
引用本文:
李东祥, 高媛媛, 张晓芳, 夏海兵. 纳米尺度的消化熟化及其在单分散纳米材料制备中的应用[J]. 化学学报,
2019, 77(4): 305-315.
doi:
10.6023/A18120512

Citation: Li Dongxiang, Gao Yuanyuan, Zhang Xiaofang, Xia Haibing. Digestive Ripening at Nanoscale and Its Application in the Preparation of Monodisperse Nanomaterials[J]. Acta Chimica Sinica, 2019, 77(4): 305-315. doi: 10.6023/A18120512
Citation: Li Dongxiang, Gao Yuanyuan, Zhang Xiaofang, Xia Haibing. Digestive Ripening at Nanoscale and Its Application in the Preparation of Monodisperse Nanomaterials[J]. Acta Chimica Sinica, 2019, 77(4): 305-315. doi: 10.6023/A18120512
纳米尺度的消化熟化及其在单分散纳米材料制备中的应用
摘要:
近年来,消化熟化方法已经被广泛采用来制备单分散纳米粒子,特别是在制备亚10 nm的小尺寸纳米粒子方面具有显著优势.但是,目前国内尚未发现关于此方法的中文文献报道,影响了部分国内学者对消化熟化方法的认识和应用.因此,从纳米尺度的消化熟化现象的发现过程和机理的提出开始,分析了沉淀反应前驱物、消化熟化剂种类、热处理温度和时间、溶剂类型以及其他因素对消化熟化现象的影响,介绍了基于颗粒表面带电与曲率效应、颗粒表面与配体分子层的相互作用以及消化熟化过程中的竞争反应平衡等因素的理论模型和研究结果,阐述了消化熟化法在制备金属纳米粒子、合金纳米粒子、金属氧化物和硫族化合物量子点以及其他纳米粒子等单分散纳米材料中的应用,最后展望了消化熟化法制备的单分散纳米粒子在纳米组装和多相催化等领域的应用.
English
Digestive Ripening at Nanoscale and Its Application in the Preparation of Monodisperse Nanomaterials
Abstract:
Recently, a digestive ripening process at nanoscale has been widely used to prepare monodisperse nanoparticles (NPs), especially for sub-10 nm small NPs, with significant advantages such as the very narrow size distribution of the obtained nanoparticles, the versatile applications for various nanoparticles and the simple operation process. However, no Chinese references are reported on digestive ripening process till now, which may limit the cognition and utility of digestive ripening method for some domestic scientists. Thus, this review starts from the discovery of the phenomenon and the proposal of mechanism for digestive ripening at nanoscale, to the analysis of influence factors including the precursor in the precipitation reaction, the digestive ripening reagent, heating treatment temperature and processing time, solvent media and so on. Then, theoretical hypothesis and the derived results are introduced based on the charged surface, the curvature effect, the interaction between NP surface and attached ligand layer, the diffusion effect and the competing reaction balance in the digestive ripening process. Subsequently, the important applications of digestive ripening method in the preparation of monodisperse nanomaterials of metal NPs, alloy NPs, quantum dots of metal oxide and metal chalcogenide, and other NPs are discussed, the obtained small metal or alloy NPs show a perfect sphere shape and a very narrow size distribution (relative standard deviation less than ±5%). Finally, the broad perspectives are proposed in the NP assembly for optical, electric and magnetic nanodevices, and the heterogeneous catalysis of monodisperse metal, alloy and semiconducotr NPs via the digestive ripening method.
-
Key words:
- nanoparticles
- / digestive ripening
- / monodisperse
- / mechanism
- / self-assembly
-
1. 引言
由于单分散纳米粒子具有独特的物理和化学性质, 其制备方法和理论研究一直受到重视[1~4], 特别是在单分散金属纳米粒子的制备方面, 新的研究结果不断出现[5~8].很久以来, 人们通常采用LaMer原理制备单分散纳米粒子体系, 即设法使晶核的形成和纳米粒子的生长两个阶段分开, 以实现快速成核和晶核同步生长[9, 10].其中, 晶种生长法能有效地避免生长过程的“二次成核”而有利于得到单分散纳米颗粒或纳米棒[11, 12].对于已经制备的尺寸多分散纳米粒子体系, 通常采用Ostwald熟化提高尺寸的均一性[13], 特别是当颗粒之间尺寸相差较大时, 小粒子的消失使颗粒尺寸趋于均一, 形成接近单分散的体系[14]. Ostwald熟化过程使固体表面积减小, 降低了体系的表面Gibbs自由能.从这一角度讲, Ostwald熟化是热力学自发过程.
值得注意的是, 在单分散纳米粒子的制备方面, 近年来报道了较多关于消化熟化方法的研究工作, 特别是在小尺寸(亚10 nm)单分散纳米粒子的制备方面表现出显著优势, 这主要包括: (1)这种方法所获得的纳米粒子的尺寸具有高度均一性, 尺寸标准偏差可小于±5%; (2)这种方法可以用于制备多种单分散纳米材料, 如金属纳米粒子、合金纳米粒子、金属氧化物和金属硫族化合物量子点以及其他纳米粒子等; (3)这种方法反应条件温和, 操作简单, 重复性好等.
自从Klabunde等[15]在研究硫醇保护的纳米金超晶格结构的过程中于2000年首次提出消化熟化机理以来, 国际上已经发表了超过120篇相关研究论文[16, 17].尽管国内学者在这一领域也开展了一些研究工作[18~22], 但是目前尚未发现关于此方法的中文文献报道, 限制了该领域部分国内学者对消化熟化方法的认识和应用.因此, 本文总结了纳米粒子消化熟化现象的发现、机理的提出、各种影响因素、理论模拟研究及其在单分散纳米材料制备中的应用, 展望了消化熟化法制备的单分散纳米粒子在纳米组装和多相催化等领域的应用.
2. 消化熟化现象的发现与机理的提出
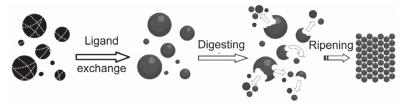
纳米粒子的消化熟化现象是在人们的科研实践中发现的. 1987年Klabunde等[23]将钯块在高温真空下蒸发并与丙酮进行共沉积, 然后加热蒸发掉溶剂制备丙酮配合的胶体钯薄膜时, 发现加热处理这种膜能够提高膜的均一性. 1995年Heath和Gelbart等利用Brust-Schiffrin方法[24]在甲苯/水两相体系中合成了较均一的金纳米粒子, 发现颗粒的尺寸由Au与硫醇的物质的量比决定[25]. 1999年Klabunde等[26, 27]向多分散金纳米粒子体系中加入季铵盐、有机硫醇、有机胺等表面活性剂作为配体改性金纳米粒子的表面, 经过加热处理后得到了尺寸均一的金纳米粒子组装的超晶格结构, 如图 1.因此, Klabunde等认为在配体存在下对金属纳米粒子进行加热处理会使颗粒尺寸变得均一.为验证这一观点, 2000年Klabunde等[15]将氯金酸溶于含有双十二烷基二甲基溴化铵(DDAB)的甲苯中, 室温下加入硼氢化钠水溶液制备了多分散的金纳米粒子(1~80 nm); 然后采用十二烷基硫醇(DDT)作为配体保护剂替换DDAB, 再用乙醇将金纳米粒子从甲苯中析出形成沉淀; 最后, 将沉淀干燥后加入含有DDT的甲苯中加热回流(110 ℃), 获得了直径为(6.2±0.3) nm的超窄尺寸分布的金纳米粒子.这种多分散纳米颗粒在配体存在下加热回流过程出现的颗粒“尺寸聚焦”现象与传统的Ostwald熟化理论相反, 于是Klabunde等[15]提出了消化熟化机理.
图 1
消化熟化的过程分为三步(如图 2)[15~17]:第一步, 向已经制备好的多分散的纳米粒子体系中加入配体分子, 配体分子会与纳米粒子表面原有的稳定剂(如表面活性剂、弱配体或溶剂分子)发生交换, 到达纳米粒子表面.第二步, 配体分子将大尺寸颗粒“破碎”为一系列不同尺寸的小颗粒, 即大颗粒被配体“消化”成小颗粒, 称为消化过程.这是因为新制备的纳米颗粒中存在较多的晶格缺陷(如孪生晶界和堆垛缺陷), 特别是大于10 nm的粒子, 由于晶体结构中存在较强的微观应力, 晶格的稳定能降低, 使得纳米颗粒反应活性提高, 因此很容易被配体诱导破坏并形成小尺寸的球状颗粒.当然, 这些小球状颗粒的尺寸仍然是多分散的.第三步, 这种消化后的小颗粒在高浓度配体的存在下经过加热处理, 能使所有颗粒尺寸趋于一致, 称为熟化过程.由于消化后的颗粒尺寸仍然是多分散的, 熟化过程中也必然存在对大颗粒的消化过程.这种具备消化和熟化功能的配体称为消化熟化剂.可以认为, 在熟化过程中, 原子或原子簇从大尺寸颗粒表面被消化熟化剂蚀刻下来, 使大颗粒的尺寸不断减小; 同时, 蚀刻下来的原子或原子簇沉积到小颗粒的表面使小颗粒尺寸变大.因此, 体系中不管是大颗粒还是小颗粒, 其尺寸都趋向于某一尺寸, 称为收敛尺寸.
图 2

从热力学角度来讲, 在消化熟化体系中, 无论是通过纳米粒子的长大来减小体系的表面积从而降低体系的总表面能, 还是通过配体分子与纳米粒子表面的结合来释放结合能从而降低体系的总能量, 两者所能达到的平衡状态本质上是由其热力学状态函数决定的.但是, 达到平衡状态的过程显然是一个动力学过程, 它受到消化熟化剂配体对大颗粒的蚀刻反应速率、蚀刻掉的原子或原子簇的扩散速率及其在小颗粒表面的沉积速率等因素的影响.因此, 消化熟化过程本质上是一个动力学控制的热力学过程[28].
关于产生消化现象的原因, Ackerson等[29]在研究消化过程中氧气和溶剂的作用时发现, 硫醇对金属纳米粒子的蚀刻消化应归因于氧存在下产生的硫自由基; Lin等[30]通过油酸和三正辛基氧膦对钴、铁、铂及其合金纳米粒子的消化熟化研究认为, 在较高的配体浓度下配体与金属原子之间的强结合力会诱导产生配体-金属配合物, 因此容易把大颗粒分解为小颗粒.
有趣的是, 这种消化熟化现象可以反过来进行[31].如图 3所示, 向尺寸和形状多分散的金纳米粒子体系(图 3a)中加入硫醇进行消化处理, 消化过程中明显地看到大颗粒生成小颗粒(图 3b); 消化后的小颗粒再经过熟化得到硫醇保护的单分散纳米粒子(图 3c)容易形成超晶格结构; 但是, 将熟化后分离出来的单分散金纳米粒子超晶格样品置于甲苯溶剂中, 再加入季铵盐表面活性剂DDAB后, 又能发现小粒子重新生成各种形状的大尺寸金纳米粒子的过程(图 3d), 样品加热回流后又变为尺寸和形状多分散的金纳米粒子(图 3a).这一现象说明, 消化熟化过程是纳米粒子在强配体存在下发生的一种特殊现象, 与传统的Ostwald熟化理论并不矛盾.
图 3
 图 3. 大尺寸多分散金纳米粒子与小尺寸单分散配体保护的金纳米粒子的相互转化过程TEM示意图[31]Figure 3. Schematic illustration by TEM for reversible change from larger polyhedral nanoparticles to smaller, monodisperse ligand-stabilized spherical particles
图 3. 大尺寸多分散金纳米粒子与小尺寸单分散配体保护的金纳米粒子的相互转化过程TEM示意图[31]Figure 3. Schematic illustration by TEM for reversible change from larger polyhedral nanoparticles to smaller, monodisperse ligand-stabilized spherical particles3. 影响消化熟化的因素
实际上, 消化熟化过程可以理解为两个竞争反应的结合:一是纳米粒子会通过长大来减小其表面积, 从而使其表面能最小化, 并最终实现体系总能量的降低; 二是过量的消化熟化剂配体能使颗粒尽可能地变小以实现配体与更多金属表面的结合, 通过释放结合能来降低体系的总能量.当纳米粒子与消化熟化剂一起在溶剂中加热处理时, 在某个温度下所建立的颗粒长大与变小两个反应的平衡会决定一个纳米粒子的收敛尺寸[17].这个收敛尺寸与纳米粒子体系的本性、消化熟化剂、温度和溶剂等许多因素有关.
3.1 沉淀反应前驱物的影响
典型的消化熟化过程中, 人们通常采用沉淀方法将消化后配体保护的金属纳米粒子从反应体系中分离出来, 并将沉淀进行洗涤纯化, 金属前驱物和还原剂的残余及其反应产物不会进入熟化过程, 因此金属前驱物和还原剂的差别对消化熟化的结果不会产生影响.但是, 由于亲水性消化熟化剂体系难以实现消化后的纳米粒子与体系的分离纯化[32], 因此, 前驱物的残余离子在熟化处理时存在于反应体系中, 从而可能对产物的形貌和性能产生影响[21, 33].例如, 使用不同的前驱物(醋酸盐、硫酸盐、硝酸盐和氯化物)利用消化熟化法制备的CuO量子点的尺寸、荧光强度和表面ζ电位等方面均存在差别[34].
3.2 消化熟化剂的影响
消化熟化剂的种类、用量和结构等因素对消化熟化的效果具有重要影响.通常使用的消化熟化剂包括能与金属纳米颗粒表面形成化学键的强相互作用配体(如硫醇、有机胺、有机酸、有机硅烷和三烷基氧膦等), 也包括仅能与金属表面发生物理吸附的弱相互作用的配体(如吡啶类、季铵盐类等).
硫醇是最常用的消化熟化剂, 其用量、烷基链长短和分子结构对消化熟化均有影响.硫醇的用量越大, 同样条件下得到的纳米粒子体系的单分散程度越高[35], 但大大过量的硫醇有可能导致形成硫醇保护的金属原子簇[36].短烷基链硫醇保护的金纳米粒子在甲苯溶剂中倾向于形成超晶格结构, 而长烷基链硫醇更容易形成单分散的粒子[37], 这可能与硫醇分子在甲苯中的溶解性有关; 硫醇的烷基链越长, 得到的金纳米颗粒的尺寸越大[38]; 单巯基长链烷基配体与双巯基及三巯基长链烷基配体的比较研究证明, 单巯基配体是更好的消化熟化剂[39].使用巯基聚乙二醇(PEG5000-SH)作为消化熟化剂可以在水相中进行消化熟化, 直接得到聚乙二醇保护的金纳米粒子[40].使用十二烷基硫醇(DDT)在室温下消化熟化蠕虫状的十二烷基胺保护的钯纳米粒子, 使其变为尺寸更为均一的球状钯纳米粒子[41, 42].
有机胺与金属表面也有较强的相互作用, 也经常用于消化熟化[43]. Jagirdar课题组[28]研究了有机胺(包括辛胺(ODA)、十二烷基胺(DDA)和十六烷基胺(HDA)等)的用量对消化熟化效果的影响.将金纳米粒子在二甲苯溶剂中于165 ℃下进行30 h的消化熟化, 其中有机胺与金属的物质的量比从10变化到30时所得样品的尺寸和尺寸分布列于表 1.可以看出, 消化熟化后的金纳米粒子的尺寸在9~12 nm之间, 相对尺寸偏差约15%~25%.但是, 这些数据不能有效反映有机胺的用量和有机胺分子中碳链的长短对金纳米粒子的消化熟化效果的影响, 这很可能是所用配体的浓度太高造成的.因此, 为获得更好的尺寸分布和最优的配体浓度, Prasad等[16]把配体与金属的物质的量比调整到2.5与20之间, 并且在更低的温度(110 ℃)下选择DDA和HDA对金纳米粒子进行消化熟化, 所得数据亦列于表 1.可以发现, 消化熟化后金纳米粒子的尺寸随配体用量的增大先略有增加, 然后又有减小; 同时, 粒子的尺寸偏差随着配体用量的增加明显减小.这与硫醇用量对消化熟化的影响一致[35].
表 1
 表 1 有机胺配体对金纳米粒子尺寸和尺寸偏差的影响Table 1. Size and size deviation of gold nanoparticles influenced by alkyl amines
表 1 有机胺配体对金纳米粒子尺寸和尺寸偏差的影响Table 1. Size and size deviation of gold nanoparticles influenced by alkyl amines 下载:
导出CSV
下载:
导出CSV
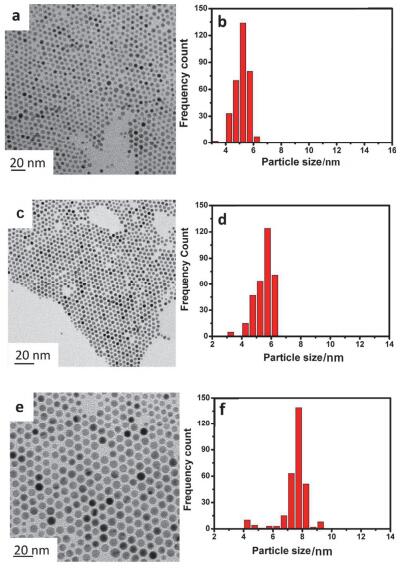
为比较硫醇和有机胺的消化熟化效果, Prasad等[44]采用十二烷基硫醇(DDT)和十二烷基胺(DDA)对银、金和钯进行了消化熟化研究, 所得样品的TEM照片如图 4.当使用DDT作消化熟化剂时, 银、金和钯的尺寸分别为(4.4±0.6) nm、(5.2±0.5) nm和(6.2±0.5) nm; 而当使用DDA作消化熟化剂时, 银、金和钯的尺寸分别为(10.2±1.2) nm、(8.0±0.6) nm和(7.1±0.6) nm.可见, 使用硫醇获得样品的尺寸明显小于有机胺, 这跟消化熟化剂与金属之间的相互作用的强弱有关.但是, 两种消化熟化剂所制备的样品的相对尺寸偏差基本一致.另外, 使用不同结构的胺类化合物作配体对金纳米粒子的消化熟化研究发现, 有机胺配体的空间稳定作用和表面吸附离子的静电屏蔽作用都对颗粒的生长和尺寸大小起到重要作用[45].
图 4
 图 4. 分别采用十二烷基醇(a~c)和十二烷基胺(d~f)消化熟化后的银(a, d)、金(b, e)和钯(c, f)纳米粒子的TEM照片[44]Figure 4. TEM images of Ag (a, d), Au (b, e) and Pd (c, f) nanoparticles after digestive ripening using dodecylthiol (a~c) and dodecylamine (d~f), respectively
图 4. 分别采用十二烷基醇(a~c)和十二烷基胺(d~f)消化熟化后的银(a, d)、金(b, e)和钯(c, f)纳米粒子的TEM照片[44]Figure 4. TEM images of Ag (a, d), Au (b, e) and Pd (c, f) nanoparticles after digestive ripening using dodecylthiol (a~c) and dodecylamine (d~f), respectively有机酸、有机硅烷和三烷基氧膦也均可以作为金属的消化熟化剂[46].使用油酸(OA)对钴纳米粒子的消化熟化研究发现, OA的用量对钴纳米粒子的尺寸效应是可逆的[47]:当n(Co):n(OA)=5:1时, 钴纳米粒子的尺寸为9.5 nm; 当n(Co):n(OA)=2:5时, 质谱研究发现形成了由2~3个Co原子为核与2~3个OA分子形成的配合物.当向9.5 nm的钴纳米粒子中继续加入OA使n(Co):n(OA)=2:5, 再进行消化熟化也形成了2~3个Co原子为核与2~3个OA分子形成的配合物; 当从已经形成Co与OA配合物的体系中去除自由的OA使n(Co):n(OA)=5:1, 再经过消化熟化又得到9.5 nm的钴纳米粒子[47].另外, 采用油酸和油胺作消化熟化剂在十八烯溶剂中对2.1和2.6 nm的混合CdSe量子点体系进行低温下的消化熟化, 可以得到2.45 nm的尺寸分布很窄的量子点[48].
消化熟化剂配体与金属表面相互作用的强弱是一项关键因素.使用弱配体4-二甲氨基吡啶对金纳米粒子进行消化熟化得到的金纳米粒子的尺寸为11.3 nm; 而相同条件下使用硫醇配体得到的金纳米粒子的尺寸为4.1 nm且单分散性更好[49].另外, 对同一金属纳米粒子的多分散体系使用强配体进行热处理时出现消化熟化现象, 而使用弱配体进行热处理时则出现Ostwald熟化现象[49], 这说明配体与金属表面的相互作用的强弱能决定系统发生何种熟化过程.
2018年, 高传博等[21]采用氯化钠和二乙胺组成双消化熟化剂体系在水相中对纳米银进行处理, 直接制备了亲水性的直径10 nm左右的单分散纳米银球, 这一过程中氯离子起到蚀刻大尺寸银颗粒的作用, 而弱配体二乙胺起到熟化过程保护剂的作用.研究发现, 这一体系通过延长消化熟化的时间即可以获得尺寸不断增大而尺寸偏差均小于10%的9~15 nm的球状银颗粒.该研究工作中, 这两种看起来毫无关联的试剂通过协同作用替代了单一的消化熟化剂, 开拓了消化熟化的新体系.
3.3 热处理温度和时间的影响
温度能够影响化学反应和扩散的速率, 采用较高的温度显然能够加快消化熟化的进程, 因此消化熟化一般在溶剂回流温度下进行[50~52].例如, 采用十二烷基硫醇(DDT)与金属的物质的量比为20时, 对多分散金纳米粒子于60 ℃、120 ℃和180 ℃下经过1 h的消化熟化后样品的尺寸分别为(4.8±0.4) nm、(5.6±0.5) nm和(7.4±0.7) nm, 其TEM照片和尺寸分布如图 5[39].可以发现, 随着温度的升高, 熟化后的纳米粒子的尺寸变大.但是, 在油酸、油胺、十八烯和十六烷基二醇存在下, 对金、铜合金纳米粒子在不同温度下进行消化熟化处理, 发现热处理温度和时间对颗粒的尺寸没有明显影响, 但是会改变合金粒子的元素组成, 温度高时Cu的含量高[53], 这可能跟Au和Cu的表面能差别有关.
图 5
 图 5. 消化熟化温度分别为60 ℃ (a, b)、120 ℃ (c, d)和180 ℃ (e, f)时金纳米粒子的尺寸和尺寸分布[39]Figure 5. Size and size distribution of gold nanoparticles influenced by the temperature in digestive ripening process
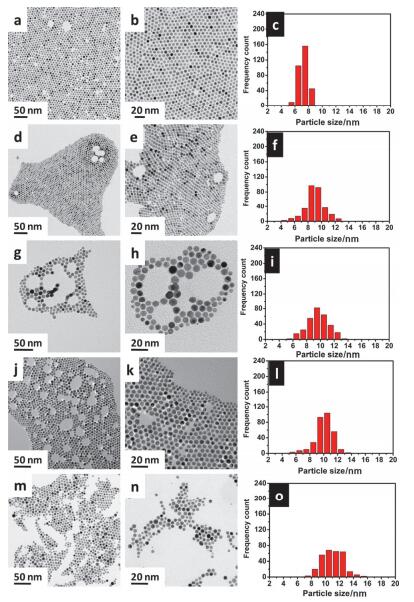
图 5. 消化熟化温度分别为60 ℃ (a, b)、120 ℃ (c, d)和180 ℃ (e, f)时金纳米粒子的尺寸和尺寸分布[39]Figure 5. Size and size distribution of gold nanoparticles influenced by the temperature in digestive ripening process需要注意的是, 消化熟化的温度和时间必须协同控制, 即要求在较低的温度下热处理较长时间, 或者在较高的温度下热处理较短时间[52].否则, 如果在较高的温度下热处理时间过长, 则纳米粒子由消化熟化转入Ostwald熟化[52, 54].例如, 多分散金纳米粒子在十六烷基硫醇和叔丁基甲苯溶剂中120 ℃下处理1 h后, 所得金纳米粒子样品的TEM照片和尺寸分布如图 6a~6c[52], 其尺寸为(7±0.7) nm; 而继续加热处理3、5、7和9 h后, 粒子的尺寸分别变为(8.8±1.4) nm、(9.4±1.5) nm、(9.9±1.3) nm和(10.8±1.5) nm, 如图 6d~6o, 样品的相对尺寸偏差变大.这是因为消化熟化虽然使颗粒尺寸分布变窄, 但是颗粒的尺寸达不到完全相同(最小偏差约±5%).因此, 继续进行热处理时体系会进入Ostwald熟化阶段, 使得本来略小的颗粒变得更小, 略大的颗粒变得越大, 体系中颗粒的尺寸变得更不均一.
图 6
 图 6. 金纳米粒子在十六烷基硫醇和叔丁基甲苯中120 ℃处理时间分别为1 h (a~c)、3 h (d~f)、5 h (g~i)、7 h (j~l)和9 h (m~o)的TEM照片和颗粒尺寸分布图[52]Figure 6. TEM micrographs and particle size histograms of gold nanoparticle systems obtained at 120 ℃ for 1 h (a~c), 3 h (d~f), 5 h (g~i), 7 h (j~l), and 9 h (m~o)
图 6. 金纳米粒子在十六烷基硫醇和叔丁基甲苯中120 ℃处理时间分别为1 h (a~c)、3 h (d~f)、5 h (g~i)、7 h (j~l)和9 h (m~o)的TEM照片和颗粒尺寸分布图[52]Figure 6. TEM micrographs and particle size histograms of gold nanoparticle systems obtained at 120 ℃ for 1 h (a~c), 3 h (d~f), 5 h (g~i), 7 h (j~l), and 9 h (m~o)研究还发现, 以十八烯为溶剂采用高温热解法制备CdS、CdSe和CuZnSnS4半导体纳米晶[55], 使用油胺作配体进行消化熟化时, 在油胺用量较少、温度较低的情况下得到颗粒尺寸较小(约5 nm), 而当油胺用量大、温度超过某一阈值后颗粒尺寸显著变大(约20 nm).
3.4 溶剂的影响
金属纳米粒子的消化熟化处理通常使用甲苯作溶剂, 长链烷基硫醇在甲苯中的溶解性好, 且回流温度较高(110 ℃).研究发现, 因为高极性溶剂可以降低带电球状金纳米粒子表面的Gibbs自由能, 消化熟化后的金纳米粒子的尺寸分布随着苯类溶剂极性的增大而减小, 通过调整溶剂的极性也可以获得相对标准偏差<±5%的高质量金属纳米粒子[22].
溶剂与配体的匹配性会影响熟化效果.如在甲苯中使用癸基硫醇作配体进行消化熟化的效果要优于在三氟甲基苯中的效果[56], 而全氟癸基硫醇作配体在三氟甲基苯中的消化熟化效果优于在甲苯中的效果.研究发现, 硫醇保护的金纳米粒子在甲苯-硫醇体系的溶解性随温度升高而增大, 且溶解行为是可逆的[57].这些金纳米粒子在烷烃中的溶解性与硫醇配体中烷基链长度和溶剂分子烷基链长度有关[58].
Prasad和Thomas等[59, 60]采用三乙胺作消化熟化剂制备CuO量子点时, 研究了多种醇和水作溶剂时所获得产物的特性, 发现溶剂的性质对量子点的尺寸、荧光性质和电动电位ζ都有影响.
3.5 其他因素的影响
还有很多因素会影响消化熟化的效果.例如, Lee等研究发现, 电场的存在能够缩短金纳米粒子消化熟化的时间, 且使硫醇保护的金纳米粒子向正极移动[50, 54].使用十二烷基硫醇(DDT)作消化熟化剂对Pd、Pt、Rh和Ru进行消化熟化时, 加入双十二烷基二甲基溴化铵(DDAB)可使颗粒的尺寸显著减小[61]; 而加入双十二烷基二甲基氯化氨(DDAC)却没有明显变化; 而且当加入1:1的DDAB和DDAC时, 消化熟化后颗粒的尺寸降低值是使用DDAB时的一半.因此, DDAB引起的尺寸降低作用应归因于溴离子.
4. 消化熟化的理论研究
自消化熟化机理提出以来, 许多科学家根据实验结果建立模型, 采用理论方法进行研究. 2007年Lee等[62, 63]同时考虑颗粒表面带电与曲率效应, 对Gibbs- Thomas方程进行修正, 建立了热力学模型来解释消化熟化和双金属的合金化过程机理; 2014年Clark[64]建立了原子或分子穿过颗粒表面的配体层的动力学模型来解释“尺寸聚焦”现象; 2017年Irzhak[65]经过研究认为, 配体层对内部核的稳定性的影响以及由纳米粒子尺寸的降低导致的配体层结构的变化决定了消化熟化过程的动力学. 2018年Thomas等突出颗粒表面与配体分子之间的软硬酸碱相互作用[34]和粒子尺寸变化过程的动力学建立了理论模型, 认为这种尺寸聚焦趋势是表面焓驱动的过程[66], 指出熟化后的颗粒收敛尺寸与两个因素对数相关, 即从大颗粒刻蚀下来的穿过颗粒表面配体层的原子或原子簇的扩散系数和颗粒固体的溶解度; 他还认为, 消化熟化只能发生一个表面电荷窗口内, 即不管颗粒尺寸多大, 如果表面电荷超过一个量值, 则颗粒非常稳定, 不发生消化熟化; 并且胶体颗粒的表面能、表面带电量和温度都会影响收敛尺寸[66].这些理论研究的结论能够解释某些实验结果.但是, 由于消化熟化过程多数采用带有长烷基链的非极性配体分子, 颗粒表面由非极性配体分子层所覆盖, 一般认为表面不带电.因此, 这些以表面电荷为主要依据的理论模型与实际不符, 需要建立更为合理的模型进行研究[16].
2017年Manzanares等[67]根据消化熟化过程中的竞争反应平衡建立了简单的统计热力学模型, 研究了各种因素对熟化后颗粒平均尺寸的影响.主要结论包括: (1)提高配体与金属的物质的量比使颗粒尺寸降低; (2)提高介质介电常数使颗粒尺寸变小, 提高颗粒表面电荷使颗粒的尺寸分布变窄; (3)配体越强, 与金属表面的结合能越高, 颗粒尺寸越小; (4)配体的烷基链越长, 颗粒尺寸越大; (5)升高热处理温度使颗粒的尺寸变大.这些结论的规律与实验结果定性地一致.但是, 从金属纳米粒子的消化熟化结果来看, 消化过程和熟化过程具有一定的相对独立性, 研究熟化过程的条件和因素对消化熟化的结果的影响应该更有意义.
5. 消化熟化法在单分散纳米材料制备中的应用
利用消化熟化法很容易地使颗粒的尺寸分布变窄, 将一个多分散的纳米粒子体系转变为单分散的体系.由于消化熟化时大量配体的存在使纳米粒子在高浓度下仍然稳定, 因此容易实现克级的纳米粒子合成[20, 68].可以发现, 消化熟化法制备的纳米粒子的尺寸多数介于3~10 nm之间, 不仅可以制备单分散金属纳米粒子、合金纳米粒子, 还可以制备单分散金属氧化物[32]与金属硫族化合物纳米粒子等[69, 70].此外, 通过对传统Turkevich方法制备金溶胶的进一步研究发现, 生成单分散金溶胶的过程也存在消化熟化[71].
5.1 金属纳米粒子的制备
通常采用长链烷基硫醇、有机胺、有机酸以及聚合物硫醇等强配体作为消化熟化剂制备单分散金属纳米粒子, 包括Au[28, 35, 46, 68, 72]、Ag[21, 73~76]、Cu[77]、Co[47, 78]、Ca[79]、Mg[80]、In[81]、Pd[20, 82~84]及Pt、Ru、Rh[61]等.其中, 某些典型的金属纳米粒子的制备条件和结果列于表 2.其中, 2010年Bigioni等[85]发表的银纳米粒子的制备工作可以作为消化熟化方法制备金属纳米粒子的代表.首先采用DDAB使硝酸银溶解在甲苯中, 当加入硼氢化钠水溶液时, DDAB能够在甲苯/水体系中形成反胶束.因此, 硼氢化钠还原硝酸银生成的银纳米粒子被胶束保护而分散于甲苯中, 所得银纳米颗粒的多分散体系的TEM照片如图 7a, 可以发现大多数银颗粒为球状粒子, 少数为棒状和三角状粒子, 并且许多颗粒的尺寸大于10 nm.油水分离后向甲苯中加入十二烷基硫醇(DDT)置换银纳米粒子表面的DDAB, 然后加入乙醇使其沉淀, 并将沉淀洗涤去除反应物、自由DDAB和过量的硫醇; 纯化后的被DDT保护的银纳米粒子沉淀大多数能重新分散于甲苯中, TEM照片如图 7b, 颗粒尺寸明显降低, 说明室温下加入硫醇后在配体置换和洗涤过程中大尺寸银纳米颗粒已经被消化为小尺寸银颗粒(约3 nm).然后, 向银纳米粒子/甲苯分散体系中加入DDT, 在110 ℃下回流3 h, 得到尺寸为(5.6±0.4) nm均一的银纳米粒子, 如图 7c[85].此外, 以十六烷基胺作保护剂对多分散钙纳米粒子在室温下搅拌处理, 可以得到直径为2~3 nm的单分散钙纳米粒子[79]; 类似地, 在室温下处理四氢呋喃溶剂化的镁粒子的多分散体系可以获得尺寸为(2.8±0.2) nm的均一镁纳米粒子[80].
表 2
表 2 消化熟化法制备金属纳米粒子的典型实验条件和结果Table 2. Typical experimental conditions and results in the preparation of metal nanoparticles by digestive ripening下载:
导出CSV
金属M 配体L M/L物质的量比 溶剂 熟化条件 平均粒径/nm ±偏差/nm 文献 Au HDT 30 甲苯 回流3 h 7.4 — [72] Au DDT 10 甲苯 回流1.5 h 4.5 0.39 [68] Au DDT — 甲苯 回流1 h 6.0 0.4 [27] Au DDT 0.39 甲苯 回流3 h 8.7 1.5 [15] Au DDT 39 甲苯 回流3 h 6.2 0.3 [15] Ag DDT 20 tbTa 回流48 h 9.6 0.5 [75] Ag DDT 20 tbT 回流48 h 6.6 1.0 [74] Ag DDT 5.1 甲苯 110 ℃ 3 h 5.6 0.4 [85] Pd DDT 30 甲苯 回流1 h 1.8 0.13 [83] Pd DDT 0.5 tbT 192 ℃ 1h 7.55 0.73 [36] Pd DDT 30 甲苯 回流 5.8 0.43 [61] Pt DDT 30 甲苯 回流 5.4 0.46 [61] Rh DDT 30 甲苯 回流 3.8 0.35 [61] Ru DDT 30 甲苯 回流 4.0 0.37 [61] Cu DDT 5.6 甲苯 110 ℃ 4 h 5.3 0.88 [77] Co HDA 2 甲苯 室温24 h 2.0 0.5 [78] In TOPO 30 DCMb 38 ℃ 24 h 5 0.6 [81] In HDA 30 DCM 38 ℃ 24 h 9 0.6 [81] In TOPO/ HDA 30 DCM 38 ℃ 4 h 5 0.6 [81] Mg HDA 2 甲苯 室温 2.8 0.2 [80] a对异丁基甲苯; b二氯甲烷. 图 7
 图 7. 银纳米粒子在消化熟化前(a)、消化后(b)和熟化后(c)的TEM照片[85]Figure 7. TEM images of silver nanoparticles before digestive ripening (a), after digestion (b) and after ripening (c)
图 7. 银纳米粒子在消化熟化前(a)、消化后(b)和熟化后(c)的TEM照片[85]Figure 7. TEM images of silver nanoparticles before digestive ripening (a), after digestion (b) and after ripening (c)另外, 金属纳米粒子与大量的强配体作用, 可以形成金属原子与配体的配合物, 也可以形成硫醇保护的金属原子簇[86].研究发现, 将不同原子数组成的大、小原子簇进行热处理时可以得到中间尺寸的金属原子簇, 例如Au23[87]、Au25[88]、Au38[89~91]、Au40[91]、Au99[92]、Au144[88]、Au333[93]、Au~500[94]和Au530[93]等.这种现象在原子簇研究中称为“尺寸聚焦”, 其原理与消化熟化本质上是一致的.
5.2 金属纳米粒子的合金化
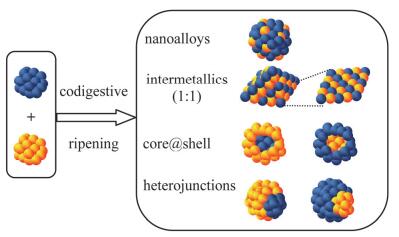
消化熟化方法可以制备各种各样的双金属纳米粒子[95]:将两种金属纳米粒子置于同一体系中, 在一定温度下进行共消化熟化, 通过金属与配体的相互作用形成配合物在反应体系中的扩散作用, 实现金属原子在不同颗粒之间的交换[30], 从而实现合金化.如图 8所示, 通过控制合适的反应条件和两种金属粒子的加入顺序和比例, 可以得到纳米复合物、均质合金、金属间化合物、核壳结构和异质结等[95].
图 8
 图 8. 两种金属纳米粒子共消化熟化制备合金纳米粒子的结构示意图Figure 8. Schematic illustration for alloy nanostructures by codigestive ripening of two metal nanoparticles
图 8. 两种金属纳米粒子共消化熟化制备合金纳米粒子的结构示意图Figure 8. Schematic illustration for alloy nanostructures by codigestive ripening of two metal nanoparticles纳米合金与结构规整的金属间化合物的制备相对简单, 其共消化熟化的结果受热力学控制.通过金属与配体的配合物的扩散, 实现不同颗粒之间的金属原子交换[30], 这种方法制备的纳米合金如Au-Pd[96, 97]、Au-Ag[19, 98]、Cu-Mg[99]、Cu-Pd[100]、Cu-Zn[101]和磁性FePt纳米粒子等[102, 103], 金属间化合物包括Au-Cu[104, 105]、Au-Sn[106]、AgIn3和AuIn2[107]等.
Jagirdar等[108]将合成的多分散金和银纳米粒子进行共消化熟化, 通过改变配体浓度、两种金属的物质的量比和热处理温度等实验条件, 制备了外壳厚度可调的Au@Ag纳米核壳结构, 并发现这种核壳结构经过紫外线照射后形成均质纳米合金. Parkin等[109]报道了一步法制备Au@Ag和Ag@Au核壳型合金纳米粒子, 且内核和外壳均可以使用不同成分的Au-Ag合金代替. Tracy等[110]向消化熟化后的8 nm金纳米粒子的DDT-甲苯体系中加入醋酸银继续加热, 通过银在金纳米粒子表面的沉积制备了Au@Ag核壳型纳米粒子, 这种粒子经过迅速高温退火也形成Au-Ag均质纳米合金.此外, Lee等[111]将经过高温消化熟化制备的铜纳米粒子加入含有银前驱物的油胺溶液中在80~150 ℃下继续加热, 得到Cu@Ag核壳型纳米粒子.
杨云等[18]结合消化熟化方法和种子生长法合成了系列核壳型双金属纳米粒子(Ag@Pd、Pd@Au、Pd@Ag、Au@Pd、Au@Ag、Pt@Au和Pt@Pd), 其中颗粒尺寸、壳层厚度和元素组成可以通过调节前驱物与种子比例控制; 如果延长消化熟化时间, 这些核壳型纳米粒子则转变为合金.此外, Jagirdar等[101]将核壳型Cu@Zn纳米粒子在对异丁基甲苯中加热回流时, 发现铜原子从内部逐渐扩散到表面聚集, 再进行配体消化后可以形成两种金属的异质结.
5.3 量子点合成
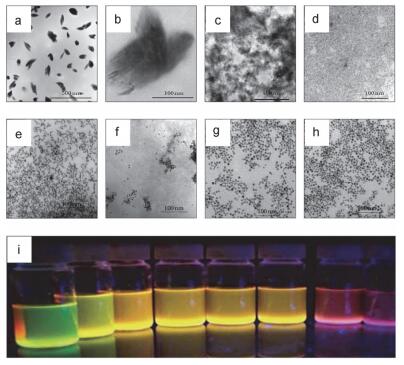
采用长链烷基硫醇、有机胺、有机酸等作为消化熟化剂, 可以制备单分散的金属氧化物或金属硫族化合物量子点体系[112].例如, Thomas等使用三乙醇胺、二乙醇胺和乙醇胺作为消化熟化剂在室温下即获得尺寸很小的ZnO量子点[32, 113]和CuO量子点[114]. Klabunde等合成了CdSe[69]、CdSe/ZnS和CdTe/ZnS等荧光量子点[70]和In2S3的半导体纳米晶[115]. Green等[116]用半胱氨酸对合成的CdTe进行消化熟化获得了橙色或绿色荧光的量子点, Sapra等[117]使用巯基丙酸作为消化熟化剂处理CdTe量子点, 其荧光发射峰的半峰宽变窄, 证明尺寸更为均一. Klabunde等[70]利用溶剂化金属原子沉积技术先将CdTe晶体块蒸发沉积形成四氢呋喃-三正辛基氧膦-油胺分散体系(TEM照片如图 9, a, b), 然后去除四氢呋喃溶剂, 在三正辛基氧膦-油胺体系中250 ℃下进行消化熟化.随着时间的延长, CdTe的大尺寸聚集体先破碎为小的粒子(图 9, c, d), 然后尺寸再增大, 形成均一的分散体系(图 9, e~h)[70].这些消化熟化时间不同的样品, 不仅颗粒尺寸不同, 而且具有不同的荧光性能(图 9, i).此外, Bossmann等[118]向溶剂化气相原子沉积技术制备的CdSe分散体系中加入联吡啶配体作为消化熟化剂直接合成了水性的CdSe量子点.
图 9
 图 9. CdTe在消化熟化前(a, b)和消化熟化时间分别为10 (c)、20 (d)、30 (e)、40 (f)、50 (g)和60 (h) min时样品TEM照片和样品紫外灯照射下的荧光照片(i)[70]Figure 9. TEM images of CdTe quantum dots before (a, b) and after digestive ripening for 10 min (c), 20 min (d), 30 min (e), 40 min (f), 50 min (g) and 60 min (h) and their corresponding optical fluorescence photo under UV illuminator (i)
图 9. CdTe在消化熟化前(a, b)和消化熟化时间分别为10 (c)、20 (d)、30 (e)、40 (f)、50 (g)和60 (h) min时样品TEM照片和样品紫外灯照射下的荧光照片(i)[70]Figure 9. TEM images of CdTe quantum dots before (a, b) and after digestive ripening for 10 min (c), 20 min (d), 30 min (e), 40 min (f), 50 min (g) and 60 min (h) and their corresponding optical fluorescence photo under UV illuminator (i)5.4 其他纳米粒子合成
Jagirdar等[119, 120]利用消化熟化法合成了Cu和Zn等金属单分散纳米粒子, 然后经过共消化熟化得到了Cu@ZnO的核壳结构; Yang等[121]把溶剂热法合成的多分散FeS2纳米晶(7.4±1.4 nm)经过消化熟化后获得了FeS2纳米晶单分散体系(5.5±0.3 nm); Viswanatha等[33]研究发现, 在油酸和油胺存在下, 经200 ℃热处理得到大尺寸的FeCoS2纳米片, 然后再升温到300 ℃, 可以转变为9 nm的高度单分散的球状纳米晶.
Kim等[122]在相同反应条件下利用油酸作消化熟化剂, 在油胺溶剂中成功制备了直径在5~8 nm之间、厚度0.8~1.4 nm的九个镧系氧化物的纳米片单分散体系(如图 10), 并且发现Co和Cd等金属离子的存在能提高纳米片的尺寸均一性. Ogale课题组[123]使用柠檬酸作为消化熟化剂处理几百纳米的Bi2O3、BiFeO3、Fe3O4和Fe2O3等金属氧化物颗粒, 获得了尺寸约5~10 nm磁性单分散纳米粒子. Jagirdar等[124]采用消化熟化法制备的金纳米粒子和CdS纳米粒子, 再经过共消化熟化可以得到Au/CdS的纳米复合物.
图 10
 图 10. 油酸/甲苯体系中消化熟化制备的镧系氧化物La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h)和Yb2O3 (i)纳米片的TEM照片[122].标尺: 20 nmFigure 10. TEM images of La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h) and Yb2O3 (i) nanodisks prepared by digestive ripening in oleic acid/toluene system. Scale bar: 20 nm
图 10. 油酸/甲苯体系中消化熟化制备的镧系氧化物La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h)和Yb2O3 (i)纳米片的TEM照片[122].标尺: 20 nmFigure 10. TEM images of La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h) and Yb2O3 (i) nanodisks prepared by digestive ripening in oleic acid/toluene system. Scale bar: 20 nm6. 总结与应用展望
消化熟化是一种制备单分散纳米粒子的高效方法, 经过近20年的研究, 其机理基本得到验证, 配体的种类、结构和用量、热处理温度和时间等诸多因素对消化熟化的影响研究也基本明确.无论是金属、合金还是金属氧化物、硫化物, 或者其他化合物, 均可以采用消化熟化方法制备, 所得纳米粒子多数小于10 nm, 粒子的尺寸分布很窄.但是, 除了高传博等[21]报道的氯化钠和二乙胺组成的双消化熟化剂体系之外, 主要的代表性的研究工作仍然局限于有机硫醇、有机胺等消化熟化体系, 因此亟待进一步拓展和建立新的消化熟化体系.同时, 尽管人们认为配体可以携带原子、原子簇或配合物离开颗粒表面进入溶剂体系, 然后沉积到小颗粒表面, 但是这种机理尚缺乏直接的证据, 因此还有待于对消化熟化的微观机理特别是熟化阶段机理进行深入研究.此外, 基于Oswald熟化的特点, 利用消化熟化-Oswald熟化两种方式结合实现单分散、大尺寸的纳米粒子的制备也值得去拓展.从纳米粒子的应用方面来讲, 采用消化熟化法制备的这种高度单分散的纳米粒子在纳米组装和多相催化等领域具有明显的优势和广阔的前景.
6.1 纳米组装
利用消化熟化法可以制备超窄尺寸分布的纳米粒子, 相对尺寸偏差可小于±5%.因此, 利用这种尺寸均一的纳米粒子进行组装时易于得到规整性好的二维膜材料或三维超晶格材料, 并且组装成功率高.目前报道的组装方法涵盖了气液界面[125, 126]、液液界面[127]和溶剂辅助的气固界面[128, 129]等二维膜构筑和溶液分散体系中三维超晶格的构筑[57, 68, 130~133].通过消化熟化法制备的硫醇保护的金纳米粒子用于构筑Langmuir单层膜[125, 126]和无支撑薄膜[134, 135], 其杨氏模量可达10 GPa数量级, 纳米粒子表面的配体之间的相互作用和配体与金属核心之间的相互作用都对膜的机械性能有很大影响[136, 137].除了硫醇保护的金属纳米粒子超晶格结构研究较多外[132], 金纳米粒子与Fe3O4纳米粒子的共自组装还可以形成AB6型的二元多态纳米晶超晶格结构[131]; 不同尺寸比例的PbTe和Ag2Te也可以组装为二元多态纳米晶超晶格薄膜[130].
重要的是, 这些纳米粒子组装体具有特殊的光学、磁性和电子学性质, 其结构规整性带来了稳定的性能, 应用前景广阔.目前, 采用溶液沉积、图案化装配、金属融合、半导体传递和绝缘纳米晶油墨等的可控制组装已经使得电磁原器件的全纳米晶制造成为现实[138, 139].因此, 研究纳米颗粒之间的相互作用, 掌握纳米颗粒组装的基本规律, 发展表面化学新技术, 设计制备具有预先设定功能和自组装指令的纳米颗粒, 然后通过组装环境的微小变化来驱动和调控自组装过程, 依靠“自下而上”的策略组装出新一代功能材料[140, 141].
6.2 多相纳米催化
金属和合金纳米粒子在多相催化领域有重要的应用地位[142~144].消化熟化法制备的小尺寸单分散纳米催化剂具有高催化效率和高选择性.这些纳米催化剂包括金属催化剂[73, 83]、合金催化剂、电催化剂[20, 100, 145]、负载催化剂[146]和半导体催化剂等.一般情况下, 消化熟化法得到的纳米粒子的尺寸均小于10 nm, 很多情况下可以小于2 nm[32, 61, 83], 这种小尺寸纳米粒子作为催化剂时具有更高的原子利用率.尽管纳米粒子在消化熟化过程中倾向于形成球状表面, 会导致高催化活性位点数目的减少, 但是这种尺寸均一、外形均一的催化剂中催化位点的活性趋于一致, 具有更高的催化选择性.另一方面, 消化熟化法使得双金属催化剂的制备和两金属的合金化更容易实现[100], 不仅可以控制双金属催化剂的结构, 而且可以控制两种金属在颗粒表面和内部的分布[110], 从而通过合金化调变催化剂的活性和选择性, 为开发新的非贵金属合金催化剂提供了方便.
-
-
[1]
Park, J.; Joo, J.; Kwon, S. G.; Jang, Y.; Hyeon, T. Angew. Chem., Int. Ed. 2007, 46, 4630. doi: 10.1002/(ISSN)1521-3773
-
[2]
严亚, 李津如, 杨云, 化学进展, 2009, 21, 971. http://www.cqvip.com/Main/Detail.aspx?id=30293476Yan, Y.; Li, J.; Yang, Y. Prog. Chem. 2009, 21, 971. http://www.cqvip.com/Main/Detail.aspx?id=30293476
-
[3]
Shi, R.; Gao, G.; Yi, R.; Zhou, K.; Qiu, G.; Liu, X. Chin. J. Chem. 2009, 27, 739. doi: 10.1002/cjoc.v27:4
-
[4]
Ji, X. H.; Song, X. N.; Li, J.; Bai, Y. B.; Yang, W. S.; Peng, X. G. J. Am. Chem. Soc. 2007, 129, 13939. doi: 10.1021/ja074447k
-
[5]
Li, H. S.; Xia, H. B.; Wang, D. Y.; Tao, X. T. Langmuir 2013, 29, 5074. doi: 10.1021/la400214x
-
[6]
Fu, Y.; Du, Y.; Yang, P.; Li, J.; Jiang, L. Sci. China B:Chem. 2007, 50, 494. doi: 10.1007/s11426-007-0085-x
-
[7]
Liu, N.; Wang, K.; Gao, Y. Y.; Li, D. X.; Lin, W. H.; Li, C. F. Colloid. Surf. A:Physicochem. Engin. Asp. 2017, 535, 251. doi: 10.1016/j.colsurfa.2017.09.017
-
[8]
Li, C. F.; Li, D. X.; Wan, G. Q.; Xu, J.; Hou, W. G. Nanoscale Res. Lett. 2011, 6, 440. doi: 10.1186/1556-276X-6-440
-
[9]
Zaiser, E. M.; LaMer, V. K. J. Colloid Sci. 1948, 3, 571. doi: 10.1016/S0095-8522(48)90050-6
-
[10]
LaMer, V. K.; Dinegar, R. H. J. Am. Chem. Soc. 1950, 72, 4847. doi: 10.1021/ja01167a001
-
[11]
Zhang, P. N.; Li, Y. J.; Wang, D. Y.; Xia, H. B. Particle Particle Sys. Character. 2016, 33, 924. doi: 10.1002/ppsc.v33.12
-
[12]
Li, D. X.; Jang, Y. J.; Lee, J.; Lee, J. E.; Kochuveedu, S. T.; Kim, D. H. J. Mater. Chem. 2011, 21, 16453. doi: 10.1039/c1jm13302k
-
[13]
Ostwald, W. Zeitsch. Phys. Chem. 1897, 22, 289.
-
[14]
Zhang, Z.; Wang, Z.; He, S.; Wang, C.; Jin, M.; Yin, Y. Chem. Sci. 2015, 6, 5197. doi: 10.1039/C5SC01787D
-
[15]
Lin, X. M.; Sorensen, C. M.; Klabunde, K. J. J. Nanopart. Res. 2000, 2, 157. doi: 10.1023/A:1010078521951
-
[16]
Shimpi, J. R.; Sidhaye, D. S.; Prasad, B. L. V. Langmuir 2017, 33, 9491. doi: 10.1021/acs.langmuir.7b00193
-
[17]
Sidhaye, D. S.; Prasad, B. L. V. New J. Chem. 2011, 35, 755. doi: 10.1039/C0NJ00359J
-
[18]
Yang, Y.; Gong, X.; Zeng, H.; Zhang, L.; Zhang, X.; Zou, C.; Huang, S. J. Phys. Chem. C 2010, 114, 256. doi: 10.1021/jp909065y
-
[19]
Ji, Y.; Yang, S.; Guo, S.; Song, X.; Ding, B.; Yang, Z. Colloid. Surf. A:Physicochem. Engin. Asp. 2010, 372, 204. doi: 10.1016/j.colsurfa.2010.10.028
-
[20]
Liu, S. L.; Han, M.; Shi, Y.; Zhang, C. Z.; Chen, Y.; Bao, J. C.; Dai, Z. H. Europ. J. Inorg. Chem. 2012, 3740.
-
[21]
Zhang, S.; Zhang, L.; Liu, K.; Liu, M.; Yin, Y.; Gao, C. Mater. Chem. Front. 2018, 2, 1328. doi: 10.1039/C8QM00077H
-
[22]
Wang, P.; Qi, X.; Zhang, X.; Wang, T.; Li, Y.; Zhang, K.; Zhao, S.; Zhou, J.; Fu, Y. Nanoscale Res. Lett. 2017, 12, 25. doi: 10.1186/s11671-016-1797-7
-
[23]
Cardenas-Trivino, G.; Klabunde, K. J.; Dale, E. B. Langmuir 1987, 3, 986. doi: 10.1021/la00078a019
-
[24]
Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D. J.; Whyman, R. J. Chem. Soc., Chem. Commun. 1994, 801.
-
[25]
Leff, D. V.; Ohara, P. C.; Heath, J. R.; Gelbart, W. M. J. Phys. Chem. 1995, 99, 7036. doi: 10.1021/j100018a041
-
[26]
Lin, X. M.; Wang, G. M.; Sorensen, C. M.; Klabunde, K. J. J. Phys. Chem. B 1999, 103, 5488. doi: 10.1021/jp990729y
-
[27]
Lin, X. M.; Sorensen, C. M.; Klabunde, K. J. Chem. Mater. 1999, 11, 198. doi: 10.1021/cm980665o
-
[28]
Bhaskar, S. P.; Vijayan, M.; Jagirdar, B. R. J. Phys. Chem. C 2014, 118, 18214. doi: 10.1021/jp505121b
-
[29]
Dreier, T. A.; Ackerson, C. J. Angew. Chem., Int. Ed. 2015, 54, 9249. doi: 10.1002/anie.201502934
-
[30]
Samia, A. C. S.; Schlueter, J. A.; Jiang, J. S.; Bader, S. D.; Qin, C.-J.; Lin, X.-M. Chem. Mater. 2006, 18, 5203. doi: 10.1021/cm0610579
-
[31]
Stoeva, S. I.; Zaikovski, V.; Prasad, B. L. V.; Stoimenov, P. K.; Sorensen, C. M.; Klabunde, K. J. Langmuir 2005, 21, 10280. doi: 10.1021/la051699v
-
[32]
Mary Jacob, N.; Thomas, T. Ceramics Int. 2014, 40, 13945. doi: 10.1016/j.ceramint.2014.05.116
-
[33]
Shetty, A.; Saha, A.; Makkar, M.; Viswanatha, R. Phys. Chem. Chem. Phys. 2016, 18, 25887. doi: 10.1039/C6CP04912E
-
[34]
Talluri, B.; Thomas, T. Chem. Phys. Lett 2017, 685, 84. doi: 10.1016/j.cplett.2017.07.041
-
[35]
Lin, M.-L.; Yang, F.; Lee, S. Colloid. Surf. A:Physicochem. Engin. Asp. 2014, 448, 16. doi: 10.1016/j.colsurfa.2014.01.081
-
[36]
Yang, Z.; Klabunde, K. J.; Sorensen, C. M. J. Phys. Chem. C 2007, 111, 18143. doi: 10.1021/jp0765751
-
[37]
Sidhaye, D. S.; Prasad, B. L. V. Chem. Mater. 2010, 22, 1680. doi: 10.1021/cm9031607
-
[38]
Prasad, B. L. V.; Stoeva, S. I.; Sorensen, C. M.; Klabunde, K. J. Langmuir 2002, 18, 7515. doi: 10.1021/la020181d
-
[39]
Sahu, P.; Shimpi, J.; Lee, H. J.; Lee, T. R.; Prasad, B. L. V. Langmuir 2017, 33, 1943. doi: 10.1021/acs.langmuir.6b03998
-
[40]
Silvestri, A.; Polito, L.; Bellani, G.; Zambelli, V.; Jumde, R. P.; Psaro, R.; Evangelisti, C. J. Colloid Interf. Sci. 2015, 439, 28. doi: 10.1016/j.jcis.2014.10.025
-
[41]
Naoe, K.; Petit, C.; Pileni, M. P. J. Phys. Chem. C 2007, 111, 16249. doi: 10.1021/jp073957y
-
[42]
Naoe, K.; Petit, C.; Pileni, M. P. Langmuir 2008, 24, 2792. doi: 10.1021/la702908y
-
[43]
Sun, Y. J.; Jose, D.; Sorensen, C.; Klabunde, K. J. Nanomaterials 2013, 3, 370. doi: 10.3390/nano3030370
-
[44]
Sahu, P.; Prasad, B. L. V. Chem. Phys. Lett 2012, 525~526, 101.
-
[45]
Mohrhusen, L.; Osmić, M. RSC Adv. 2017, 7, 12897. doi: 10.1039/C6RA27454D
-
[46]
Prasad, B. L. V.; Stoeva, S. I.; Sorensen, C. M.; Klabunde, K. J. Chem. Mater. 2003, 15, 935. doi: 10.1021/cm0206439
-
[47]
Samia, A. C. S.; Hyzer, K.; Schlueter, J. A.; Qin, C.-J.; Jiang, J. S.; Bader, S. D.; Lin, X.-M. J. Am. Chem. Soc. 2005, 127, 4126. doi: 10.1021/ja044419r
-
[48]
Kholmicheva, N.; Yang, M. R.; Moroz, P.; Eckard, H.; Vore, A.; Cassidy, J.; Pushina, M.; Boddy, A.; Porotnikov, D.; Anzenbacher, P.; Zamkov, M. J. Phys. Chem. C 2018, 122, 23623. doi: 10.1021/acs.jpcc.8b09215
-
[49]
Griffin, F.; Fitzmaurice, D. Langmuir 2007, 23, 10262. doi: 10.1021/la061261a
-
[50]
Su, Y. Y.; Yang, F. Q.; Lee, S. Mater. Res. Exp. 2015, 2, 055007. doi: 10.1088/2053-1591/2/5/055007
-
[51]
Sahu, P.; Prasad, B. L. V. Nanoscale 2013, 5, 1768. doi: 10.1039/C2NR32855K
-
[52]
Sahu, P.; Prasad, B. L. V. Langmuir 2014, 30, 10143. doi: 10.1021/la500914j
-
[53]
Destro, P.; Colombo, M.; Prato, M.; Brescia, R.; Manna, L.; Zanchet, D. RSC Adv. 2016, 6, 22213. doi: 10.1039/C6RA02027E
-
[54]
Lin, M. L.; Yang, F. Q.; Peng, J. S.; Lee, S. J. Appl. Phys. 2014, 115, 054312. doi: 10.1063/1.4863784
-
[55]
Razgoniaeva, N.; Yang, M.; Garrett, P.; Kholmicheva, N.; Moroz, P.; Eckard, H.; Royo Romero, L.; Porotnikov, D.; Khon, D.; Zamkov, M. Chem. Mater. 2018, 30, 1391. doi: 10.1021/acs.chemmater.7b05165
-
[56]
Shimpi, J. R.; Chaudhari, V. R.; Prasad, B. L. V. Langmuir 2018, 34, 13680. doi: 10.1021/acs.langmuir.8b02699
-
[57]
Powell, J. A.; Schwieters, R. M.; Bayliff, K. W.; Herman, E. N.; Hotvedt, N. J.; Changstrom, J. R.; Chakrabarti, A.; Sorensen, C. M. RSC Adv. 2016, 6, 70638. doi: 10.1039/C6RA15822F
-
[58]
Lohman, B. C.; Powell, J. A.; Cingarapu, S.; Aakeroy, C. B.; Chakrabarti, A.; Klabunde, K. J.; Law, B. M.; Sorensen, C. M. Phys. Chem. Chem. Phys. 2012, 14, 6509. doi: 10.1039/c2cp40645d
-
[59]
Talluri, B.; Prasad, E.; Thomas, T. J. Mol. Liq. 2018, 265, 771. doi: 10.1016/j.molliq.2018.05.069
-
[60]
Talluri, B.; Prasad, E.; Thomas, T. Superlatt. Microstruct. 2018, 113, 600. doi: 10.1016/j.spmi.2017.11.044
-
[61]
Seth, J.; Prasad, B. L. V. Nano Res. 2016, 9, 2007. doi: 10.1007/s12274-016-1091-0
-
[62]
Lee, D.-K.; Park, S.-I.; Lee, J. K.; Hwang, N.-M. Acta Mater. 2007, 55, 5281. doi: 10.1016/j.actamat.2007.05.048
-
[63]
Lee, D. K.; Hwang, N. M. Scripta Mater. 2009, 61, 304. doi: 10.1016/j.scriptamat.2009.04.008
-
[64]
Clark, M. D. J. Nanopart. Res. 2014, 16, 2264. doi: 10.1007/s11051-014-2264-y
-
[65]
Irzhak, V. I. Russ. J. Phys. Chem. A 2017, 91, 1503.
-
[66]
Thomas, T.; Sethuraman, S.; Satyam, D.; Kumar, D.; Kannadasan, B.; Anderson, A.; Prashant, S.; Vijayakrishnan, R.; Khan, S.; Das, P.; Kumar, M.; Bisi, K.; Chinta, Y.; Talluri, B. Appl. Surf. Sci. 2018, 448, 248. doi: 10.1016/j.apsusc.2018.04.134
-
[67]
Manzanares, J. A.; Peljo, P.; Girault, H. H. J. Phys. Chem. C 2017, 121, 13405. doi: 10.1021/acs.jpcc.7b04234
-
[68]
Stoeva, S.; Klabunde, K. J.; Sorensen, C. M.; Dragieva, I. J. Am. Chem. Soc. 2002, 124, 2305. doi: 10.1021/ja012076g
-
[69]
Cingarapu, S.; Yang, Z.; Sorensen, C. M.; Klabunde, K. J. Chem. Mater. 2009, 21, 1248. doi: 10.1021/cm802831m
-
[70]
Cingarapu, S.; Yang, Z.; Sorensen, C. M.; Klabunde, K. J. J. Nanomater. 2012, 2012, 312087.
-
[71]
Uppal, M. A.; Kafizas, A.; Lim, T. H.; Parkin, I. P. New J. Chem. 2010, 34, 1401. doi: 10.1039/b9nj00745h
-
[72]
Sidhaye, D. S.; Prasad, B. L. V. Chem. Phys. Lett 2008, 454, 345. doi: 10.1016/j.cplett.2008.02.053
-
[73]
Han, M.; Liu, S. L.; Nie, X. P.; Yuan, D.; Sun, P. P.; Dai, Z. H.; Bao, J. C. RSC Adv. 2012, 2, 6061. doi: 10.1039/c2ra20119d
-
[74]
Smetana, A. B.; Klabunde, K. J.; Sorensen, C. M. J. Colloid Interf. Sci. 2005, 284, 521. doi: 10.1016/j.jcis.2004.10.038
-
[75]
Zhang, Q.; Xie, J.; Yang, J.; Lee, J. Y. ACS Nano 2009, 3, 139. doi: 10.1021/nn800531q
-
[76]
Li, P.; Peng, Q.; Li, Y. Chem. Eur. J. 2011, 17, 941. doi: 10.1002/chem.v17.3
-
[77]
Shaik, A. H.; Chakraborty, J. RSC Adv. 2015, 5, 85974. doi: 10.1039/C5RA16508C
-
[78]
Arora, N.; Jagirdar, B. R. J. Mater. Chem. 2012, 22, 20671. doi: 10.1039/c2jm33712f
-
[79]
Sanyal, U.; Datta, R.; Jagirdar, B. R. RSC Adv. 2012, 2, 259. doi: 10.1039/C1RA00468A
-
[80]
Kalidindi, S. B.; Jagirdar, B. R. Inorg. Chem. 2009, 48, 4524. doi: 10.1021/ic9003577
-
[81]
Cingarapu, S.; Yang, Z.; Sorensen, C. M.; Klabunde, K. J. Inorg. Chem. 2011, 50, 5000. doi: 10.1021/ic200232b
-
[82]
Naoe, K.; Kataoka, M.; Kawagoe, M. Colloid. Surf. A:Physicochem. Engin. Asp. 2010, 364, 116. doi: 10.1016/j.colsurfa.2010.05.004
-
[83]
Seth, J.; Kona, C. N.; Das, S.; Prasad, B. L. V. Nanoscale 2015, 7, 872. doi: 10.1039/C4NR04239E
-
[84]
Jose, D.; Jagirdar, B. R. J. Solid State Chem. 2010, 183, 2059. doi: 10.1016/j.jssc.2010.07.013
-
[85]
Shankar, R.; Wu, B. B.; Bigioni, T. P. J. Phys. Chem. C 2010, 114, 15916. doi: 10.1021/jp911316e
-
[86]
Jose, D.; Matthiesen, J. E.; Parsons, C.; Sorensen, C. M.; Klabunde, K. J. J. Phys. Chem. Lett. 2012, 3, 885. doi: 10.1021/jz201640e
-
[87]
Muhammed, M. A. H.; Verma, P. K.; Pal, S. K.; Kumar, R. C. A.; Paul, S.; Omkumar, R. V.; Pradeep, T. Chem. Eur. J. 2009, 15, 10110. doi: 10.1002/chem.v15:39
-
[88]
Qian, H.; Jin, R. Chem. Mater. 2011, 23, 2209. doi: 10.1021/cm200143s
-
[89]
Qian, H.; Zhu, Y.; Jin, R. ACS Nano 2009, 3, 3795. doi: 10.1021/nn901137h
-
[90]
Qian, H. Pure Appl. Chem. 2014, 86, 27. doi: 10.1515/pac-2014-5011
-
[91]
Nimmala, P. R.; Jupally, V. R.; Dass, A. Langmuir 2014, 30, 2490. doi: 10.1021/la404618r
-
[92]
Nimmala, P. R.; Dass, A. J. Am. Chem. Soc. 2014, 136, 17016. doi: 10.1021/ja5103025
-
[93]
Qian, H.; Zhu, Y.; Jin, R. Proc. Nat. Acad. Sci. 2012, 109, 696. doi: 10.1073/pnas.1115307109
-
[94]
Kumara, C.; Zuo, X.; Ilavsky, J.; Chapman, K. W.; Cullen, D. A.; Dass, A. J. Am. Chem. Soc. 2014, 136, 7410. doi: 10.1021/ja502327a
-
[95]
Bhattacharya, C.; Arora, N.; Jagirdar, B. R. Langmuir 2019, ASAP, doi: 10.1021/acs.langmuir.1028b02208.
-
[96]
Jose, D.; Jagirdar, B. R. J. Phys. Chem. C 2008, 112, 10089. doi: 10.1021/jp802721s
-
[97]
Liu, F.-K.; Chang, Y.-C. Chromatographia 2011, 74, 767. doi: 10.1007/s10337-011-2139-7
-
[98]
Zhang, Q. B.; Xie, J. P.; Liang, J.; Lee, J. Y. Adv. Funct. Mater. 2009, 19, 1387. doi: 10.1002/adfm.v19:9
-
[99]
Kalidindi, S. B.; Jagirdar, B. R. Chem. Asian J. 2009, 4, 835. doi: 10.1002/asia.v4:6
-
[100]
Chen, D.; Xu, L.; Liu, H.; Yang, J. Green Energy & Environment 2019, doi: 10.1016/j.gee.2018.1009.1002.
-
[101]
Bhaskar, S. P.; Jagirdar, B. R. J. All. Comp. 2017, 694, 581. doi: 10.1016/j.jallcom.2016.09.318
-
[102]
Heller, H.; Ahrenstorf, K.; Broekaert, J. A. C.; Weller, H. Phys. Chem. Chem. Phys. 2009, 11, 3257. doi: 10.1039/b822306h
-
[103]
Chokprasombat, K.; Koyvanich, K.; Sirisathitkul, C.; Harding, P.; Rugmai, S. Trans. Indian Inst. Metal. 2016, 69, 733. doi: 10.1007/s12666-015-0545-5
-
[104]
Smetana, A. B.; Klabunde, K. J.; Sorensen, C. M.; Ponce, A. A.; Mwale, B. J. Phys. Chem. B 2006, 110, 2155. doi: 10.1021/jp0539932
-
[105]
Destro, P.; Cantaneo, D. A.; Meira, D. M.; Honorio, G. D.; da Costa, L. S.; Bueno, J. M. C.; Zanchet, D. Europ. J. Inorg. Chem. 2018, 3770.
-
[106]
Arora, N.; Jagirdar, B. R. Phys. Chem. Chem. Phys. 2014, 16, 11381. doi: 10.1039/C4CP00249K
-
[107]
Arora, N.; Jagirdar, B. R.; Klabunde, K. J. J. All. Comp. 2014, 610, 35. doi: 10.1016/j.jallcom.2014.04.190
-
[108]
Bhattacharya, C.; Jagirdar, B. R. J. Phys. Chem. C 2018, 122, 10559. doi: 10.1021/acs.jpcc.8b00874
-
[109]
Uppal, M. A.; Ewing, M. B.; Parkin, I. P. Eur. J. Inorg. Chem. 2011, 2011, 4534. doi: 10.1002/ejic.v2011.29
-
[110]
Shore, M. S.; Wang, J.; Johnston-Peck, A. C.; Oldenburg, A. L.; Tracy, J. B. Small 2011, 7, 230. doi: 10.1002/smll.v7.2
-
[111]
Chee, S. S.; Lee, J. H. J. Mater. Chem. C 2014, 2, 5372. doi: 10.1039/C4TC00509K
-
[112]
Heroux, D.; Ponce, A.; Cingarapu, S.; Klabunde, K. J. Adv. Funct. Mater. 2007, 17, 3562. doi: 10.1002/adfm.v17:17
-
[113]
Jacob, N. M.; Thomas, T. RSC Adv. 2015, 5, 15154. doi: 10.1039/C4RA05778C
-
[114]
Talluri, B.; Prasad, E.; Thomas, T. Superlatt. Microstruct. 2018, 116, 122. doi: 10.1016/j.spmi.2018.02.010
-
[115]
Cingarapu, S.; Ikenberry, M. A.; Hamal, D. B.; Sorensen, C. M.; Hohn, K.; Klabunde, K. J. Langmuir 2012, 28, 3569. doi: 10.1021/la203624p
-
[116]
Green, M.; Harwood, H.; Barrowman, C.; Rahman, P.; Eggeman, A.; Festry, F.; Dobson, P.; Ng, T. J. Mater. Chem. 2007, 17, 1989 doi: 10.1039/b615871d
-
[117]
Mittal, M.; Sapra, S. Pramana-J. Phys. 2015, 84, 1049. doi: 10.1007/s12043-015-1007-7
-
[118]
Kalita, M.; Cingarapu, S.; Roy, S.; Park, S. C.; Higgins, D.; Jankowiak, R.; Chikan, V.; Klabunde, K. J.; Bossmann, S. H. Inorg. Chem. 2012, 51, 4521. doi: 10.1021/ic202252m
-
[119]
Bhaskar, S. P.; Jagirdar, B. R. J. Chem. Sci. 2012, 124, 1175. doi: 10.1007/s12039-012-0317-2
-
[120]
Kalidindi, S. B.; Jagirdar, B. R. J. Phys. Chem. C 2008, 112, 4042. doi: 10.1021/jp7100896
-
[121]
Yoder, T. S.; Cloud, J. E.; Leong, G. J.; Molk, D. F.; Tussing, M.; Miorelli, J.; Ngo, C.; Kodambaka, S.; Eberhart, M. E.; Richards, R. M.; Yang, Y. Chem. Mater. 2014, 26, 6743. doi: 10.1021/cm5030553
-
[122]
Jeong, J.; Kim, N.; Kim, M. G.; Kim, W. Chem. Mater. 2016, 28, 172. doi: 10.1021/acs.chemmater.5b03616
-
[123]
Shaikh, P. A.; Banerjee, A.; Game, O.; Kolekar, Y.; Kale, S.; Ogale, S. Phys. Chem. Chem. Phys. 2013, 15, 5091. doi: 10.1039/c3cp43425g
-
[124]
Bhaskar, S. P.; Karthika, M. S.; Jagirdar, B. R. Chemistryselect 2018, 3, 6638. doi: 10.1002/slct.201801157
-
[125]
Schultz, D. G.; Lin, X.-M.; Li, D.; Gebhardt, J.; Meron, M.; Viccaro, J.; Lin, B. J. Phys. Chem. B 2006, 110, 24522. doi: 10.1021/jp063820s
-
[126]
Griesemer, S. D.; You, S. S.; Kanjanaboos, P.; Calabro, M.; Jaeger, H. M.; Rice, S. A.; Lin, B. Soft Matter 2017, 13, 3125. doi: 10.1039/C7SM00319F
-
[127]
Shaik, A. H.; Reddy, D. S. Mater. Res. Exp. 2017, 4, 035043. doi: 10.1088/2053-1591/aa5e5b
-
[128]
He, J.; Lin, X.-M.; Divan, R.; Jaeger, H. M. Small 2011, 7, 3487. doi: 10.1002/smll.v7.24
-
[129]
He, J.; Lin, X.-M.; Chan, H.; Vuković, L.; Král, P.; Jaeger, H. M. Nano Lett. 2011, 11, 2430. doi: 10.1021/nl200841a
-
[130]
Urban, J. J.; Talapin, D. V.; Shevchenko, E. V.; Kagan, C. R.; Murray, C. B. Nat. Mater. 2007, 6, 115. doi: 10.1038/nmat1826
-
[131]
Ye, X.; Chen, J.; Murray, C. B. J. Am. Chem. Soc. 2011, 133, 2613. doi: 10.1021/ja108708v
-
[132]
García-Barrasa, J.; López-de-Luzuriaga, J. M.; Monge, M.; Soulantica, K.; Viau, G. J. Nanopart. Res. 2011, 13, 791. doi: 10.1007/s11051-010-0079-z
-
[133]
Stoeva, S. I.; Prasad, B. L. V.; Uma, S.; Stoimenov, P. K.; Zaikovski, V.; Sorensen, C. M.; Klabunde, K. J. J. Phys. Chem. B 2003, 107, 7441. doi: 10.1021/jp030013+
-
[134]
He, J.; Kanjanaboos, P.; Frazer, N. L.; Weis, A.; Lin, X.-M.; Jaeger, H. M. Small 2010, 6, 1449. doi: 10.1002/smll.v6:13
-
[135]
Wang, Y.; Chan, H.; Narayanan, B.; McBride, S. P.; Sankaranarayanan, S. K. R. S.; Lin, X.-M.; Jaeger, H. M. ACS Nano 2017, 11, 8026. doi: 10.1021/acsnano.7b02676
-
[136]
Wang, Y.; Kanjanaboos, P.; Barry, E.; McBride, S.; Lin, X.-M.; Jaeger, H. M. Nano Lett. 2010, 14, 826.
-
[137]
Lin, X. M.; Jaeger, H. M.; Sorensen, C. M.; Klabunde, K. J. J. Phys. Chem. B 2001, 105, 3353. doi: 10.1021/jp0102062
-
[138]
Zhu, B.; Gong, S.; Cheng, W. Chem. Soc. Rev. 2019, doi: 10.1039/C1038CS00609A.
-
[139]
Kagan, C. R. Chem. Soc. Rev. 2019, doi: 10.1039/C1038CS00629F.
-
[140]
Boles, M. A.; Engel, M.; Talapin, D. V. Chem. Rev. 2016, 116, 11220. doi: 10.1021/acs.chemrev.6b00196
-
[141]
Ortega, S.; Ibáñez, M.; Liu, Y.; Zhang, Y.; Kovalenko, M. V.; Cadavid, D.; Cabot, A. Chem. Soc. Rev. 2017, 46, 3510. doi: 10.1039/C6CS00567E
-
[142]
王艺霖, 王敏杰, 李静, 魏子栋, 化学学报, 2019, 77, 84. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346896.shtmlWang, Y.; Wang, M.; Li, J.; Wei, Z. Acta Chim. Sinica 2019, 77, 84. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346896.shtml
-
[143]
Li, N. Chin. J. Chem. 2016, 34, 1129. doi: 10.1002/cjoc.v34.11
-
[144]
Cao, J.; Zhu, Z.; Zhao, W.; Xu, J.; Chen, Z. Chin. J. Chem. 2016, 34, 1086. doi: 10.1002/cjoc.v34.11
-
[145]
Scanlon, M. D.; Smirnov, E.; Stockmann, T. J.; Peljo, P. Chem. Rev. 2018, 118, 3722. doi: 10.1021/acs.chemrev.7b00595
-
[146]
Seth, J.; Dubey, P.; Chaudhari, V. R.; Prasad, B. L. V. New J. Chem. 2018, 42, 402. doi: 10.1039/C7NJ03753H
-
[1]
-

图 3 大尺寸多分散金纳米粒子与小尺寸单分散配体保护的金纳米粒子的相互转化过程TEM示意图[31]
Figure 3 Schematic illustration by TEM for reversible change from larger polyhedral nanoparticles to smaller, monodisperse ligand-stabilized spherical particles
图 4 分别采用十二烷基醇(a~c)和十二烷基胺(d~f)消化熟化后的银(a, d)、金(b, e)和钯(c, f)纳米粒子的TEM照片[44]
Figure 4 TEM images of Ag (a, d), Au (b, e) and Pd (c, f) nanoparticles after digestive ripening using dodecylthiol (a~c) and dodecylamine (d~f), respectively
图 5 消化熟化温度分别为60 ℃ (a, b)、120 ℃ (c, d)和180 ℃ (e, f)时金纳米粒子的尺寸和尺寸分布[39]
Figure 5 Size and size distribution of gold nanoparticles influenced by the temperature in digestive ripening process
图 6 金纳米粒子在十六烷基硫醇和叔丁基甲苯中120 ℃处理时间分别为1 h (a~c)、3 h (d~f)、5 h (g~i)、7 h (j~l)和9 h (m~o)的TEM照片和颗粒尺寸分布图[52]
Figure 6 TEM micrographs and particle size histograms of gold nanoparticle systems obtained at 120 ℃ for 1 h (a~c), 3 h (d~f), 5 h (g~i), 7 h (j~l), and 9 h (m~o)
图 7 银纳米粒子在消化熟化前(a)、消化后(b)和熟化后(c)的TEM照片[85]
Figure 7 TEM images of silver nanoparticles before digestive ripening (a), after digestion (b) and after ripening (c)
图 8 两种金属纳米粒子共消化熟化制备合金纳米粒子的结构示意图
Figure 8 Schematic illustration for alloy nanostructures by codigestive ripening of two metal nanoparticles
图 9 CdTe在消化熟化前(a, b)和消化熟化时间分别为10 (c)、20 (d)、30 (e)、40 (f)、50 (g)和60 (h) min时样品TEM照片和样品紫外灯照射下的荧光照片(i)[70]
Figure 9 TEM images of CdTe quantum dots before (a, b) and after digestive ripening for 10 min (c), 20 min (d), 30 min (e), 40 min (f), 50 min (g) and 60 min (h) and their corresponding optical fluorescence photo under UV illuminator (i)
图 10 油酸/甲苯体系中消化熟化制备的镧系氧化物La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h)和Yb2O3 (i)纳米片的TEM照片[122].标尺: 20 nm
Figure 10 TEM images of La2O3 (a), Eu2O3 (b), Gd2O3 (c), Tb2O3 (d), Dy2O3 (e), Ho2O3 (f), Er2O3 (g), Tm2O3 (h) and Yb2O3 (i) nanodisks prepared by digestive ripening in oleic acid/toluene system. Scale bar: 20 nm
表 1 有机胺配体对金纳米粒子尺寸和尺寸偏差的影响
Table 1. Size and size deviation of gold nanoparticles influenced by alkyl amines
 下载: 导出CSV
下载: 导出CSV
表 2 消化熟化法制备金属纳米粒子的典型实验条件和结果
Table 2. Typical experimental conditions and results in the preparation of metal nanoparticles by digestive ripening
金属M 配体L M/L物质的量比 溶剂 熟化条件 平均粒径/nm ±偏差/nm 文献 Au HDT 30 甲苯 回流3 h 7.4 — [72] Au DDT 10 甲苯 回流1.5 h 4.5 0.39 [68] Au DDT — 甲苯 回流1 h 6.0 0.4 [27] Au DDT 0.39 甲苯 回流3 h 8.7 1.5 [15] Au DDT 39 甲苯 回流3 h 6.2 0.3 [15] Ag DDT 20 tbTa 回流48 h 9.6 0.5 [75] Ag DDT 20 tbT 回流48 h 6.6 1.0 [74] Ag DDT 5.1 甲苯 110 ℃ 3 h 5.6 0.4 [85] Pd DDT 30 甲苯 回流1 h 1.8 0.13 [83] Pd DDT 0.5 tbT 192 ℃ 1h 7.55 0.73 [36] Pd DDT 30 甲苯 回流 5.8 0.43 [61] Pt DDT 30 甲苯 回流 5.4 0.46 [61] Rh DDT 30 甲苯 回流 3.8 0.35 [61] Ru DDT 30 甲苯 回流 4.0 0.37 [61] Cu DDT 5.6 甲苯 110 ℃ 4 h 5.3 0.88 [77] Co HDA 2 甲苯 室温24 h 2.0 0.5 [78] In TOPO 30 DCMb 38 ℃ 24 h 5 0.6 [81] In HDA 30 DCM 38 ℃ 24 h 9 0.6 [81] In TOPO/ HDA 30 DCM 38 ℃ 4 h 5 0.6 [81] Mg HDA 2 甲苯 室温 2.8 0.2 [80] a对异丁基甲苯; b二氯甲烷.
下载: 导出CSV
-















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 62
- 文章访问数: 3487
- HTML全文浏览量: 674
 下载:
下载:
