图 1.
氟取代效应对Strecker反应活性的影响
Figure 1.
Fluorine effect on reactivity of Strecker reactions

氢键(hydrogen bond)是连接在一个分子或分子片段的X―H键的H原子与另一原子或原子团(同一分子或另一分子)之间形成的一种弱静电相互作用.氢键作用具有方向性和饱和性, 可存在于分子间或者分子内, 通常用X―H…Y―Z表示, 其中“…”代表氢键, X―H称为氢键供体(H-bond donor), Y或Y―Z称为氢键受体(H-bond acceptor).氢键受体可以是一个原子或阴离子Y, 也可以是一个分子片段或分子Y―Z[1, 2].氢键相互作用在化学、物理、生命科学等领域都扮演着重要角色, 其研究至今已有一百余年的历史, 也是研究最为广泛的一种弱相互作用[3~8].值得一提的是, 电负性大的N和O原子可以和醇、酰胺、磺酰胺、磷酰胺、(硫)脲、四方酸等多种氢键给体形成有效的氢键作用[9, 10].这一性质也成功用于活化羰基、亚胺、硝基以及环氧等不同官能团来实现的高效反应, 甚至不对称催化合成[11].
与之形成鲜明对比的是, 尽管氟元素是周期表中电负性最大的元素, 氟负离子与氢键给体可以形成很强的氢键, 但利用有机氟化合物的C―F键与氢键给体之间的相互作用来调控反应的活性和选择性则很少研究, 目前才刚起步.事实上, 对于C―F键能否形成C―F…H―X氢键作用学术界一直存在争议.氟原子的半径为1.47 Å, 介于H(1.20 Å)和O(1.52 Å)之间.而C―F键则是一种很强的化学键, 其解离能高达105.4 kcal/mol, 远大于C―H键(98.8 kcal/mol)和C―C键(83.1 kcal/mol), 因此含氟有机化合物性质较稳定.由于F原子的高电负性, 使得C―F键呈高度极化(Cδ+―Fδ-)形式, 同时存在三对孤对电子.看似可作为一个较好的氢键受体, 实际上由于F原子的高电负性及d+C―δ-F键较强的静电性, 约束了孤对电子, 致使其可极化性低, 质子亲和力弱, 使得C―F键仅能作为弱氢键受体[12, 13], 一些C―F…H―C弱氢键作用强度仅达0.4~1.7 kcal/mol[12].
早在1939年, Pauling[14]就指出: “与碳相连的氟原子总体而言不具有显著的作为氢键给体来形成氢键的能力, 这不同于通常认为的由于碳和氟的电负性差别大使得C―F键可作为良氢键给体”.随后几十年, C―F…H―X作用是否存在一直有争议, 直到1997年, Dunitz和Taylor还发表了题为《Organic Fluorine Hardly Ever Accepts Hydrogen Bonds》的文章.他们通过对剑桥结构数据库(CSD)及布鲁克海文蛋白质数据库(Brookhaven Protein Data Bank)中含氟化合物的晶体结构的统计分析指出, 有机氟很难作为一个氢键受体[15], 但也有少量例子如氟取代苯与水形成的C―F…H―O氢键作用强度在2.8~3.3 kcal/mol左右, 属弱氢键范围.
随着研究的深入, 越来越多的证据支持在晶体结构、溶液和气相中存在C―F…H―X氢键作用(X一般为电负性较强元素如氧和氮):即C―F键可作为氢键受体与X―H键形成氢键相互作用.这一作用的键能为1~4 kcal/mol, 略强于C―F…H―C弱氢键作用, 比常规氢键(如ROH…O=C, 键能约为5~10 kcal/mol)要弱, 但C―F…H―O和C―F…H―N作用不能被忽视[16~20].目前, 人们认为当氟和氢之间的距离小于2.50 Å时, 存在C―F…H―X相互作用.如果这一距离小于2.30 Å以及F…H―X键角大于120o, 则氢键作用很强.
随着对C―F…H―X氢键作用认识的深入, 人们开始探索这种弱相互作用的应用.例如, 上海有机所黎占亭等[21]成功利用C―F…H―X相互作用来进行自组装研究.随后, 这种弱相互作用也逐渐被发现能影响有机化学反应的活性和选择性. 2011年, 我们和四川大学王欣课题组[22]合作, 通过实验和理论计算研究, 提出三氟和二氟甲基酮亚胺的C―F键与硫脲催化剂的N―H键之间存在相互作用, 从而显著影响反应.这是首例报道的C―F…H―X作用对反应的影响.随后, 陆续有新的C―F…H―X作用影响反应的例子报道, 并且显示这种弱相互作用不仅能影响反应的选择性和活性, 而且还能通过理性设计来实现其他方法难以获得的高活性和选择性.尽管成功例子还不多, 但已显示底物与催化剂之间、底物与溶剂之间、以及底物之间形成C―F…H―X相互作用、甚至反应中间体也能形成这种作用, 进而对反应活性和选择性产生深远影响.这些结果不仅显示了利用C―F…H―X相互作用来设计新型手性催化剂方面的潜力, 同时也说明这种弱作用在选择性引入氟原子或含氟基团方面的应用价值.众所周知, 在有机分子中选择性引入含氟基团可以影响其构象、理化性质、药学活性等, 在医药、农药、材料等领域具有广泛应用, 是有机合成化学的前沿研究热点[23].
尽管Paquin等[24]最近对有机氟化物中的C(sp2)―F、C(sp3)―F键作为氢键受体形成氢键的能力及具体性质等进行了综述, 但关于C―F…H―X相互作用对有机反应的活性及选择性的影响目前尚未有专题报道.因此, 我们拟综述C―F…H―X相互作用调控有机反应的活性和选择性的实例, 以期为从事有机合成及有机氟化学的研究人员提供借鉴和参考.
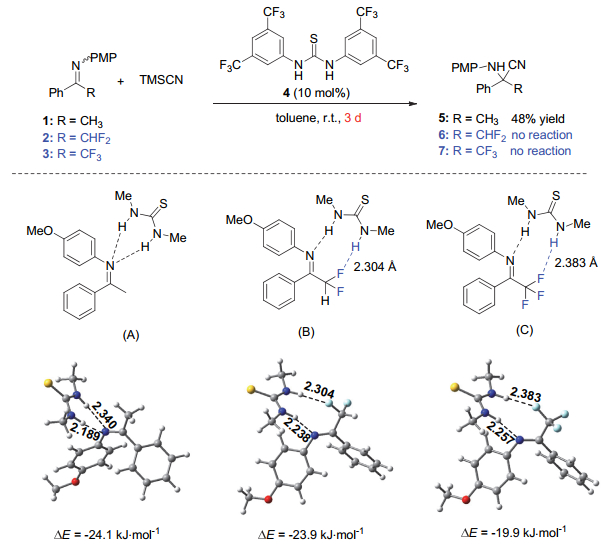
2011年, 我们[22]在研究有机催化的酮亚胺与三甲基氰硅烷(TMSCN)的Strecker反应时, 发现在非手性硫脲4的作用下, 非氟代的甲基酮亚胺1反应3 d能以48%的产率获得目标产物5, 而α-CF2H或CF3取代的酮亚胺2和3则不能发生反应(图 1).这一实验结果有悖于拉电子取代基CF2H和α-CF3可以增加酮亚胺的亲电性来促进氰化反应的常识.针对这一不寻常的实验现象, 我们与四川大学王欣教授合作进行了理论计算研究, 进而认为是源于C―F…H―X氢键相互作用.非氟代酮亚胺1的N原子同时与硫脲催化剂的两个N―H键形成双氢键作用[25], 从而能有效稳定反应过渡态中氮上产生的负电荷, 降低反应能垒, 使得反应进行(A, 图 1).而氟代酮亚胺2和3则与催化剂形成不同的识别模式:其α-F原子和亚胺的N原子分别与硫脲的两个N―H键形成氢键相互作用(氟和氢之间的距离均小于2.50 Å, B和C), 其分子间相互作用能分别为4.8和5.7 kcal/mol.由于C―F…H―N相互作用的形成, 不能很好地稳定反应过渡态中氮上产生的负电荷, 从而导致反应过渡态的能垒高, 反应难以进行.这是首次报道的C―F…H―N氢键相互作用对有机化学反应的影响.
而在氢化奎宁衍生的双功能脲催化剂8实现的不对称催化反应中, 也发现了显著的氟取代效应[22].如图 2所示, 在相同的条件下, 非氟代酮亚胺1仅反应得到外消旋的氨基氰化合物5, 而α-CF2H或CF3酮亚胺2和3则能分别以87%和94%的ee值得到相应的氟代Cα-四取代氨基氰6和7.尽管氟原子在反应立体选择性控制方面的作用还有待进一步探索, 我们根据图 1所示的结果, 认为C―F…H―N相互作用是一种可能的因素.
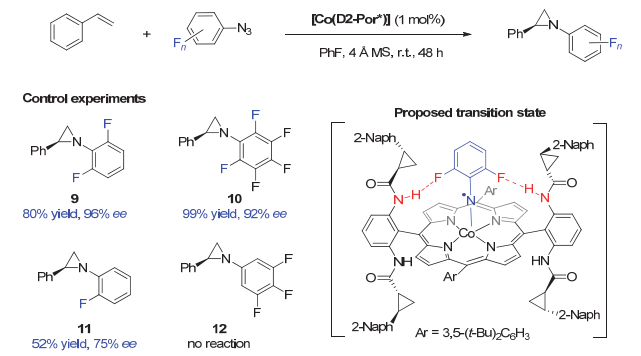
2013年, Zhang等[26]巧妙地利用底物上的氟原子与手性配体上的酰胺N―H键之间的C―F…H―N相互作用, 实现了高对映选择性的钴催化的苯乙烯与芳基叠氮的氮杂环丙烷化反应(图 3).对照实验显示, 叠氮化合物芳基上的氟原子的位置和个数对反应活性及对映选择性具有明显影响.邻位2, 6-二氟取代和全氟取代的芳基叠氮能分别以96% ee值和92% ee值获得相应的目标产物9和10. 2-单氟取代芳基叠氮生成的产物11的产率和对映选择性大幅下降.当使用邻位无氟取代的芳基叠氮时, 反应则完全不进行.基于实验结果, 作者认为手性Co催化剂中的酰胺N―H键与芳基叠氮邻位氟原子之间形成的C―F…H―N氢键相互作用, 使得反应过渡态更为有序, 能在降低能垒的同时, 更好地控制反应的对映选择性.
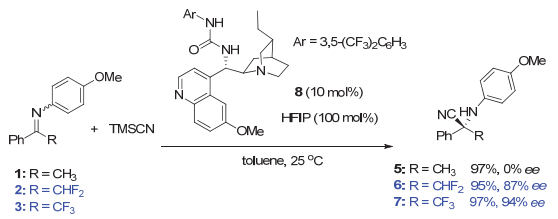
同年, 马军安等[27]利用他们发展的糖类衍生的叔胺-硫脲催化剂18实现了β-酮酸与氟代酮亚胺的不对称脱羧Mannich反应, 以优秀的产率和对映选择性获得各种3, 4-二氢喹唑啉-2(1H)-酮衍生物.在研究过程中, 他们发现该反应存在明显的氟取代效应(图 4A).在10 mol%催化剂的作用下, α-CF3和α-CF2H取代酮亚胺13和14分别高效高选择性地获得相应的产物19(99%产率, 99% ee)和20(91%产率, 93% ee); 而甲基和苯基取代酮亚胺15和16在相同条件下则完全不反应.虽然作者推测产生这一明显反应活性差异的原因可能是酮亚胺中三氟甲基和二氟甲基的强拉电子性质使相应的酮亚胺活性更高; 但也可能是氟代酮亚胺与催化剂之间的C―F…H―C相互作用促使反应顺利进行, 因为理论计算的过渡态模型中, 三氟甲基酮亚胺的氟原子与催化剂的环己基骨架存在一定的C―F…H―C相互作用(图 4B), 其中氟原子与环己基骨架上两个氢原子之间距离分别为2.499和2.670 Å[27].
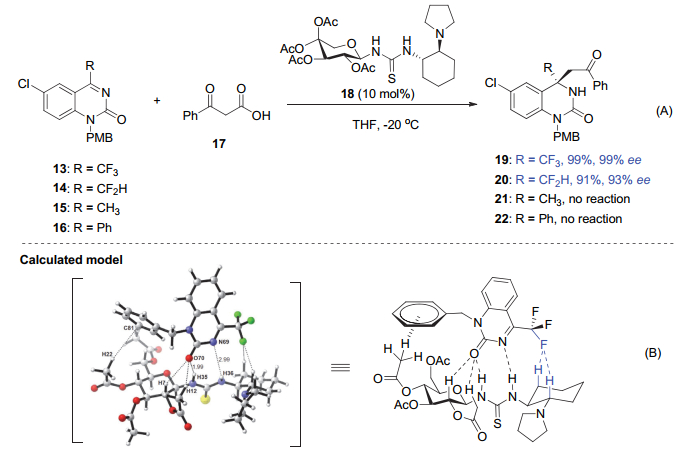
2016年, Hoveyda等[28]在手性氨基酚催化的烯丙基硼试剂与三氟甲基酮的不对称烯丙基加成反应中观察到了明显的氟取代效应(图 5).在2.5 mol%的25催化下, 三氟甲基苯乙酮23能以高达92% ee得到R构型的产物26, 而苯乙酮则在相同条件下以36% ee得到S构型的产物27.这暗示催化剂25与三氟甲基酮和非三氟甲基酮的作用模式可能不同.基于理论计算的结果, 他们认为导致产物绝对构型翻转的可能原因之一是三氟甲基苯乙酮与手性催化剂形成的过渡态Ⅰ中, 带正电荷的铵盐的N―H键与邻近的F原子之间的距离为4.10 Å.尽管没有形成C―F…H―N氢键作用, 但存在有一定的静电相互作用, 从而减轻了羰基氧上孤对电子与氟原子上孤对电子的排斥力, 使得三氟甲基苯乙酮与催化剂的识别方式不同于苯乙酮与催化剂的.
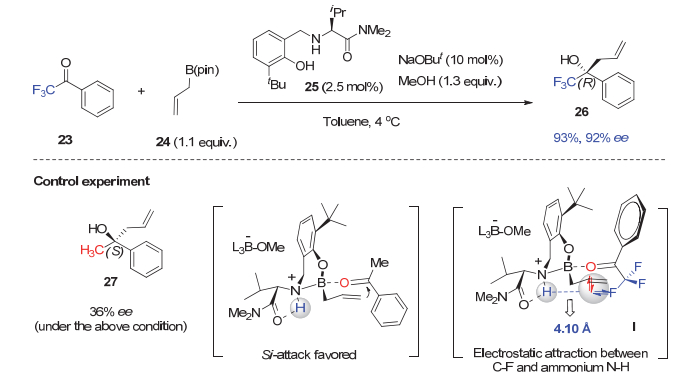
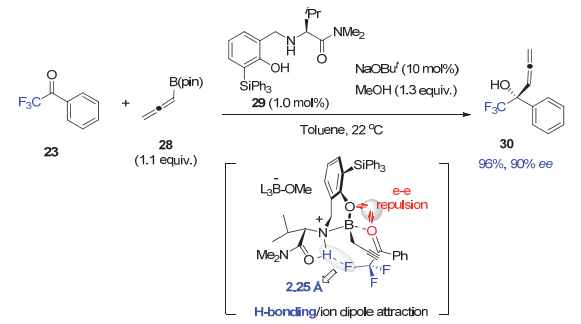
而在联烯基硼酸酯28与三氟甲基酮的高对映选择性加成反应中, 在酚羟基邻位具有位阻更大的三苯基硅基的催化剂29可以比上述催化剂25取得更高的选择性(图 6)[28].理论计算显示, 由于三苯基硅基比叔丁基的位阻更大, 就迫使三氟苯乙酮与催化剂采取如下的识别模式.在这一最优过渡态中, 虽然催化剂29的酚氧原子与三氟甲基酮的氧原子存在电子-电子排斥作用, 但是, 由于三氟甲基酮的C―F键与催化剂铵盐上的N―H键之间存在氢键相互作用(氟与氢之间距离仅为2.25 Å), 稳定了这一过渡态, 并取得优异的对映选择性.
除了含氟底物的C―F键与催化剂氢键给体的C―F…H―X相互作用影响反应外, 含氟底物与氢键给体溶剂之间也可能存在C―F…H―X相互作用, 并被用于实现一些无需外加催化剂的高效转化.
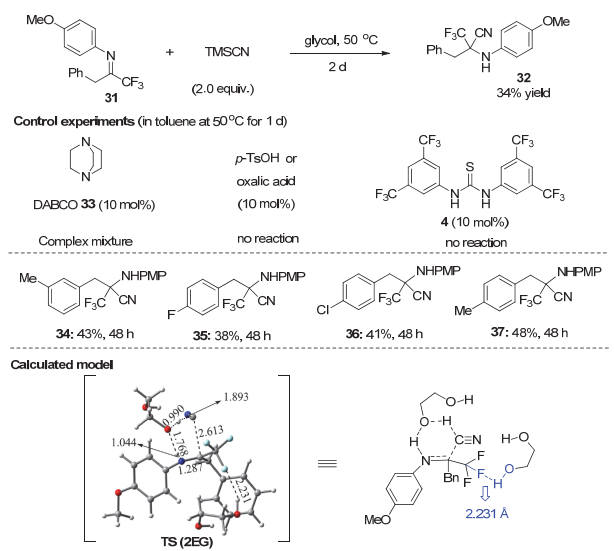
在研究α-CF3酮亚胺的Strecker反应时, 我们发现虽然叔胺作为催化剂可以取得较好的底物适用范围, 但苄基取代的α-CF3酮亚胺31在叔胺作为碱的反应条件下反应很混乱, 可能是因为碱性条件下易发生烯胺-亚胺异构化的原因.因此, 这一类底物的Strecker反应意外地具有一定难度.除了Brønsted碱如DABCO等导致反应混乱外, Brønsted酸不论p-TsOH或草酸, 还是硫脲4都不能催化该反应(图 7).分析认为这类底物的反应需要在中性条件下进行, 同时又需克服C―F…H―X作用的影响.基于对醇促进的无需催化剂的Strecker反应和Mannich反应的研究, 我们设想在醇作为溶剂时, 无处不在的氢键可以克服C―F…H―O作用的不利影响, 同时也有足够的氢键作用来活化亚胺和稳定反应过渡态.没有外加的碱, 苄基取代的α-CF3酮亚胺的异构化也会被尽量减少.最后, 我们[29a]发现乙二醇作为溶剂, 可以实现无催化剂的α-CF3和不同苄基取代的酮亚胺的Strecker反应, 以中等产率得到目标产物, 当然这一体系也适用于其他α-CF2H和α-CF3酮亚胺.最近, 王欣等[29b]对这一特殊的反应性进行了理论计算研究, 首先优化了系列三氟甲基酮亚胺31与乙二醇相互作用的氢键复合物.研究结果发现溶剂与底物之间存在较强的C―F…H―O氟氢键作用, 其分子间相互作用能在1.5~6 kcal/mol之间, 最强的氟氢键强度达5.7 kcal/mol.在此基础上考虑氟氢键促进该反应的机理发现在二分子乙二醇与31作用下, 反应的自由能垒由47.5 kcal/mol大幅降低至24.7 kcal/mol; 其中一分子乙二醇与亚胺的N原子和HCN同时作用, 乙二醇作为质子传递[30]的桥梁从而降低反应能垒; 而另一分子乙二醇则与酮亚胺的氟原子形成较强的C―F…H―O氢键作用(氟和氢原子的距离为2.231 Å)来稳定反应的过渡态并进一步降低反应的能垒(从30.2降至24.7 kcal/mol).在乙二醇为溶剂的情况下, 氢键给体足够多, 既可通过与亚胺形成氢键作用来稳定过渡态的负电荷, 同时还能形成C―F…H―O氢键作用来促进反应.
2013年, Paquin等[31]利用苄氟类底物与溶剂水之间的C―F…H―X氢键相互作用实现了高效的SN2亲核取代反应(图 8).众所周知, C―F键的键能远高于其它C―X键(C―F: 513.8; C―Cl: 394.9; C―Br: 318.0; C―I: 253.1, 单位均为kJ/mol).因此C―F键的断裂明显难于其他碳-卤键[31], 氟代烷烃的SN2取代反应通常需使用金属催化剂来活化C―F键, 而中性或碱性条件下的方法很少.
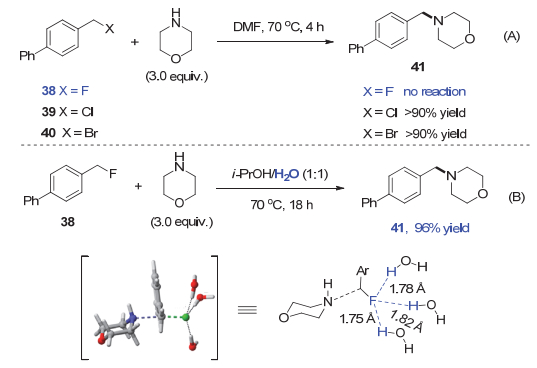
他们发现4-苯基苄氟38与吗啉的亲核取代反应在DMF、DMSO和CH3CN等溶剂中完全不进行, 而相应的苄氯和苄溴化合物39与40在相同条件下均能顺利反应(图 8A).这一结果符合氯或溴代烷在SN2取代反应中的活性远大于氟代烷的常识.然而, 当反应在水中进行时, 苄氟38与吗啉的反应有7%的转化率.由于苄氟化合物在水中的溶解性不好, 作者尝试使用混合溶剂, 最终发现反应在i-PrOH/H2O(1:1, V/V)的混合溶剂中可以顺利进行, 以96%的产率得到产物41(图 8B).理论计算结果表明苄氟38、苄氯39和苄溴40与水的相互作用能ΔEint分别为-11.1, -7.0和-6.4 kcal/mol, 显示水对C―F键的活化效果优于对C―Cl和C―Br键.另外, 在三个水分子的协同作用下, 苄氟的形变能ΔEdist也由39.6降低为31.6 kcal/mol, 而相应苄氯和苄溴的ΔEdist则变化没有这么明显.因此, 作者认为溶剂水分子与苄氟38之间的C―F…H―O相互作用, 在活化C―F键的同时稳定了反应的过渡态(图 8B).
随后, Paquin等[32]进一步利用溶剂六氟异丙醇(HFIP)与C―F键形成的C―F…H―O氢键作用来活化苄氟, 实现了与芳烃43的Friedel-Crafts反应.在HFIP与二氯甲烷的混合溶剂中, 该反应能以50%~99%产率合成了各种结构的1, 1-二芳基甲烷类化合物44(图 9).
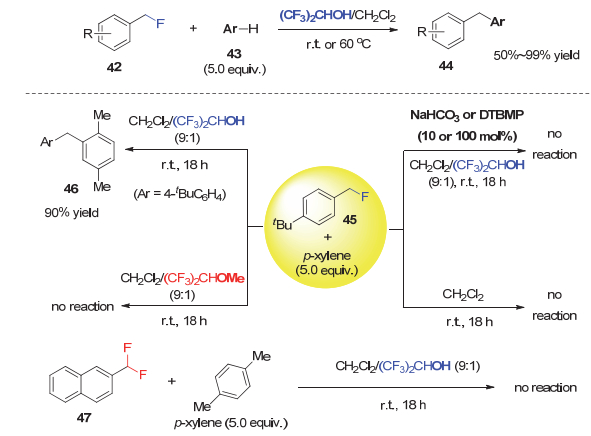
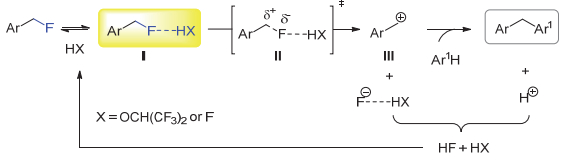
对照实验显示, 当混合溶剂中的HFIP被相应的六氟异丙基甲基醚替换时, 或仅使用CH2Cl2作为溶剂时, 傅克烷基化反应并不发生.这说明HFIP的羟基对反应的顺利进行至关重要.外加催化或当量的NaHCO3或2, 6-二叔丁基-4-甲基吡啶(DTBMP), 均导致反应不能进行, 显示反应中产生的副产物HF对反应起促进作用.此外, 苄基二氟化物47在标准条件下不能发生反应, 这可能因为CHF2是比CH2F更弱的氢键给体[33, 34], 不能通过C―F…H―O氢键作用被有效活化.在此基础上, 作者认为氟原子是作为氢键受体参与反应:反应首先是苄氟的C―F键与HFIP的羟基或者HF通过C―F…H―X氢键作用形成复合物Ⅰ(X=OCH(CF3)2或F), 继而C―F键离子化生成过渡态Ⅱ, 之后分解成碳正离子Ⅲ.随后Ⅲ与富电子芳环反应, 生成1, 1-二芳基甲烷.同时产生的副产物HF起到促进作用(图 10).
2014年, 我们在研究氟代烯醇硅醚参与的反应时[35], 利用二氟烯醇硅醚在水油界面形成的C―F…H―O氢键作用, 实现了无需外加催化剂的“在水上”二氟烯醇硅醚48与醛、活泼酮以及3-烯基氧化吲哚的亲核加成反应, 为具有较高应用价值的α, α-二氟酮羰基取代的醇类化合物和3-二氟烷基取代的季碳氧化吲哚类提供了一个高效合成方法(图 11)[36].反应观察到明显的二氟取代效应.例如, 对氯苯甲醛51与二氟烯醇硅醚52在水为溶剂和50 ℃的条件下, 经过10 h就能以85%的分离产率得到目标产物53. 在相同条件下, 单氟烯醇硅醚54只能以26%的核磁产率得到产物55, 非氟烯醇硅醚57完全没有反应.二氯烯醇硅醚56不能得到目标产物的事实说明52的反应性能并非源于二氟亚甲基的拉电子作用.其次, 反应的非均相性也很重要, 因为在均相的条件下, 如THF、THF/H2O(7:1, V/V)或甲醇作为溶剂时, 反应仅以16%、21%和29%得到目标产物.此外, 反应在无溶剂条件下不进行, 说明在水上反应的高效并非是因为反应物有效浓度很高所致.
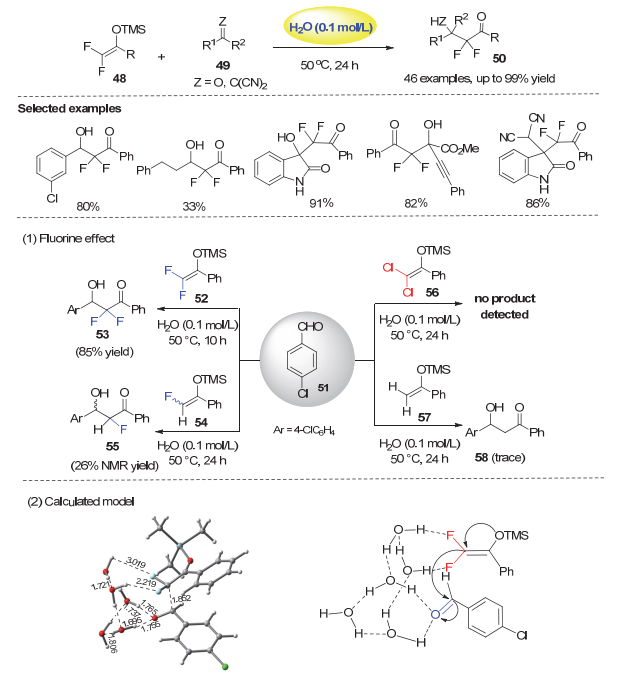
利用五水分子模型的理论计算表明, 在五个水分子形成的氢键网络中, 二氟烯醇硅醚52和4-氯苯甲醛51与水分子均存在氢键相互作用:硅醚的两个C―F键分别与两个水分子形成C―F…H―O相互作用(其中一个C―F键形成的氢键较强, 氟和氢的距离仅为2.219 Å), 同时醛的C=O键也与水分子形成两个O…H氢键.这种协同的氢键作用, 不仅可以活化醛基, 稳定过渡态中氧上产生的负电荷, 同时组织反应以类似于分子内的方式进行, 使得反应的能垒从没有催化剂时的41.6 kcal/mol大幅降低至23.3 kcal/mol, 从而无需催化剂也能高效进行.
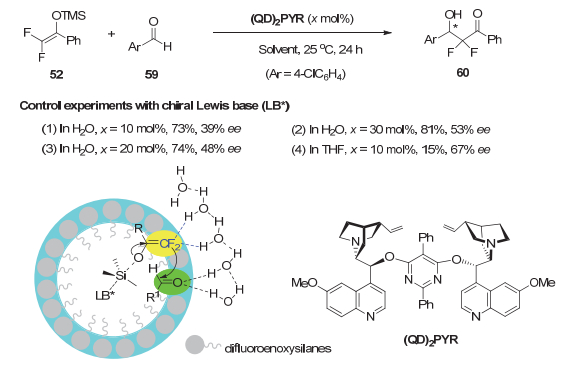
进一步设想在水油两相界面的氢键网络与两个反应组分作用的同时, 可以添加手性Lewis碱从油滴的憎水内部去活化氟代烯醇硅醚[37], 从而开展不对称催化研究.最后, 发现金鸡纳碱衍生物(QD)2PYR能取得较好结果.当催化剂用量从10 mol%增加为30 mol%时, ee值从39%提高到53%, 这可能因为外消旋的背景反应随着手性催化剂的增加得到了抑制.而当反应在THF中进行时, 相同条件下仅有15%的产率, 尽管ee值略高(图 12).这进一步说明“在水上”反应条件下, C―F…H―O相互作用对反应的促进作用.
除了含氟底物与催化剂、溶剂形成的C―F…H―X氢键相互作用对反应活性和选择性有重要的影响以外, 最近研究人员也发现底物与底物之间的C―F…H―X氢键作用也能明显加速反应.例如, Raines等[38] 2016年报道了α-重氮酰胺61与4, 4, 4-三氟巴豆酸乙酯62的环加成反应.在乙腈/水(1:1, V/V)的混合溶剂中, 该反应能迅速以当量的产率和9:1的比例得到环加成产物63和64.经测定, 该反应的速率常数k1为0.105 L·mol-1·s-1, 而非氟代丙烯酸酯65和巴豆酸酯66替换氟代底物 62时, 反应的速率显著下降, 速率常数k2和k3分别为1.6×10-3 L·mol-1·s-1[39]和5.8×10-7 L·mol-1·s-1.氟代底物62的反应速率分别快了102和105倍, 表明其F原子对反应的高速率起到了显著影响(图 13).作者随后通过计算对这一实验现象进行了解释, 他们推测α-重氮酰胺61的N―H键与三氟巴豆酸乙酯62的C―F键形成了C―F…H―N氢键作用, 从而加速了反应的进行.
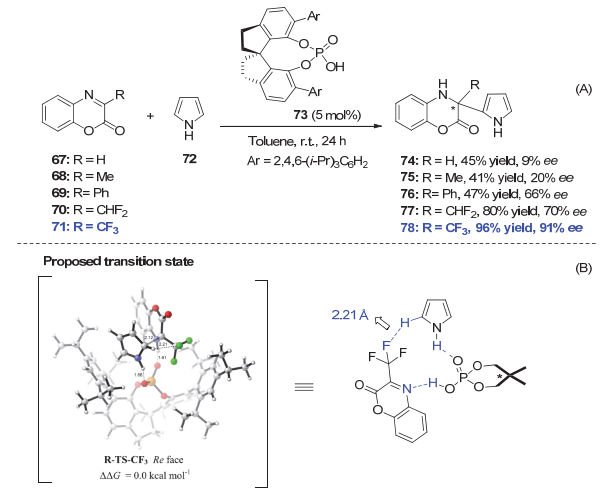
此外, 林旭锋等[40]在手性磷酸73催化的苯并噁嗪酮与吡咯aza-Friedel-Crafts反应中也发现了显著的氟效应(图 14). R为氢、甲基、苯基取代的亚胺仅能以较低的产率和对映选择性得到相应的产物.当R为CF2H时, 反应产率和ee值均有所提高.而CF3取代的亚胺71的反应产率和ee值分别提高到96%和91%.通过理论计算, 他们认为手性磷酸与三氟甲基酮亚胺71以及吡咯72通过氢键作用形成了有利的反应过渡态.手性磷酸作为双功能催化剂分别与吡咯的N―H键以及亚胺71形成氢键作用, 使得反应以类似于分子内的方式进行.而亚胺71上的三氟甲基上的F与吡咯2-位H也形成C―F…H―C氢键相互作用(氟和氢原子的距离为2.21 Å), 引导吡咯从Re方向进攻.
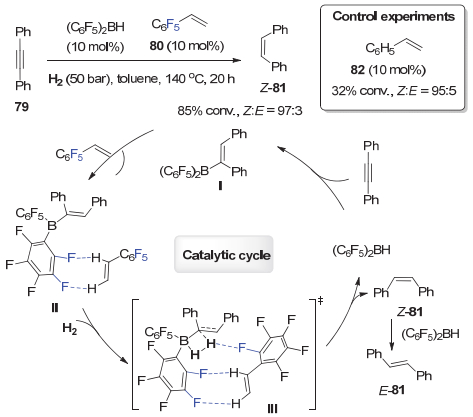
除了含氟底物可以与催化剂和溶剂, 以及含氟底物之间可以形成C―F…H―X相互作用来影响反应外, 反应中间体也能在适当条件下形成这一弱氢键相互作用.例如, 2015年, 杜海峰等[41]在研究(C6F5)2BH催化的炔烃的选择性氢化时, 发现加入10 mol%的全氟苯乙烯80能明显加速反应.通过调节(C6F5)2BH的用量和改变反应时间, 能以优秀的产率和选择性分别得到各种Z-式和E-式烯烃.对照实验揭示了明显的氟效应:当在10 mol%的氟代烯烃80的作用下, 炔烃79在140 ℃下反应20 h, 能以85%转化率、97:3的Z/E比氢化为烯烃81; 形成鲜明对照的是, 使用10 mol%非氟代苯乙烯82只有32%的转化率和95:5 Z/E比, 与没有烯烃添加剂时29%的转化率相类似, 这说明添加剂80中氟原子对反应的高效性非常重要(图 15).基于以上对照实验结合DFT理论计算, 他们提出了图 15所示的可能反应机理.炔烃与(C6F5)2BH首先发生硼氢化反应生成烯基硼物种Ⅰ, 其与全氟苯乙烯通过C―F…H―C氢键作用形成复合物Ⅱ. 在氢气作用下, 复合物Ⅱ通过过渡态Ⅲ发生氢解反应, 得到顺式烯烃Z-81, 同时再生催化剂(C6F5)2BH.延长反应时间, 顺式烯烃Z-81在(C6F5)2BH的作用下能高选择性地转化成反式烯烃E-81.作者认为过渡态Ⅲ中, 全氟苯乙烯参与形成的C―F…H―C相互作用, 不仅可以稳定反应过渡态, 还有助于氢解过程中H2的H―H键断裂, 进而加速反应.这也是首例C―F…H―C相互作用对反应活性和选择性影响的例子.
我们在利用丁氏螺缩酮双膦配体(S, S, S)-SKP[42]研究金催化的重氮氧化吲哚与烯烃的环丙烷化反应时[43], 发现反应在氟取代溶剂中进行时的速率, 比在结构类似的非氟代溶剂中进行时明显要高很多, 而且选择性也略高[44].例如, α-单氟甲基取代的苯乙烯的反应在氟苯或者三氟甲苯中进行时, 比在氯苯或者甲苯中进行的速率均要高几乎一个数量级, 并且分离产率和产物的ee值也要高(图 16).其次, 重氮氧化吲哚的N―H键对反应的选择性有明显影响.当用甲基保护的重氮氧化吲哚时, 反应的对映选择性明显下降[45].这些显著差别无法用介电常数或者配位能力等通常认为影响环丙烷化反应的溶剂性质来进行解释.
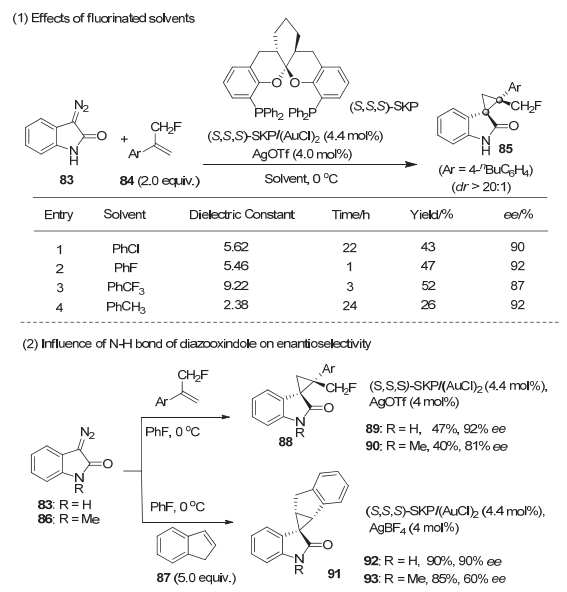
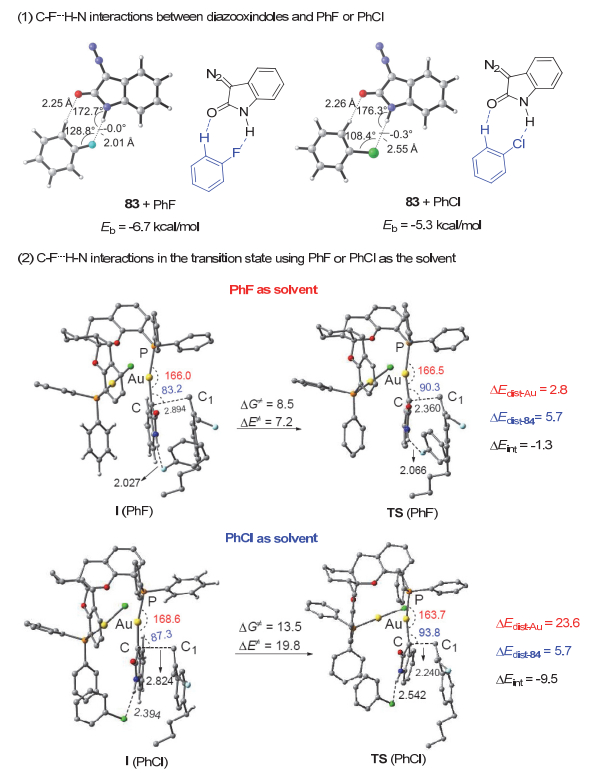
通过与南京大学马晶课题组合作进行理论计算研究, 反应在氟代溶剂进行时很高效可能是溶剂的C―F键可以和无保护的重氮氧化吲哚以及其被金分解后产生的金卡宾物种的N―H键之间, 形成C―F…H―N氢键相互作用[44].无保护的重氮氧化吲哚与氟苯和氯苯通过氢键相互作用形成的复合物如图 17所示.氟苯和重氮氧化吲哚之间的氢键作用很强, 因为氢和氟的距离仅为2.01 Å, 氟苯复合物的相互作用能为-6.7 kcal/mol, 氟氢键约比氯氢键强1.4 kcal/mol.金分解重氮氧化吲哚产生的卡宾中间体与烯烃84的反应是决速步骤, 而这一步反应在氟苯中的活化能为8.5 kcal/mol, 远低于在氯苯中的13.5 kcal/mol, 预示反应在氯苯和氟苯中的速率分别为4.3×1022s-1和1.3×1019 s-1, 大致符合实验结果.利用反应路径的形变-结合能模型(distortion/interaction model[46])对反应在氟苯和氯苯中的过渡态进行分析发现:当氟苯为溶剂时, C―F…H―N相互作用使得反应过渡态TS (PhF)中, 金卡宾中间体与烯烃反应需克服的形变能(ΔEdist-Au), 远小于氯苯为溶剂时的反应过渡态TS(PhCl)中所需的形变能(2.8 vs 23.6 kcal/mol).而反应在氟苯中进行时的结合能(ΔEint)明显低于在氯苯中的(-1.3 vs -9.5 kcal/mol)结合能.其中的形变能的差异主要来自金卡宾中间体的形变能(ΔEdist-Au), 而烯烃84的形变能(ΔEdist-84)在氟苯和氯苯中均为5.7 kcal/mol.
自20世纪20年代初氢键的概念被正式提出以来, 关于氢键的研究已有一百余年的历史.期间人们对氢键的性质、特点、类型等进行了广泛深入的研究, 对其理解和认识也在不断加深和完善.同时, 也在不断发现新形式的氢键, 如有机氟化物形成的C―F…H―X氢键.有关氢键相互作用的研究已成为化学领域的一个重要研究方向.
长久以来, 学术界对于C―F键能否作为氢键受体一直存在争议.随着理论、实验及计算模拟技术的发展与进步, 人们对C―F…H―X氢键相互作用的认识逐渐深入, 也有越来越多的证据支持C―F…H―X氢键的客观存在.同时, 随着有机氟化学的发展及新型氟化试剂的不断出现, 大量含氟分子不断涌现, 其在药物化学、物理学、材料学、生物学等领域的地位也日趋重要, 这也为C―F…H―X氢键在有机合成中的具体应用奠定了坚实基础.在此, 结合自己课题组的研究工作, 我们对截止目前C―F…H―X形成的氢键在有机合成中的应用实例进行了概括总结, 并分为以下几类: (1)底物与催化剂之间; (2)底物与溶剂之间; (3)底物与底物之间; (4)反应中间体形成的.这些实例中的C―F…H―X氢键一般作用强度在1~4 kcal/mol范围内, 属弱氢键作用, 但在一些反应中却能起到降低反应能垒促进反应的重要作用.这些氟氢键的实例涵盖了参与反应的所有成分(底物、溶剂、催化剂、添加物), 使读者能更直观地看到C―F…H―X氢键在有机合成的实际应用以及所表现出来的优势.正如凡事都有两面性一样, 这些应用中有些C―F…H―X氢键对反应起到了促进作用, 可以提高反应的活性或对映选择性; 有些C―F…H―X氢键则起到抑制作用, 降低反应活性, 甚至使反应不能发生.在今后的研究工作中, 我们应该充分利用C―F…H―X氢键的积极作用, 以其为工具或辅助手段探索多种类型的有机反应, 并尽可能提高反应的活性及对映选择性.相信随着人们对C―F…H―X氢键认识的不断加深, 其在有机合成领域的应用将会不断涌现.
综上所述, 在了解C―F…H―X氢键作用在有机合成中的重要应用的同时, 也应记住对于有机氟化合物形成的C―F…H―X氢键作用的研究依然刚起步.如C―F…H―X氢键的成键本质、成键规律等理论研究还有待完善; 实验技术、表征手段等也需逐渐加强.尽管如此, 但我们相信, 随着科学技术的日趋进步及研究人员的不懈努力, C―F…H―X氢键的理论和实验研究在将来必将迎来更具实质性的进展, 并将在有机合成、药物化学、生命科学等重要领域创造出更广阔的应用前景.
Arunan, E.; Desiraju, G. R.; Klein, R. A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D. C.; Crabtree, R. H.; Dannenberg, J. J.; Hobza, P.; Kjaergaard, H. G.; Legon, A. C.; Mennucci, B.; Nesbitt, D. J. Pure Appl. Chem. 2011, 83, 1619. doi: 10.1351/PAC-REP-10-01-01
Desiraju, G. R. Angew. Chem., Int. Ed. 2011, 50, 52. doi: 10.1002/anie.v50.1
Latimer, W. M.; Rodebush, W. H. J. Am. Chem. Soc. 1920, 42, 1419. doi: 10.1021/ja01452a015
Robertson, J. M. Nature 1935, 136, 755. https://www.nature.com/articles/136755c0
Taylor, R.; Kennard, O. J. Am. Chem. Soc. 1982, 104, 5063. doi: 10.1021/ja00383a012
Katz, B. A.; Spencer, J. R.; Elrod, K.; Luong, C.; Mackman, R. L.; Rice, M.; Sprengeler, P. A.; Allen, D.; Janc, J. J. Am. Chem. Soc. 2002, 124, 11657. doi: 10.1021/ja020082m
(a) Bondar, A. N.; White, S. H. BBA-Biomembranes 2012, 1818, 942. (b) Zhang, J.; Chen, P. C.; Yuan, B. K.; Ji, W.; Cheng, Z. H.; Qiu, X. H. Science 2013, 342, 611.
For the recent works on H-bonding interaction from Chinese research group: (a) Wang, M.; Cheng, C. Q.; Song, J. T.; Wang, J.; Zhou, X. G.; Xiang, H. F.; Liu, J. Chin. J. Chem. 2018, 36, 698. (b) Zhu, X. W.; Cui, X. Y.; Cai, W. S.; Shao, X. G. Acta Chim. Sinica 2018, 76, 298(in Chinese). (朱雪薇, 崔晓宇, 蔡文生, 邵学广, 化学学报, 2018, 76, 298.). (c) Sun, G. J.; Nie, C. B.; Zhao, X.; Li, Z. T. Chin. J. Org. Chem. 2017, 37, 1757(in Chinese). (孙广军, 聂承斌, 赵新, 黎占亭, 有机化学, 2017, 37, 1757.)
Desiraju, G. R. Acc. Chem. Res. 1991, 24, 290. doi: 10.1021/ar00010a002
Jeffrey, G. A. Cryst. Rev. 1995, 4, 213. doi: 10.1080/08893119508039923
For a review on H-bond donor catalysis, see: Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713. For bifucntional catalysis with H-bond donors, see amide based organocatalysts: (a) Liu, X. H.; Lin, L. L.; Feng, X. M. Chem. Commun. 2009, 41, 6145; with enamine catalysis: (b) Kano, T.; Maruoka, K. Chem. Commun. 2008, 43, 5465; with tertiary phosphine catalysis: (c) Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005; (d) Xu, L. W. ChemCatChem 2013, 5, 2775; (e) Wang, S. X.; Han, X. Y.; Zhong, F. R.; Wang, Y. Q.; Lu, Y. X. Synlett 2011, 19, 2766; with NHCs catalysis: (f) Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314; (g) Sun, L. H.; Liang, Z. Q.; Jia, W. Q.; Ye, S. Angew. Chem., Int. Ed., 2013, 52, 5803; (h) Lv, H.; Jia, W. Q.; Sun, L. H.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 8607; with PTC catalysis: (i) Novacek, J.; Waser, M. Eur. J. Org. Chem. 2013, 4, 637; (j) Shirakawa, S.; Maruoka, K. Tetrahedron Lett. 2014, 55, 3833; with metal catalysis: (k) Song, J.; Guo, C.; Chen, P. H.; Yu, J.; Luo, S. W.; Gong, L. Z. Chem. Eur. J. 2011, 17, 7786; (l) Lang, K.; Park, J.; Hong, S. Angew. Chem., Int. Ed. 2012, 51, 1620. with phosphoramide: (m) Ding, M.; Zhou, F.; Liu, Y. L.; Wang, C. H.; Zhao, X. L.; Zhou, J. Chem. Sci. 2011, 2, 2035. (n) Gao, W. M.; Yu, J. S.; Zhao, Y. L.; Liu, Y. L.; Zhou, F.; Wu, H. H.; Zhou, J. Chem. Commun. 2014, 50, 15179.
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308. doi: 10.1039/B711844A
Howard, J. A. K.; Hoy, V. J.; O'Hagan, D.; Smith, G. T. Tetrahedron 1996, 52, 12613. doi: 10.1016/0040-4020(96)00749-1
Pauling, L. The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 1939, p. 28.
Dunitz, J. D.; Taylor, R. Chem. Eur. J. 1997, 3, 89. doi: 10.1002/(ISSN)1521-3765
Shimoni, L.; Glusker, J. P. Struct. Chem. 1994, 5, 383. doi: 10.1007/BF02252897
Thalladi, V. R.; Weiss, H. C.; Bla1ser, D.; Boese, R.; Nangia, A.; Desiraju, G. R. J. Am. Chem. Soc. 1998, 120, 8702.
Thakur, T. S.; Kirchner, M. T.; Bläser, D.; Boese, R.; Desiraju, G. R. CrystEngComm 2010, 12, 2079. doi: 10.1039/b925082d
Anzahaee, M. Y.; Watts, J. K.; Alla, N. R.; Nicholson, A. W.; Damha, M. J. J. Am. Chem. Soc. 2011, 133, 728. doi: 10.1021/ja109817p
Schneider, H. J. Chem. Sci. 2012, 3, 1381. doi: 10.1039/c2sc00764a
Zhao, X.; Wang, X. Z.; Jiang, X. K.; Chen, Y. Q.; Li, Z. T.; Chen, G. J. J. Am. Chem. Soc. 2003, 125, 15128. doi: 10.1021/ja037312x
Liu, Y. L.; Shi, T. D.; Zhou, F.; Zhao, X. L.; Wang, X.; Zhou, J. Org. Lett. 2011, 13, 3826. doi: 10.1021/ol201316z
(a) Pesenti, C.; Viani, F. ChemBioChem 2004, 5, 590. (b) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881. (c) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359. (d) Wang, J.; Liu, H. Chin. J. Org. Chem. 2011, 31, 1785(in Chinese). (王江, 柳红, 有机化学, 2011, 31, 1785.) (e) Ojima, I. J. Org. Chem. 2014, 44, 6358. (f) Liu, Y. L.; Yu, J. S.; Zhou, J. Asian J. Org. Chem. 2013, 2, 194. (g) Lin, J. H.; Xiao, J. C. Tetrahedron Lett. 2014, 55, 6147. (h) Ni, C. F.; Zhu, L. G.; Hu, J. B. Acta Chim. Sinica 2015, 73, 90(in Chinese). (倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90.) (i) Champagne, P. A.; Desroches, J.; Hamel, J. D.; Vandamme, M.; Paquin, J. F. Chem. Rev. 2015, 115, 9073. (j) Zhang, J.; Jin, C. F.; Zhang, Y. J. Chin. J. Org. Chem. 2014, 34, 662(in Chinese). (张霁, 金传飞, 张英俊, 有机化学, 2014, 34, 662.) (k) Swallow, S. Progress in Medicinal Chemistry 2015, 54, 65. (l) Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315. (m) Wang, G. M.; Zhu, Z. D.; Chen, Z. Q.; Xu, Z. J.; Zhu, W. L. Acta Pharmaceutica Sinica 2018, 53, 701(in Chinese). (王桂敏, 朱正诞, 陈照强, 徐志建, 朱维良, 药学学报, 2018, 53, 701.) (n) Ni, C. F.; Hu, J. B. Chem. Soc. Rev. 2016, 45, 5441. (o) Cahard, D.; Bizet, V. Chem. Soc. Rev. 2014, 43, 135. (p) Fustero, S.; Fuentes, A. S.; Barrio, P.; Haufe, G. Chem. Rev. 2015, 115, 871.
Champagne, P. A.; Desroches, J.; Paquin, J. F. Synthesis 2014, 47, 306. doi: 10.1055/s-00000084
Vachal, P.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 10012. doi: 10.1021/ja027246j
Jin, L. M.; Xu, X.; Lu, H. J.; Cui, X.; Wojtas, L.; Zhang, X. P. Angew. Chem., Int. Ed. 2013, 52, 5309. doi: 10.1002/anie.201209599
Yuan, H. N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J. A. Angew. Chem., Int. Ed. 2013, 52, 3869. doi: 10.1002/anie.v52.14
Lee, K. A.; Silverio, D. L.; Torker, S.; Robbins, D. W.; Haeffner, F.; Mei, F. W.; Hoveyda, A. H. Nat. Chem. 2016, 8, 768. doi: 10.1038/nchem.2523
(a) Liu, Y. L.; Zeng, X. P.; Zhou, J. Chem. Asian J. 2012, 7, 1759. (b) Liu, H. X.; Tao, Z.; Xie, Q.; Zhou, J.; Wang, X. Comput. Theor. Chem. 2018, 1142, 57. (c) Liu, Y. L.; Zhou, F.; Cao, J. J.; Ji, C. B.; Ding, M.; Zhou, J. Org. Biomol. Chem. 2010, 8, 3847. (d) Ji, C. B.; Cao, Z. Y.; Wang, X.; Wu, D. Y.; Zhou, J. Chem. Asian J. 2013, 8, 877.
For the related proton-transfer process, see: (a) Xia, Y. Z.; Liang, Y.; Chen, Y. Y.; Wang, M.; Jiao, L.; Huang, F.; Liu, S.; Li, Y. H.; Yu, Z. X. J. Am. Chem. Soc. 2007, 129, 3470. (b) Shi, F. Q.; Li, X.; Xia, Y. Z.; Zhang, L. M.; Yu, Z. X. J. Am. Chem. Soc. 2007, 129, 15503. (c) Li, X.; Ye, S. Y.; He, C.; Yu, Z. X. Eur. J. Org. Chem. 2008, 25, 4296.
Champagne, P. A.; Pomarole, J.; Thérien, M. È.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J. F. Org. Lett. 2013, 15, 2210. doi: 10.1021/ol400765a
Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J. F. Angew. Chem., Int. Ed. 2014, 53, 13835. doi: 10.1002/anie.201406088
Rosenberg, R. E. J. Phys. Chem. A 2012, 116, 10842. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23057647
Dalvit, C.; Invernizzi, C.; Vulpetti, A. Chem. Eur. J. 2014, 20, 11058. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM25044441
For our efforts in selective fluoroalkylation using fluorinated enol silyl ethers, see: (a) Liu, Y. L.; Zhou, J. Chem. Commun. 2012, 48, 1919. (b) Liu, Y. L.; Zhou, J. Acta Chim. Sinica 2012, 70, 1451(in Chinese). (刘运林, 周剑, 化学学报, 2012, 70, 1451.) (c) Liu, Y. L.; Liao, F. M.; Niu, Y. F.; Zhao, X. L.; Zhou, J. Org. Chem. Front. 2014, 1, 742. (d) Liao, F. M.; Liu, Y. L.; Yu, J. S.; Zhou, F.; Zhou, J. Org. Biomol. Chem. 2015, 13, 8906. (e) Yu, J. S.; Zhou, J. Org. Biomol. Chem. 2015, 13, 10968. (f) Yu, J. S.; Liao, F. M.; Gao, W. M.; Liao, K.; Zuo, R. L.; Zhou, J. Angew. Chem., Int. Ed. 2015, 54, 7381. (g) Yu, J. S.; Zhou, J. Org. Chem. Front. 2016, 3, 298. (h) Zeng, X. P.; Zhou, J. J. Am. Chem. Soc. 2016, 138, 8730. (i) Liao, F. M.; Cao, Z. Y.; Yu, J. S.; Zhou, J. Angew. Chem., Int. Ed. 2017, 56, 2459. (j) Hu, X. S.; Du, Y.; Yu, J. S.; Liao, F. M.; Ding, P. G.; Zhou, J. Synlett 2017, 28, 2194. (k) Liao, F. M.; Gao, X. T.; Hu, X. S.; Xie, S. L.; Yu, J. S.; Zhou, J. Sci. Bull. 2017, 62, 1504. (l) Liao, F. M.; Du, Y.; Zhou, F.; Zhou, J. Acta Chim. Sinica 2018, 76, DOI: 10.6023/A18060238(inChinese).(廖富民,杜溢,周锋,周剑,化学学报,2018,76,DOI: 10.6023/A18060238).
Yu, J. S.; Liu, Y. L.; Tang, J.; Wang, X.; Zhou, J. Angew. Chem., Int. Ed. 2014, 53, 9512. doi: 10.1002/anie.201404432
For the use of chiral Lewis base to activate silyl reagents, see: (a) Tian, S. K.; Deng, L. J. Am. Chem. Soc. 2001, 123, 6195. (b) Tian, S. K.; Hong, R.; Deng, L. J. Am. Chem. Soc. 2003, 125, 9900. (c) Fuerst, D. E.; Jacobsen, E. N. J. Am. Chem. Soc. 2005, 127, 8964. (d) Wang, J.; Hu, X. L.; Jiang, J.; Gou, S. H.; Huang, X.; Liu, X. H.; Feng, X. M. Angew. Chem., Int. Ed. 2007, 46, 8468. (e) Wang, J.; Wang, W. T.; Li, W.; Hu, X. L.; Shen, K.; Tan, C.; Liu, X. H.; Feng, X. M. Chem. Eur. J. 2009, 15, 11642. (f) Liu, Y. L.; Zhou, J. Chem. Commun. 2013, 49, 4421. (g) Zhao, Y. L.; Cao, Z. Y.; Zeng, X. P.; Shi, J. M.; Yu, Y. H.; Zhou, J. Chem. Commun. 2016, 52, 3943. For a review: Liu, Y. L.; Zhou, J. Synthesis 2015, 47, 1210.
Aronoff, M. R.; Gold, B.; Raines, R. T. Tetrahedron Lett. 2016, 57, 2347. doi: 10.1016/j.tetlet.2016.04.020
Aronoff, M. R.; Gold, B.; Raines, R. T. Org. Lett. 2016, 18, 1538. doi: 10.1021/acs.orglett.6b00278
Lou, H. Q.; Wang, Y. T.; Jin, E. Z.; Lin, X. F. J. Org. Chem. 2016, 81, 2019. doi: 10.1021/acs.joc.5b02848
Liu, Y. B.; Hu, L. R.; Chen, H.; Du, H. F. Chem. Eur. J. 2015, 21, 3495. doi: 10.1002/chem.201405388
(a) Wang, X. M.; Han, Z. B.; Wang, Z.; Ding, K. L. Angew. Chem., Int. Ed. 2012, 51, 936. (b) Wang, X. M.; Meng, F. Y.; Wang, Y.; Han, Z. B.; Chen, Y. J.; Liu, L.; Wang, Z.; Ding, K. L. Angew. Chem., Int. Ed. 2012, 51, 9276.
(a) Cao, Z. Y.; Wang, X. M.; Tan, C.; Zhao, X. L.; Zhou, J.; Ding, K. L. J. Am. Chem. Soc. 2013, 135, 8197. Also see: (b) Cao, Z. Y.; Zhou, F.; Zhou, J. Acc. Chem. Res. 2018, 61, 1443. (c) Cao, Z. Y.; Zhou, J. Org. Chem. Front. 2015, 2, 849.
Cao, Z. Y.; Wang, W. M.; Liao, K.; Wang, X.; Zhou, J.; Ma, J. Org. Chem. Front. 2018, 5, 2960. doi: 10.1039/C8QO00842F
We also reported a Hg-catalyzed cyclopropanation of diazooxindoles with alkenes, and found that the N-methyl group of diazooxindole had no negative influence on enantioselectivity, possibly because the reaction solvent is not fluorinated solvents, see: Cao, Z. Y.; Zhou, F.; Yu, Y. H.; Zhou, J. Org. Lett. 2013, 15, 42.
(a) Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2007, 129, 10646. (b) van Zeist, W. J.; Bickelhaupt, F. M. Org. Biomol. Chem. 2010, 8, 3118. (c) Bickelhaupt, F. M.; Houk, K. N. Angew. Chem., Int. Ed. 2017, 56, 10070.

图 1 氟取代效应对Strecker反应活性的影响
Figure 1 Fluorine effect on reactivity of Strecker reactions

图 2 氟取代效应对Strecker反应对映选择性的影响
Figure 2 Fluorine effect on enantioselectivity of Strecker reaction

图 4 β-酮酸与氟代酮亚胺的不对称脱羧Mannich反应
Figure 4 Asymmetric decarboxylative Mannich reaction of β-ketoacid to α-CF3 ketimines

图 5 烯丙基硼试剂对三氟甲基酮的不对称加成反应
Figure 5 Asymmetric addition of allyl boron reagents to α-CF3 ketones

图 6 氟代酮与联烯基硼试剂的不对称加成反应
Figure 6 Asymmetric addition of allenyl boron reagents to α-CF3 ketones

图 7 乙二醇促进的无催化剂的氟代酮亚胺的Strecker反应
Figure 7 Catalyst-free Strecker reaction of α-CF3 ketimines in glycol

图 8 4-苯基苄氟与吗啉的SN2亲核取代反应
Figure 8 SN2 substitution of morpholine to 4-phenylbenzyl fluoride

图 9 苄基氟化物与芳基化物的Friedel-Crafts反应
Figure 9 Friedel-Crafts reactions between benzyl fluoride and arene

图 10 HFIP促进的Friedel-Crafts反应的可能机理
Figure 10 Possible mechanism for HFIP promoted alkylation reaction

图 11 无催化剂的“在水上”二氟烯醇硅醚的亲核加成反应
Figure 11 "On-water" catalyst-free reaction of difluoroenoxysilane

图 12 手性碱催化的“在水上”不对称aldol反应
Figure 12 Chiral base catalyzed asymmetric "on water" aldol reaction

图 14 氟效应对aza-Friedel-Crafts反应的影响
Figure 14 Fluorine effect on the reaction of aza-Friedel-Crafts

图 16 金催化的环丙烷化反应中的氟代溶剂效应
Figure 16 The influence of fluorinated solvents in Au-catalyzed cyclopropanation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:




 下载:
下载: