Received Date:
02 October 2018 Available Online:
15 February 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 41673096, 41772243, 51578398) and National Postdoctoral Program for Innovative Talents (BX201700172)
Abstract:
The unique "core-shell" structure endows nanoscale zero-valent iron (nZVI) rich aquatic surface chemistry properties. Transformation of surface chemistry and crystal phase of nZVI affect its reactivity and environmental transport and fate. Recent advances on the surface chemistry and phase transformation of nZVI in aqueous media are highlighted in this paper focusing on a basic theory of nZVI for pollution control and environmental application. Surface chemistry and phase of both fresh and reacted nZVI are presented. The structure, composition and properties of nanoparticles are determined not only by reaction time but also by environmental conditions. Specifically, the influences of dissolved oxygen, hydraulic conditions (static and stirring), types and concentrations of heavy metals (U(Ⅵ), Cr(Ⅵ), Se(Ⅳ)) and anions (NO3-, SO42-, HPO42- and HCO3-) are investigated. In addition, the effect of surface modification with polyelectrolytes, including anionic polyacrylamide (APAM) and carboxymethylcellulose sodium (CMC), on microstructure, morphology and composition of nanoparticles in aqueous phase was discussed. Results demonstrate that environmental conditions have significant impacts on particles structure, composition and properties, consequently on nZVI performance for pollutant removal. After corrosion under different aqueous conditions, the core-shell structured nZVI are distorted and the metallic iron core is transformed into different iron oxides/hydroxides, such as γ-Fe2O3, Fe3O4 and γ-FeOOH. These iron (hydr)oxides exhibit different surface complexation and affinity proprieties, thus eventually affecting the pollutant removal performance and the environmental fate of reaction products. More research on the effect of dynamic structure transformation by different types of pollutants, and a reaction model between the surface chemistry/phase transformation and removal performance are needed to deepen our understanding on nZVI surface chemistry, and develop more effective technologies of environmental applications.
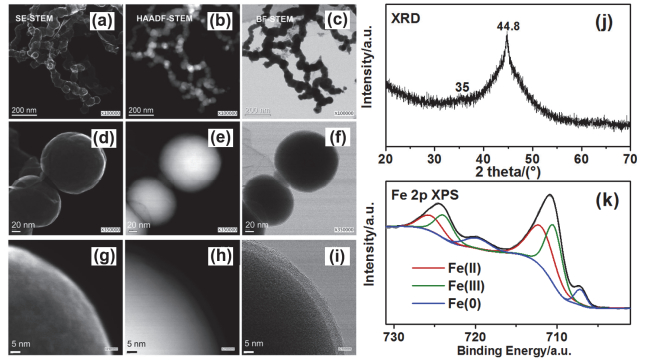
图 1.
纳米零价铁的表面化学和晶相特征:扫描透射电镜图[13]、X射线衍射图和X射线光电子能谱分析. (a, d, g)二次电子(SE)图像, (b, e, h) STEM高角度环形暗场像(HAADF), (c, f, i) STEM明场像(BF), (j) X射线衍射图, (k) Fe 2p的X射线光电子能谱分析
Figure 1.
Surface chemistry and crystallization phase characteristic of fresh nZVI, including STEM [13], XRD and XPS: (a, d and g) are secondary electron (SE) images, (b, e and h) are high-angle annular dark field (HAADF) images, (c, f and i) are bright field (BF) images, (j) is the XRD spectrum and (k) is the XPS response of Fe2p core levels of fresh nZVI
图 3.
纳米零价铁在充氧扰动和无氧扰动条件下腐蚀3 d. (a)充氧条件下腐蚀后颗粒的TEM图, (b)无氧扰动条件下腐蚀后颗粒的TEM图、(c) XRD, (d)拉曼[11, 13]
M: Fe3O4; L: γ-FeOOH
Figure 3.
nZVI corroded anaerobically and aerobically in DI water for 3 days. (a) TEM images for particles aerobically corrosion; (b) TEM image for particles anaerobically corrosion (c) XRD; (d) Raman spectra [11, 13]
Figure 4.
nZVI corroded in static and stirring condition for 30 d and 1 d. (a) TEM image of stirring 1 d particles, (b) TEM image of static 30 d particles, (c) XRD, (d) pH and Eh trends as a function of reaction time after adding corroded particles into water[11, 12]
Figure 6.
STEM-XEDS mapping color overlays of spent nZVI after reactions with Ag (a) and Au (b). (c) STEM-XEDS mapping of uranium reaction after 1 h with nZVI; STEM-XEDS mapping of nZVI after corroding anaerobically in Cr(Ⅵ) for 24 h (d) 10 mg/L, (e) 100 mg/L; (f) STEM mappings of reduced selenium in the spent nZVI: Fe+O+Se color overlay; (g) STEM mappings of selenium nano-needles in the spent nZVI: Fe+O+Se color overlay; (h) XRD pattern of nZVI ater reactions with Se(Ⅳ) (100 mg/L) at different pH[26, 32, 33, 35]
Figure 7.
(a) XRD patterns of nZVI corroded anaerobically in different anions (10 mmol/L) for 30 d; (b) TEM images of nZVI corroded anaerobically in HCO3- solution [41].
Peaks are Fe3O4 (M), Fe4(PO4)2OH6•xH2O (D), Fe163+O16(OH, SO4)12~13•10~12H2O (S), α-FeO(OH) (G)
Wang, C. Y.; Chen, Z. Y.; Cheng, B.; Zhu, Y. R.; Liu, H. J. Mater. Sci. Eng. B1999, 60, 223. doi: 10.1016/S0921-5107(99)00032-X
[16]
Liu, A. R.; Zhang, W. X. Analyst2014, 139, 4512. doi: 10.1039/C4AN00679H
[17]
Wang, C.; Baer, D. R.; Amonette, J. E.; Engelhard, M. H.; Antony, J.; Qiang, Y. J. Am. Chem. Soc. 2009, 131, 8824. doi: 10.1021/ja900353f
[18]
Nurmi, J. T.; Tratnyek, P. G.; Sarathy, V.; Baer, D. R.; Amonette, J. E.; Pecher, K.; Wang, C.; Linehan, J. C.; Matson, D. W.; Penn, R. L. Environ. Sci. Technol. 2005, 39, 1221. doi: 10.1021/es049190u
[19]
Yan, W.; Herzing, A. A.; Kiely, C. J.; Zhang, W. X. J. Contam. Hydrol. 2010, 118, 96. doi: 10.1016/j.jconhyd.2010.09.003
[20]
Liu, Y.; Majetich, S. A.; Tilton, R. D.; Sholl, D. S.; Lowry, G. V. Environ. Sci. Technol. 2005, 39, 1338. doi: 10.1021/es049195r
[21]
Kim, H. S.; Kim, T.; Ahn, J. Y.; Hwang, K. Y.; Park, J. Y.; Lim, T. T.; Hwang, I. Chem. Eng. J. 2012, 197, 16. doi: 10.1016/j.cej.2012.05.018
[22]
Kanel, S. R.; Greneche, J. M.; Choi, H. Environ. Sci. Technol. 2006, 40, 2045. doi: 10.1021/es0520924
[23]
Ling, L.; Zhang, W. X. Environ. Sci. Technol. Lett. 2014, 1, 209. doi: 10.1021/ez4002054
[24]
Yan, W. L.; Herzing, A. A.; Li, X. Q.; Kiely, C. J.; Zhang, W. X. Environ. Sci. Technol. 2010, 44, 4288. doi: 10.1021/es100051q
Jiemvarangkul, P.; Zhang, W. X.; Lien, H. L. Chem. Eng. J. 2011, 170, 482. doi: 10.1016/j.cej.2011.02.065
[43]
Johnson, R. L.; Nurmi, J. T.; O'Brien Johnson, G. S.; Fan, D.; O'Brien Johnson, R. L.; Shi, Z.; Salter-Blanc, A. J.; Tratnyek, P. G.; Lowry, G. V. Environ. Sci. Technol. 2013, 47, 1573. doi: 10.1021/es304564q
[44]
Liu, J.; Liu, A. R.; Zhang, W. X. Chem. Eng. J. 2016, 303, 26832.
Su, Y. M.; Adeleye, A. S.; Keller, A. A.; Huang, Y. X.; Dai, C. M.; Zhou, X. F.; Zhang, Y. L. Water Res. 2015, 74, 47. doi: 10.1016/j.watres.2015.02.004
图 1
纳米零价铁的表面化学和晶相特征:扫描透射电镜图[13]、X射线衍射图和X射线光电子能谱分析. (a, d, g)二次电子(SE)图像, (b, e, h) STEM高角度环形暗场像(HAADF), (c, f, i) STEM明场像(BF), (j) X射线衍射图, (k) Fe 2p的X射线光电子能谱分析
Figure 1
Surface chemistry and crystallization phase characteristic of fresh nZVI, including STEM [13], XRD and XPS: (a, d and g) are secondary electron (SE) images, (b, e and h) are high-angle annular dark field (HAADF) images, (c, f and i) are bright field (BF) images, (j) is the XRD spectrum and (k) is the XPS response of Fe2p core levels of fresh nZVI
图 3
纳米零价铁在充氧扰动和无氧扰动条件下腐蚀3 d. (a)充氧条件下腐蚀后颗粒的TEM图, (b)无氧扰动条件下腐蚀后颗粒的TEM图、(c) XRD, (d)拉曼[11, 13]
Figure 3
nZVI corroded anaerobically and aerobically in DI water for 3 days. (a) TEM images for particles aerobically corrosion; (b) TEM image for particles anaerobically corrosion (c) XRD; (d) Raman spectra [11, 13]
Figure 4
nZVI corroded in static and stirring condition for 30 d and 1 d. (a) TEM image of stirring 1 d particles, (b) TEM image of static 30 d particles, (c) XRD, (d) pH and Eh trends as a function of reaction time after adding corroded particles into water[11, 12]
Figure 6
STEM-XEDS mapping color overlays of spent nZVI after reactions with Ag (a) and Au (b). (c) STEM-XEDS mapping of uranium reaction after 1 h with nZVI; STEM-XEDS mapping of nZVI after corroding anaerobically in Cr(Ⅵ) for 24 h (d) 10 mg/L, (e) 100 mg/L; (f) STEM mappings of reduced selenium in the spent nZVI: Fe+O+Se color overlay; (g) STEM mappings of selenium nano-needles in the spent nZVI: Fe+O+Se color overlay; (h) XRD pattern of nZVI ater reactions with Se(Ⅳ) (100 mg/L) at different pH[26, 32, 33, 35]
图 7
(a) 纳米零价铁在浓度为10 mmol/L不同阴离子无氧溶液中反应30 d XRD; (b)纳米零价铁在浓度为10 mmol/L HCO3-溶液中腐蚀后TEM图[41]
Figure 7
(a) XRD patterns of nZVI corroded anaerobically in different anions (10 mmol/L) for 30 d; (b) TEM images of nZVI corroded anaerobically in HCO3- solution [41].

 下载:
下载:









 下载:
下载:










 下载:
下载:
