Received Date:
06 December 2018 Available Online:
15 March 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21673061) and the Open Project of the State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology (No. QAK201503)
Abstract:
Carbonate radical, nitrate radical, phosphate radical and sulfate radical are all important intermediates of chemical reactions with oxidizing ability. They have a significant effect on the transfer of pollutants in natural environment. In this review, the redox potential, modes of production, detection methods of these radicals and the mechanisms of their reactions with organic compounds are introduced. It can be found that:these four radicals have different reaction rates with organic compounds because of their various redox potential; Carbonate radical is not a scavenger of hydroxyl radical. For some easily oxidized compounds, carbonate radical shows higher oxidizing ability than hydroxyl radical; Hydroxyl radicals can be converted into other four types of radicals. Meanwhile, these four types of radicals react with organic matters by electron transfer, hydrogen abstraction and addition, which is basically consistent with hydroxyl radicals. It can be predicted that the mechanism of organic compounds degradation by these four types of free radicals is similar with that of hydroxyl radicals. In the future, it is necessary to study the mutual conversion principles between these free radicals and hydroxyl radicals and the degradation mechanism of these radicals when reacting with some representative organic compounds.
Stenman, D.; Carlsson, M.; Reitberger, T. J. Wood. Chem. Technol. 2005, 24, 83. doi: 10.1081/WCT-200026553
[2]
Canonica, S.; Kohn, T.; Mac, M.; Real, F. J.; Wirz, J.; Von, G. U. Environ. Sci. Technol. 2005, 39, 9182. doi: 10.1021/es051236b
[3]
Dell'Arciprete, M. L.; Soler, J. M.; Santos-Juanes, L.; Arques, A.; Mártire, D. O.; Furlong, J. P.; Gonzalez, M. C. Water Res. 2012, 46, 3479. doi: 10.1016/j.watres.2012.03.051
[4]
Medinas, D. B.; Cerchiaro, G.; Trindade, D. F.; Augusto, O. Iubmb. Life. 2007, 59, 255. doi: 10.1080/15216540701230511
Brusa, M. A.; Grela, M. A. Phys. Chem. Chem. Phys. 2003, 5, 3294. doi: 10.1039/b302296j
[47]
Criado, S.; Marioli, J. M.; Allegretti, P. E.; Furlong, J.; Nieto, F. J. R.; Mártire, D. O.; Garcia, N. A. J. Photochem. Photobiol. B 2001, 65, 74. doi: 10.1016/S1011-1344(01)00239-1
[48]
Kumar, M. R.; Adinarayana, M. J. Chem. Sci. 2000, 112, 551. doi: 10.1007/BF02709288
[49]
Kumar, M. R.; Rao, M. T.; Adinarayana, M. Indian J. Biochem. Bio. 2000, 37, 13.
[50]
Huber, J. R.; Hayon, E. J. Phys. Chem. 1968, 71, 3820.
[51]
Black, E. D.; Hayon, E. J. Phys. Chem. 1970, 74, 3199. doi: 10.1021/j100711a007
[52]
Ma, J.; Schmidhammer, U.; Mostafavi, M. J. Phys. Chem. B 2015, 119, 7180.
[53]
Caregnato, P.; Bertolotti, S. G.; Gonzalez, M. C.; Mártire, D. O. Photochem. Photobiol. 2005, 81, 1526. doi: 10.1562/2005-07-07-RA-603
Clément, J. L.; Gilbert, B. C.; Ho, W. F.; Jackson, N. D.; Newton, M. S.; Silvester, S.; Timmins, G. S.; Tordo, P.; Whitwood, A. C. J. Chem. Soc., Perkin Trans. 21998, 8, 1715.
[76]
Chawla, O. P.; Fessenden, R. W. J. Phys. Chem. 1975, 79, 2693. doi: 10.1021/j100591a020
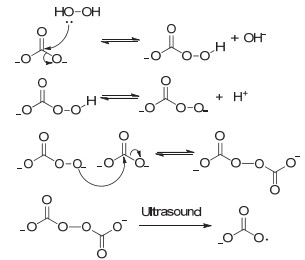
图 1
碳酸根离子和过氧化氢体系下碳酸根自由基可能的产生方式
Figure 1
A possible mechanism for carbonate radicals generation in the presence of carbonate ion and hydrogen peroxide

 下载:
下载:






 下载:
下载:



 下载:
下载:
