-
[1]
(a) Royles, B. J. L. Chem. Rev. 1995, 95, 1981; (b) O'Hagan, D. Nat. Prod. Rep. 2000, 17, 435. (c) Lewis, J. R. Nat. Prod. Rep. 2001, 18, 95. (d) Fish, P. V.; Andrews, M. D.; Jonathan Fray, M.; Stobie, A.; Wakenhut, F.; Whitlock, G. A. Bioorg. Med. Chem. Lett. 2009, 19, 2829.
-
[2]
(a) Fache, F.; Schulz, E.; Tommasino, M. L.; Lemaire, M. Chem. Rev. 2000, 100, 2159. (b) Notz, W.; Tanaka, F.; Barbas, C. F. Acc. Chem. Res. 2004, 37, 580. (c) Erkkila, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416. (d) Caputo, C. A.; Jones, N. D. Dalton Trans. 2007, 4627.
-
[3]
(a) Pettit, G. R.; Goswami, A.; Cragg, G. M.; Schmidt, J. M.; Zou, J. C. J. Nat. Prod. 1984, 47, 913. (b) Su, B.; Cai, C.; Deng, M.; Wang, Q. J. Agric. Food. Chem. 2016, 64, 2039.
-
[4]
Oloff, S.; Mailman, R. B.; Tropsha, A. J. Med. Chem. 2005, 48, 7322. doi: 10.1021/jm049116m
-
[5]
Burnett, D. A.; Hart, D. J. J. Org. Chem. 1987, 52, 5662. doi: 10.1021/jo00235a004
-
[6]
Lebon, F.; Pegurier, C.; Ledecq, M.; Mathieu, B.; Bosman, N.; Frycia, A.; Lengele, S.; Dhurke, K.; Kanduluru, A. K.; Meunier, S.; Wagner, A.; Wolff, C.; Provins, L. Bioorg. Med. Chem. Lett. 2012, 22, 3978. doi: 10.1016/j.bmcl.2012.04.097
-
[7]
For selected reviews, see: (a) Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765. (b) Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484. (c) Pellissier, H. Tetrahedron 2007, 63, 3235. (d) Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749.
-
[8]
For selected racemic examples, see: (a) Ney, J. E.; Wolfe, J. P. Angew. Chem., Int. Ed. 2004, 43, 3605. (b) Ney, J. E.; Hay, M. B.; Yang, Q.; Wolfe, J. P. Adv. Synth. Catal. 2005, 347, 1614. (c) Bertrand, M. B.; Leathen, M. L.; Wolfe, J. P. Org. Lett. 2007, 9, 457. (d) Bertrand, M. B.; Neukom, J. D.; Wolfe, J. P. J. Org. Chem. 2008, 73, 8851. (e) Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., 3rd; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519. (f) Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., 3rd; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293. (g) Fumagalli, G.; Boyd, S.; Greaney, M. F. Org. Lett. 2013, 15, 4398. (h) Sahoo, B.; Hopkinson, M. N.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 5505. (i) Fornwald, R. M.; Fritz, J. A.; Wolfe, J. P. Chem.-Eur. J. 2014, 20, 8782. (j) Alicea, J.; Wolfe, J. P. J. Org. Chem. 2014, 79, 4212. (k) Faulkner, A.; Scott, J. S.; Bower, J. F. J. Am. Chem. Soc. 2015, 137, 7224. (l) Peterson, L. J.; Wolfe, J. P. Adv. Synth. Catal. 2015, 357, 2339. (m) Garad, D. N.; Mhaske, S. B. Org. Lett. 2016, 18, 3862. (n) Yang, H.-B.; Pathipati, S. R.; Selander, N. ACS Catal. 2017, 7, 8441. (o) Yang, M.-N.; Yan, D.-M.; Zhao, Q.-Q.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2017, 19, 5208. (p) Zhang, R.; Xu, Q.; Shi, M. Acta Chim. Sinica 2012, 70, 1593. (张睿, 徐琴, 施敏, 化学学报, 2012, 70, 1593.) (q) Zhang, J.; Wu, Z.; Xie, F.; Zhang, W. Chin. J. Org. Chem. 2018, 38, 1319. (张金刚, 吴正兴, 谢芳, 张万斌, 有机化学, 2018, 38, 1319.)
-
[9]
For selected reviews, see: (a) Heinrich, M. R. Chem. Eur. J. 2009, 15, 820. (b) Hari, D. P.; Koenig, B. Angew. Chem., Int. Ed. 2013, 52, 4734. (c) Kindt, S.; Heinrich, M. R. Synthesis 2016, 48, 1597.
-
[10]
For selected examples, see: (a) Sahoo, B.; Hopkinson, M. N.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 5505. (b) Hari, D. P.; Hering, T.; Koenig, B. Angew. Chem., Int. Ed. 2014, 53, 725. (c) Kindt, S.; Wicht, K.; Heinrich, M. R. Org. Lett. 2015, 17, 6122.
-
[11]
(a) Mai, D. N.; Wolfe, J. P. J. Am. Chem. Soc. 2010, 132, 12157. (b) Babij, N. R.; Wolfe, J. P. Angew. Chem., Int. Ed. 2013, 52, 9247. (c) Hopkins, B. A.; Wolfe, J. P. Chem. Sci. 2014, 5, 4840. (d) White, D. R.; Hutt, J. T.; Wolfe, J. P. J. Am. Chem. Soc. 2015, 137, 11246. (e) Garlets, Z. J.; Parenti, K. R.; Wolfe, J. P. Chem.-Eur. J. 2016, 22, 5919.
-
[12]
Zhang, W.; Chen, P.; Liu, G. Angew. Chem., Int. Ed. 2017, 56, 5336. doi: 10.1002/anie.201700889
-
[13]
(a) Neukom, J. D.; Perch, N. S.; Wolfe, J. P. J. Am. Chem. Soc. 2010, 132, 6276. (b) Neukom, J. D.; Perch, N. S.; Wolfe, J. P. Organometallics 2011, 30, 1269.
-
[14]
Zhu, R.; Buchwald, S. L. J. Am. Chem. Soc. 2015, 137, 8069. doi: 10.1021/jacs.5b04821
-
[15]
Lin, J.-S.; Dong, X.-Y.; Li, T.-T.; Jiang, N.-C.; Tan, B.; Liu, X.-Y. J. Am. Chem. Soc. 2016, 138, 9357. doi: 10.1021/jacs.6b04077
-
[16]
Lin, J.-S.; Wang, F.-L.; Dong, X.-Y.; He, W.-W.; Yuan, Y.; Chen, S.; Liu, X.-Y. Nat. Commun. 2017, 8, 14841. doi: 10.1038/ncomms14841
-
[17]
Wang, F.-L.; Dong, X.-Y.; Lin, J.-S.; Zeng, Y.; Jiao, G.-Y.; Gu, Q.-S.; Guo, X.-Q.; Ma, C.-L.; Liu, X.-Y. Chem 2017, 3, 979. doi: 10.1016/j.chempr.2017.10.008
-
[18]
(a) Wang, F.; Wang, D.; Wan, X.; Wu, L.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2016, 138, 15547. (b) Wang, D.; Wang, F.; Chen, P.; Lin, Z.; Liu, G. Angew. Chem., Int. Ed. 2017, 56, 2054. (c) Wu, L.; Wang, F.; Wan, X.; Wang, D.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2017, 139, 2904.
-
[19]
Cheng, Y.-F.; Dong, X.-Y.; Gu, Q.-S.; Yu, Z.-L.; Liu, X.-Y. Angew. Chem., Int. Ed. 2017, 56, 8883. doi: 10.1002/anie.v56.30

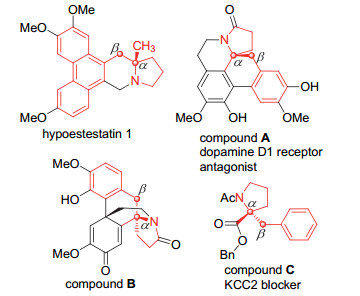
 下载:
下载:
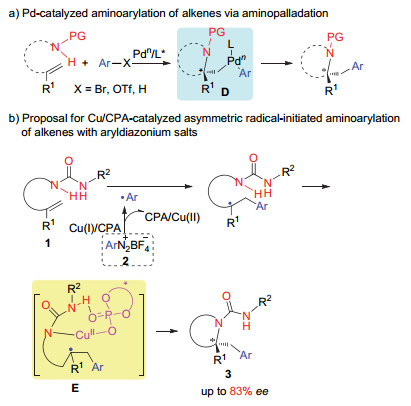




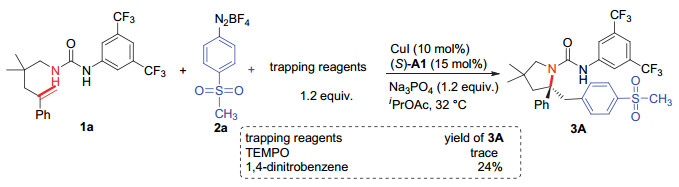
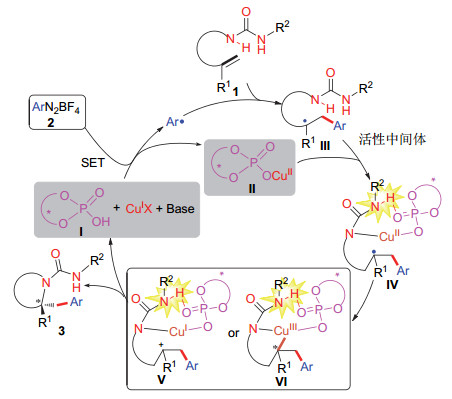

 下载:
下载:





 下载:
下载:
