, Y. Y.; Dai, W.-L; Xie, S. H.; Fan, K. N. Chem. Commun. 2013, 49, 5195;(b) Ye, R.-P.; Lin, L.; Li, Q. H.; Zhou, Z. F.; Wang, T. T.; Russell, C.; Adidharma, H.; Xu, Z. H; Yao, Y.-G.; Fan, M. H. Catal. Sci. Technol. 2018, 8, 3428.
(a) Yu, H. B.; Mao, Z. W.; Le, Y.; Peng, H.; Dai, W. Sci. China: Chem. 2014, 44, 1685(于海波, 毛祖旺, 乐毅, 彭晖, 戴伟, 中国科学: 化学, 2014, 44, 1685);(b) Zhou, G. B.; Wang, H.; Pei, Y.; Qiao, M. H.; Sun, B.; Zong, B. N. Acta Chim. Sinica 2017, 75, 321. (周功兵, 王浩, 裴燕, 乔明华, 孙斌, 宗保宁, 化学学报, 2017, 75, 321.)
Yin, A. Y.; Wen, C.; Guo, X. Y.; Dai, W.-L.; Fan, K. N. J. Catal. 2011, 280, 77. doi: 10.1016/j.jcat.2011.03.006
(a) Kim, N. D.; Oh, S.; Joo, J. B; Jung, K.; Yi, J. H. Top Catal. 2010, 53, 517;(b) Xiao, Z. H.; Ma, Z. Q.; Wang, X. K.; Williams, C.; Liang, C. H. Ind. Eng. Chem. Res. 2011, 50, 2031.
Tsoncheva, T.; Järn, M.; Paneva, D.; Dimitrov, M.; Mitov, I. Micropor. Mesopor. Mat. 2011, 137, 56. doi: 10.1016/j.micromeso.2010.08.021
Dragoi, B.; Ungureanu, A.; Chirieac, A.; Hulea, V.; Royer, S.; Dumitriu, E. Catal. Sci. Technol. 2013, 3, 2319. doi: 10.1039/c3cy00198a
Wang, J. Q.; Chernavskii, P.; Khodakov, A.; Wang, Y. J. Catal. 2012, 286, 51. doi: 10.1016/j.jcat.2011.10.012
尹安远, 郭秀英, 戴维林, 范康年, 化学学报, 2009, 67, 1731. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract329845.shtmlYin, A. Y.; Guo, X. Y.; Dai, W. -L.; Fan, K. N. Acta Chim. Sinica 2009, 67, 1731. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract329845.shtml
Yin, A. Y.; Guo, X. Y.; Dai, W.-L.; Fan, K. N. J. Phys. Chem. C 2009, 113, 11003.
(a) Wang, Y.; Shen, Y. L.; Zhao, Y. J.; Lv, J.; Wang, S. P.; Ma, X. B. ACS Catal. 2015, 5, 6200;(b) Zhao, Y. J.; Li, S. M.; Wang, Y.; Shan, B.; Zhang, J.; Wang, S. P.; Ma, X. B. Chem. Eng. J. 2017, 313, 759.
郭小玲, 陈霄, 苏党生, 梁长海, 化学学报, 2018, 76, 22. doi: 10.3866/PKU.WHXB201706302Guo, X. L.; Chen, X.; Su, D. S.; Liang, C. H. Acta Chim. Sinica 2018, 76, 22. doi: 10.3866/PKU.WHXB201706302
Liang, C. H.; Ma, Z. Q.; Ding, L.; Qiu, J. S. Catal. Lett. 2009, 130, 169. doi: 10.1007/s10562-009-9844-y
Zheng, X. L.; Lin, H. Q.; Zheng, J. W.; Duan, X. P.; Yuan, Y. Z. ACS Catal. 2013, 3, 2738. doi: 10.1021/cs400574v
Zhang, M. H.; Li, G. M.; Jiang, H. X.; Zhang, J. Y. Catal. Lett. 2011, 141, 1104. doi: 10.1007/s10562-011-0635-x
Yurieva, T. M.; Plyasova, L. M.; Makarova, O. Y.; Krieger, T. A. J. Mol. Catal. A:Chem. 1996, 113, 455. doi: 10.1016/S1381-1169(96)00272-5
Khassin, A. A.; Kustova, G. N.; Jobic, H.; Yurieva, T. M.; Chesalov, Y. A.; Filonenko, G. A.; Plyasova, L. M.; Parmon, V. N. Phys. Chem. Chem. Phys. 2009, 11, 6090. doi: 10.1039/b821381j
Xiao, Z. H.; Wang, X. K.; Xiu, J. H.; Wang, Y. M.; Williams, C.; Liang, C. H. Catal. Today 2014, 234, 200. doi: 10.1016/j.cattod.2014.02.025
(a) Choi, K.; Vannice, M. J. Catal. 1991, 131, 22;(b) Fisher, I. A.; Bell, A. T. J. Catal. 1998, 178, 153;(c) Zhu, S. H.; Gao, X. Q.; Zhu, Y. L.; Fan, W. B.; Wang, J. G.; Li, Y. W. Catal. Sci. Technol. 2015, 5, 1169.
Ge, X.; Zou, H.; Wang, J.; Shen, J. Y. React. Kinet. Catal. Lett. 2005, 85, 253. doi: 10.1007/s11144-005-0268-4
Boccuzzi, F.; Chiorino, A.; Tsubota, S.; Haruta, M. J. Phys. Chem. 1996, 100, 3625. doi: 10.1021/jp952259n
(a) Gong, J. L.; Yue, H. R.; Zhao, Y. J.; Zhao, S.; Zhao, L.; Lv, J.; Wang, S. P.; Ma, X. B. J. Am. Chem. Soc. 2012, 134, 13922;(b) Yue, H. R.; Zhao, Y. J.; Ma, X. B.; Gong, J. L. Chem. Soc. Rev. 2012, 41, 4218;(c) Zheng, J. W.; Zhou, J. F.; Lin, H. Q.; Duan, X. P.; Williams, C.; Yuan, Y. Z. J. Phys. Chem. C 2015, 119, 13758.
Garcilaso, V.; Barrientos, J.; Bobadilla, L. F.; Laguna, O. H.; Boutonnet, M.; Centeno, M. A.; Odriozola, J. A. Renew. Energ. 2019, 132, 1141. doi: 10.1016/j.renene.2018.08.080
Bechara, R.; Wrobel, G.; Daage, M.; Bonnelle, J. P. Appl. Catal. 1985, 16, 15. doi: 10.1016/S0166-9834(00)84066-X
He, Z.; Lin, H. Q.; He, P.; Yuan, Y. Z. J. Catal. 2011, 277, 54. doi: 10.1016/j.jcat.2010.10.010
周功兵, 谭晓荷, 窦镕飞, 裴燕, 范康年, 乔明华, 孙斌, 宗保宁, 中国科学:化学, 2014, 44, 121. http://www.cnki.com.cn/Article/CJFDTotal-JBXK201401014.htmZhou, G. B.; Tan, X. H.; Dou, R. F.; Pei, Y.; Fan, K. N.; Qiao, M. H.; Sun, B.; Zong, B. N. Sci. China:Chem. 2014, 44, 121. http://www.cnki.com.cn/Article/CJFDTotal-JBXK201401014.htm

图式 1 CO2经由碳酸二乙酯制甲醇转化路径
Scheme 1 Strategy of CO2 to methanol based on diethyl carbonate

图 1 Crx-Cu/SiO2催化剂对碳酸二乙酯加氢产物甲醇收率随液时空速的变化曲线.反应条件: T=503 K (a), T=483 K (b), p(H2)=2.5 MPa, mcat.=0.8 g
Figure 1 MeOH yield over various Cux-Cr/SiO2 catalysts with different LHSV of DEC. Reaction conditions: T=503 K (a), T=483 K (b), p(H2)=2.5 MPa, mcat.=0.8 g

图 2 焙烧后(a)及还原后(b) Crx-Cu/SiO2催化剂的XRD图
Figure 2 XRD patterns of the calcined (a) and reduced (b) Crx-Cu/SiO2 catalysts

图 3 还原后Cr0.03-Cu/SiO2催化剂的TEM (a), HRTEM (a内插图)图像及EDX能谱图(b)
Figure 3 TEM (a), HRTEM (inset of a) and EDX analysis (b) of the reduced Cr0.03-Cu/SiO2 catalyst

图 5 还原后Crx-Cu/SiO2催化剂的Cu 2p (a)和Cr 2p (b) XPS谱图; 还原后Crx-Cu/SiO2催化剂的Cu LMM俄歇电子能谱图(c); Crx-Cu/SiO2催化剂表面Cu/Si和Cr/Si物质的量比随Cr含量的变化趋势(d)
Figure 5 Cu 2p (a) and Cr 2p (b) XPS of the reduced Crx-Cu/SiO2 catalysts; Cu LMM (c) XAES of the reduced Crx-Cu/SiO2 catalysts; Surface Cu/Si and Cr/Si molar ratio of Crx-Cu/SiO2 catalysts (d)

图 6 Cu/SiO2和Cr0.03-Cu/SiO2催化剂吸附30 min (a)和氩气吹扫30 min (b)后的原位CO红外图谱.温度: T=313 K
Figure 6 In-situ DRIFTS spectra of CO adsorption on Cu/SiO2 and Cr0.03-Cu/SiO2 catalysts before (a) and after Ar purging (b). T=313 K
表 1 Crx-Cu/SiO2催化剂的物理化学性质
Table 1. Physicochemical parameters of the Crx-Cu/SiO2 catalysts
| Catalyst | SBET/(m2•g-1) | Vpore/(cm3•g-1) | Dpore/nm | dCua/nm | DCub/% | SCub/(m2•gcat-1) |
| Cu/SiO2 | 368 | 1.03 | 11.8 | 10.7 | 31.5 | 40.9 |
| Cr0.005-Cu/SiO2 | 350 | 0.75 | 9.5 | 8.3 | 51.0 | 66.2 |
| Cr0.01-Cu/SiO2 | 196 | 0.84 | 19.5 | 7.6 | 52.8 | 68.5 |
| Cr0.03-Cu/SiO2 | 185 | 0.87 | 21.1 | 7.5 | 46.3 | 60.1 |
| Cr0.05-Cu /SiO2 | 183 | 0.77 | 19.4 | 8.9 | 32.5 | 42.2 |
| aCu average diameter of particle size calculated from XRD; bDispersion and surface area of Cu determined by N2O titration. | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 还原后Crx-Cu/SiO2催化剂表面铜的价态分布
Table 2. Chemical state of surface copper on the reduced Crx-Cu/SiO2 catalysts
| Catalyst | B. E.a/eV Cu 2p | K. E.b/eV | Cu(Ⅰ)/(Cu(0)+Cu(Ⅰ))c | |
| Cu(Ⅰ) | Cu(0) | |||
| Cu/SiO2 | 933.5 | 912.7 | 916.6 | 0.358 |
| Cr0.005-Cu/SiO2 | 933.6 | 912.8 | 916.4 | 0.291 |
| Cr0.01-Cu/SiO2 | 933.4 | 912.8 | 916.6 | 0.363 |
| Cr0.03-Cu/SiO2 | 933.5 | 912.8 | 916.5 | 0.387 |
| Cr0.05-Cu /SiO2 | 933.4 | 912.6 | 916.7 | 0.383 |
| a Binding energy; bKinetic energy; c Calculated from XAES of the reduced Crx-Cu/SiO2 catalysts. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: