引用本文:
金维则, 陆国林, 李永军, 黄晓宇. 耐极端条件含氟材料的研究进展[J]. 化学学报,
2018, 76(10): 739-748.
doi:
10.6023/A18080340

Citation: Jin Weize, Lu Guolin, Li Yongjun, Huang Xiaoyu. Recent Advances in Fluorine-containing Materials with Extreme Environment Resistance[J]. Acta Chimica Sinica, 2018, 76(10): 739-748. doi: 10.6023/A18080340
Citation: Jin Weize, Lu Guolin, Li Yongjun, Huang Xiaoyu. Recent Advances in Fluorine-containing Materials with Extreme Environment Resistance[J]. Acta Chimica Sinica, 2018, 76(10): 739-748. doi: 10.6023/A18080340
耐极端条件含氟材料的研究进展
摘要:
耐极端条件材料是指能在普通碳氢材料不耐受的极端环境(如:高温≥ 200℃、低温≤-50℃、紫外辐射1000 h等)下正常使用的材料.而电负性最强的氟元素与碳形成的碳氟键是最强的分子键之一,引入氟原子的材料也展示出特别优异的各项性能,因此含氟化合物在耐极端条件材料方面的应用研究得到了广泛的关注.本文对耐极端条件含氟材料的发展历史、研究现状与发展趋势进行了综述.首先,对氟化方法,包括碳-氟键形成及断裂、氟对碳-碳键形成的影响以及多氟芳基化方法等进行了介绍.接着,介绍了满足锂同位素萃取的苛刻条件的含氟萃取剂,应用于航空、航天、汽车等领域的含氟耐热材料,以及应用于电子设备、通信领域的高性能含氟低介电常数材料的相关研究.将来如何进一步发展优化耐极端条件含氟材料,并将这些研究成果投入大规模使用是科研人员努力的方向.
English
Recent Advances in Fluorine-containing Materials with Extreme Environment Resistance
Abstract:
The extreme environment resistance materials can be normally used under severe conditions (e.g. T ≤ -50℃ or T ≥ 200℃, 1000 h exposed to UV light etc.), which common hydrocarbon materials cannot tolerate. It was found that fluorine atoms can effectively enhance the extreme environment resistance property of materials. The reason why fluorine atoms have such ability is mainly due to two key factors:first, fluorine and carbon elements are in the same cycle of the periodic table, the electronegativity of fluorine is large (4.0) and its atomic radius is small; second, polarization of fluorine atom is extremely low. The C-F bond is the strongest chemical single bond (≥ 116 kcal/mol) in which the carbon atom participates. It is a short bond and highly polarized. This paper makes brief introduction to the development and present situation of fluorine-containing materials with extreme environment resistance. In the field of fluorination methods, the history of fluorine chemistry since 1970s, the researches on the formation and fracture of carbon-fluorine bond, the influence of fluorine on the formation of carbon-carbon bond and the related researches on polyfluoro-arylation methods are introduced. This paper also introduces the important results of fluorine-containing materials in lithium isotope extraction, thermo-stable fluoropolymers which can be applied in aviation, aerospace, automobile and other fields, as well as the preparation of high-performance fluorine-containing materials with low dielectric constant in electronic equipment and communication fields. In the future, how to further develop and optimize the fluorine-containing materials with extreme environment resistance and put the research results into large-scale use is the working direction for researchers.
-
Key words:
- extreme environment resistance
- / fluorine-containing material
- / nanomaterial
- / polymer
- / synthesis
-
1. 引言
材料发展是人类社会进步的重要基础. 20世纪以来, 科学技术飞速发展, 每一项重大突破都和新材料密不可分.其中, 有机纳米材料是一类具有广泛应用并且不可替代的重要新材料.有机纳米材料的粒子尺度在1~100 nm[1], 与金属和非金属纳米材料相比, 具有加工性能好、质轻、结构可控等优点.近几年来, 有机纳米材料在飞速发展的同时, 还面临着一个很严重的技术瓶颈, 就是一般的有机纳米材料在极端条件下无法保持原有性状.这是由有机纳米材料中碳-氢键的键能相对较弱(约99 kcal/mol), 容易被氧化等特点所造成的[2].极端条件是指普通碳氢材料不能耐受的使用环境, 如:高温(≥200 ℃)、低温(≤-50 ℃)、强酸强碱(浓度5 mol/L)、紫外辐射(UV1000 h)、高频(5 GHz)等.在核工业、航空航天等许多领域中, 具有高耐受能力的材料都必不可少, 材料性能的优良直接关系到一个国家在该行业所能达到的技术高度, 因此, 耐极端条件材料成为了各国科研界的重要研究方向之一.
学术研究表明, 将氟原子引入材料中能有效增强材料耐极端条件的性能.美国杜邦公司研发的含氟聚酰亚胺Kapton FCR就是一种性能良好的耐高频材料[3].氟原子之所以具备这样的能力主要是由于两个关键因素:一是, 氟元素与碳元素处于元素周期表的同一周期, 电负性大(4.0)、原子半径小; 二是, 氟原子可极化性极低.碳-氟键是碳原子参与的最强化学单键(一般大于116 kcal/mol), 它键短, 并且高度极化.诺贝尔化学奖得主Barry Sharpless教授指出: “(在化学和材料科学中)真正能代替碳-氟键的化学键根本不存在, 就连稍微接近一点的都找不到(…an honest surrogate for a C-F unit does not exist--nothing even comes close.)”[4].因此, 研究含氟的耐极端条件材料具有非常重要的科研价值和实用意义.
本文综述了氟化技术的研究进展, 可用于核工业、通信以及航天领域的含氟耐极端条件材料的最新研究动态, 详细介绍了多个科研团队在该领域的重要成果, 为耐极端条件含氟材料的未来发展方向提供借鉴.
2. 高选择性氟化方法
新氟化试剂的开发是有机氟化学研究中永恒的主题, 它为化学合成中氟原子的引入提供了有效的手段.发展条件温和、环境友好、使用方便的氟化试剂是目前氟化学研究的新趋势. 20世纪70年代, Olah发展了吡啶合氟化氢这一有效氟试剂(Olah试剂), 促进了各种氟化试剂的相继开发.目前商品化亲电氟化试剂有NFSI、DFI、Selectfluor和DeoxoFluor等.商品化亲核三氟甲基试剂有Ruppert-Prakash试剂(TMSCF3), 亲电三氟甲基试剂有Umemoto试剂[5]和Togni试剂[6]等.美国的Lanxess公司和3M公司开发的氟化试剂—全氟丁基磺酰氟(PBSF)目前已达几万吨的生产规模.中国科学院上海有机化学研究所陈庆云院士等开发的Chen试剂也已达几百公斤级的生产规模[7, 8].然而, 目前大部分有机氟试剂的合成都是以氟气为起始原料开始, 大量制备中需要特种设备, 费用昂贵, 而且环境污染比较严重.因此, 开发简单实用高效和绿色的有机氟试剂, 对于有机氟化学的发展具有重要的现实意义.
碳-氟键的生成是有机氟化学的核心问题.对于芳烃的碳-氟键生成, 经典的方法是将氟原子选择性地引入芳环化合物指定位置的Balz-Schiemann反应, 但是该方法使用了有害危险的重氮盐.氟卤直接交换法是另一种合成一些特殊氟化芳环化合物可靠的方法, 然而该方法只能应用于活化的芳环底物.近年来, 许多化学家致力于过渡金属催化的芳烃碳-氟键的生成研究, 是目前有机化学研究的热点之一.美国杜邦公司的Grushin[9]研究了不同配体络合的含氟钯键的钯络合物的基元反应, 发现碳-氟键的还原消除反应较慢.在Grushin工作的基础上, 美国麻省理工学院的Buchwald教授[10]发现了富电子和高空间位阻的膦配体与钯形成的络合物能有效地催化芳烃碳-氟键的生成, 该工作发表在Science上, 是有机氟化学研究领域的一个重大发现.美国的Scripps研究所的余金权教授[11]和Michigan大学的Sanford教授[12]实现了钯催化的芳烃碳-氢键活化来生成碳-氟键.哈佛大学的Ritter教授[13]利用高价钯络合物, 实现了碳-硼键的激活和与亲电氟化试剂的重组生成碳-氟键.此外, Ritter小组[14]还发展了银催化的芳烃碳-氟键的生成.美国的Dimagno教授[15]利用反应活性高的无水TBAF实现了温和条件下的亲核取代生成碳-氟键, 德国的Beller教授[16]则用Grignard试剂与亲电氟化试剂的亲核取代生成碳-氟键.对于烷烃的碳-氟键形成, 一般采用亲核性氟化试剂如DAST和DeoxoFluoro进行脱氧氟化, 但该法所用试剂存在昂贵、腐蚀性大, 不安全等局限.利用有机氟盐如TBAF, 以及氟化氢吡啶或者三乙胺盐与亲电试剂如环氧化合物或者烯烃进行反应是另外一种方式, 然而该方法在底物上受到了限制.除了利用亲核氟化试剂外, 利用亲电试剂如N-氟二苯磺酰亚胺(NFSI)和Selectfluoro等, 与高活性的亲核试剂如烯醇负离子进行反应也是一种行之有效的方法, 但依然存在底物受限的缺点.最近, 国内外学者通过自由基化学也发展了一系列反应温和的直接氟化方法.美国的Boger教授[17]利用Selectfluoro在铁的参与下与烯烃反应, 将氟原子引入到烷烃上.普林斯顿大学的Groves教授[18]通过卟啉锰催化剂成功实现了对烷烃C—H键的选择性氟化.上海有机化学研究所的李超忠研究员[19]发展了卓有成效的Ag催化脱羧氟化反应, 为烷烃的氟化提供了新思路.天津大学的唐向阳和王光伟教授[20]开发了一种有效且通用的铜催化方法来构建3, 3-二氟-γ-内酰胺衍生物, 该方法的底物范围广, 氮取代前体易于制备, 反应条件温和, 并能高效地在一步反应中实现环化.南京大学的朱成建教授[21]通过可见光诱导, 以二氟溴乙酸乙酯为烷基化试剂对高炔丙醇进行二氟烷基化, 这是一种简便的通过串联自由基二氟烷基化和1, 4-芳基迁移过程获得官能二氟化烯烃的方法.
含氟芳烃的亲核取代脱氟和Lewis酸条件下碳-氟键断裂在过去几十年里已被系统研究, 而金属催化的温和条件下选择性断裂碳-氟键并官能团化是近年来的研究热点[22].以色列的Milstein小组[23], 美国的McNeill小组[24]和日本的Murai小组[25]报道了铑催化的含氟芳烃或烯烃脱氟氢化反应.德国的Herrmann小组[26], Braun小组和英国的Perutz小组[27], 法国的Mongin小组[28]实现了镍催化的含氟芳烃的碳-氟键断裂并与格氏试剂重组.德国的Radius小组[29]则报道了镍催化的含氟芳烃的碳-氟键断裂并与苯硼酸的重组.钯催化的含氟芳烃的碳-氟键断裂并与苯硼酸的重组也有报道.英国的Widdowson[30], 美国Pfizer公司的Yu[31], 德国的Sanford[32]和日本的Mikami[33]均实现了这一转化.
长碳链的全氟烷基取代的有机化合物同时具有疏水性和疏油性, 其聚合物在表面处理领域应用广泛. 90年代中期发现长碳链的全氟烷基有机物在生态环境中不易降解, 同时可能对人有致癌作用.长碳链的全氟烷基有机物中的碳-氟键断裂降解很不容易, 国际上相关的研究刚刚起步.美国的Ozerov小组[34]在Science上发表了杂硼原子簇催化的长碳链全氟烷基有机物中碳-氟键断裂降解的文章, 引起了广泛的关注.
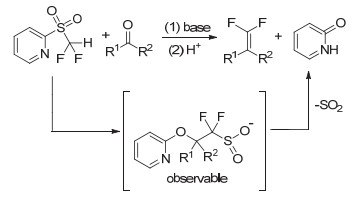
由于氟原子的特性, 含氟化合物参与的碳-碳键的形成反应往往与不含氟的化合物差别较大.卤代芳烃的三氟甲基化反应中的氟效应是目前有机氟化学研究的一个重点. Grushin[35, 36]系统研究了不同配体络合的含三氟甲基钯络合物的基元反应, 发现碳-三氟甲基的还原消除反应比碳-甲基还原消除反应慢很多, 但在一定条件下产率较高, 这些研究为实现催化的卤代芳烃的三氟甲基化提供了非常有价值的基础. Buchwald[37]随后发现了富电子和大位阻的磷配体与钯形成的络合物可有效催化氯代芳烃的三氟甲基化反应, 这是有机氟化学研究领域的重大发现.但该反应的催化剂用量较大, 如何提高催化剂的活性是该领域的一个挑战.铜参与或催化的卤代芳烃三氟甲基化是制备三氟甲基取代芳烃的主要方法, 但是反应局限于芳基溴、碘代物的三氟甲基化.我国学者在卤代芳烃的三氟甲基化方面也做出了重要贡献, 上海有机化学研究所的陈庆云院士发展了一个三氟甲基化试剂, 国际上称为Chen试剂[7, 8].卿凤翎小组[38, 39]实现了铜参与下的末端炔烃或苯基硼酸与亲核三氟甲基化试剂CF3SiMe3的氧化偶联反应, 反应的适用范围较广, 这是一种选择性地向有机分子引入三氟甲基的新方法.沈其龙小组[40, 41]发展了铜催化下亲电三氟甲基化Togni试剂与苯基、烯基硼酸的三氟甲基化方法.虽然这些研究导致了一些新颖三氟甲基化反应的发现, 但是该反应作用机理仍然不清楚.此外, 含一氟亚甲基和二氟亚甲基的有机物参与的碳-碳键生成的反应与不含氟有机物的反应差别很大.这方面美国的Prakash教授和上海有机化学研究所的胡金波研究员做了许多有特色的工作[42~44].如胡金波小组由2-巯基吡啶反应制备了二氟甲基(2-吡啶基)砜试剂, 该试剂能高效地将醛酮类物质反应得到偕二氟烯烃(图 1).这种新型二氟烯化方法操作简单, 应用范围广, 可大规模应用于有机合成领域.二氟甲基(2-吡啶基)砜试剂在国际上被同行称为“胡试剂”(Hu reagent)[43~45].
图 1
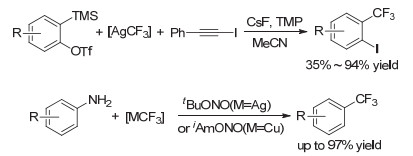
多氟芳基化是制备多氟芳烃的一种重要技术.多氟芳烃是另一类重要的含氟化合物, 在材料科学中具有十分重要的应用.例如:多氟芳烃结构作为一种重要结构单元也存在于许多功能材料中, 如液晶材料、光电材料和太阳能电池等.传统方法主要是通过官能团化多氟芳烃进行多步转化引入氟芳基, 但这种方法存在底物适用性窄, 原子不经济、官能团兼容性差等缺陷.因此, 发展高效简捷向有机分子中引入氟芳基的方法很重要, 是含氟功能分子的重要需求[46~49].胡金波小组[50]在2013年首次用AgCF3一步法实现了芳炔的三氟甲基化, 为合成邻三氟甲基碘代芳烃提供了有效途径, 解决了邻三氟甲基碘代芳烃通过一般的三氟甲基化方法难合成的问题(图 2). Sandmeyer反应是一种经典的人名反应, 该反应通过用亲核试剂置换其重氮盐将芳基胺转化为取代的芳烃.北京大学王剑波小组[51]和中国科学技术大学傅尧小组[52]在2013年分别首次报道了用AgCF3和CuCF3进行Sandmeyer式三氟甲基化反应的成果(图 2).
图 2

3. 有机含氟萃取材料
随着世界人口数量的激增, 人类社会对能源的需求量呈指数上升.目前, 世界上使用的能源有85%为非可再生资源, 包括:石油、天然气和煤炭.按目前的速度, 地球上的石油储量仅能维持人们继续消耗几十年, 石油危机使得对新能源和可再生能源的开发迫在眉睫.现在商业化运行的原子能电站是基于铀同位素的可控裂变反应堆.由于全球铀资源极其有限, 而且铀裂变反应的产物是长寿命的放射碎片, 一旦泄露或处理不当将给人类带来极大的灾难.因此, 开发可控聚变堆代替裂变堆日益受到世界各国的广泛关注.聚变反应是利用氘(D)和氚(T)聚合成氦(4He)核时放出的巨大核能:
D+T → 4He (3. 52 MeV)+n (14. 06 MeV)
自然界中几乎不存在氚, 必须依靠中子轰击6Li来产生和不断增殖:
6Li+n → T+4He
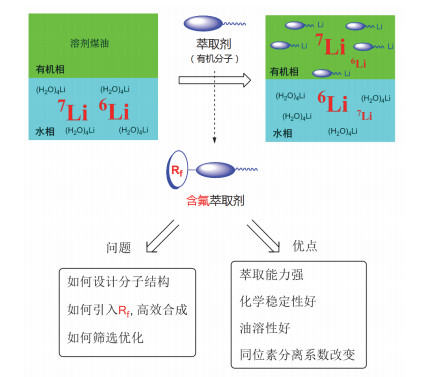
聚变反应堆可获得的能量受到锂同位素资源限制, 因此锂成了人类未来最终的战略资源.锂(Li)是最轻的金属, 其最大用途在于其可以提供新的能源.据估算, 1 kg锂含有的能量大约相当于20000吨优质煤的燃烧.元素锂在自然界有6Li和7Li两种稳定同位素, 其丰度分别约为7.5%和92.5%. 6Li和7Li在核反应中均有着极其重要的作用. 6Li被中子轰击后生成氚和氦, 使得聚变堆中氚能不断增殖, 所以6Li是必不可少的核聚变堆燃料. 7Li则被用来作为核聚变反应堆的堆心冷却剂和导热的载热剂, 7Li还可以作为钍堆熔盐介质.将元素锂的同位素6Li和7Li分离的过程称为锂的同位素分离.因为6Li在核发电和氢弹生产中具有非常重要的作用, 世界各国对锂同位素的分离都在紧张秘密进行.现在分离方法很多, 大致可分为化学法和物理法.化学法包括锂汞齐法[53]、离子交换色层分离[54~58]、萃取[59]、分级结晶和分级沉淀等; 物理法包括电磁法、熔盐电解法、电子迁移[60]、分子蒸馏和激光分离[61, 62]等.目前已用于工业生产的方法只有锂汞齐法.锂汞齐法要使用大量金属汞, 受汞量不足限制难以大规模提高生产能力, 不能满足民用核能的需求.另外, 汞对人体和环境的危害也是制约其发展的一个重要因素.因此, 最近国际上开始兴起了利用有机萃取剂材料通过有机和水两相的快速平衡, 以及串级萃取工艺来实现锂同位素的分离.
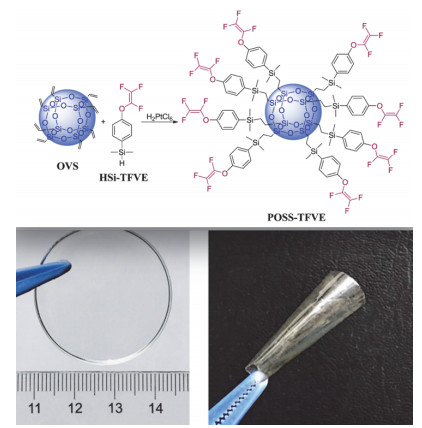
目前已知的有机萃取剂处理包括冠醚、二酮、偶氮、酰胺等几大类.这些有机萃取剂材料有些在同位素分离系数方面已经接近汞齐法水平, 但是这些有机萃取剂材料的一大缺点是耐腐蚀能力较弱, 在长时间串级萃取过程中萃取剂容易发生降解, 从而使得这些萃取剂材料最终不能用于实用化生产过程中.到目前为止, 国际上还很少有采用有机含氟萃取材料来分离锂同位素的报道.近年来, 上海有机化学研究所采用氟化技术, 首次在锂同位素分离类有机萃取剂材料中引入氟原子, 极大地提高了有机萃取剂材料的耐腐蚀能力以及抗氧化能力, 成功实现了15级串级萃取分离锂同位素, 从实验上再一次证实了氟原子引入对于提高有机材料耐极端条件性能的重要性.胡金波小组在现有锂同位素萃取剂的基础上, 引入含氟基团, 得到了一种新型含氟锂同位素萃取剂(图 3)[63~67].含氟基团可调节锂原子的配位环境和锂氧键的力常数, 提高了萃取能力和同位素分离能力, 所以通过萃取剂中氟原子的调控, 可提高萃取体系的综合分离性能及长期化学稳定性.含氟萃取剂还具有化学稳定性好和油溶性好的特点.随后, 该小组通过分子设计、合成、再优化制得高效含氟萃取剂, 进一步开展工艺研究, 得到了最优化的工艺体系[68~71], 实现了锂同位素的多级萃取富集.突破了新型液液两相萃取工艺技术, 最终应用于锂同位素分离、锂离子萃取分离的工程装置中.
图 3
 图 3. 含氟萃取剂材料在锂同位素分离中的应用Figure 3. Application of fluorine-containing extractant materials in lithium isotope separation
图 3. 含氟萃取剂材料在锂同位素分离中的应用Figure 3. Application of fluorine-containing extractant materials in lithium isotope separation4. 有机含氟高分子纳米材料
4.1 含氟耐热材料
有机含氟高分子纳米材料因为其优异的耐热性、耐腐蚀性、耐候性和电绝缘性等特点, 已被广泛应用于航空、航天、汽车、电子信息等领域, 对国家科技的进步、社会经济的发展和人民生活水平的提高具有不可替代的地位.因此, 研究有机含氟高分子纳米材料的制备及其热学、电学性能, 对于发展高性能绝缘材料和耐热材料有着重要的意义.
耐热性方面, 多项研究表明氟原子的引入能使材料的耐热性有显著提高.美国科罗拉多矿业学院Sellinger小组[72]发表了一种性能优异能耐极端条件的塑料闪烁体用于脉冲形状甄别体系, 他们用双酚A二甲基丙烯酸酯(BPA-DM)和双酚AF二甲基丙烯酸酯(BPAF-DM)作为交联剂合成闪烁体, BPAF-DM中的氟原子可以改善闪烁体的热学性能和疏水性.
美国Dow公司于1993年研发了一类新型含氟聚合物即全氟环丁基芳基醚聚合物[73], 这类聚合物主链上含有芳香基团和全氟环丁基结构, 它不仅具有低的介电常数、优异的化学稳定性、光透明性和热稳定性, 还具有普通含氟高分子材料所不具备的溶剂溶解性和熔融性(该类聚合物的Tg一般在150~300 ℃之间), 解决了含氟高分子材料加工性差的问题.
中国科学院上海有机化学研究所卿凤翎小组[74]应用点击化学的方法, 合成了含有全氟环丁基芳基醚结构的高性能聚合物.他们将聚乙二醇叠氮化物和1, 2-双(4-乙炔基苯氧基)全氟环丁烷在铜催化作用80 ℃下进行Huisgen 1, 3-偶极环加成, 得到了交替共聚物, 并且产率很高.随后, 他们[75]又通过点击化学方法, 利用1, 2-双(4-叠氮甲基苯氧基)全氟环丁烷和双乙炔基化合物合成了一种含有全氟环丁基芳基醚和三唑结构的新型线性芳香醚聚合物.这两种新型聚合物都具有优异的热稳定性, 在常规有机溶剂中表现出良好的溶解性.
目前国内各类飞机的舷窗均使用普通的聚甲基丙烯酸甲酯(PMMA)或聚碳酸酯(PC)树脂, 其最高耐热温度达到130 ℃, 可满足目前各类飞机的需求.但是随着国家国防事业的发展, 研发飞行速度超过5倍音速的超高速飞行器已开始进行紧锣密鼓的布局, 如此高的飞行速度必然导致摩擦升温, 对舷窗所使用的材料的耐热温度提出了新的要求. PMMA是透明性最好的聚合物材料之一, 透光率高达92%.其具有许多优良的性能:如优异的光学性能, 较高且均衡的机械强度, 良好的加工性能及电绝缘性能, 且价格低, 原料易得, 有很大的成本优势.但是普通PMMA有机玻璃的耐热性差, 热变形温度(即玻璃化转变温度Tg)低于130 ℃, 热分解温度低于225 ℃, 另外其不耐有机溶剂, 从而大大限制了PMMA有机玻璃的应用.通过将甲基丙烯酸甲酯与少量第二单体(如马来酸酐、甲基丙烯酸环己基酯和甲基丙烯酸金刚烷酯等)共聚, 可改善PMMA树脂的耐热性.尽管通过这种方法得到的共聚物, 耐热性能有所改善, 但是其它性能不可避免地受到影响.也有报道合成结构特殊的氟代和三氟甲基丙烯酸酯类单体, 用于提高聚甲基丙烯酸酯类聚合物的耐热性能(Tg可达到142 ℃), 但是单体合成困难且成本高, 而且由于双键的碳上连入了含氟基团, 聚合行为复杂, 很难大量使用.
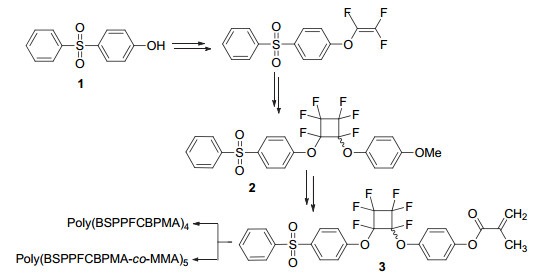
上海有机化学研究所黄晓宇小组[76]合成了一种新型含有全氟环丁基芳基醚和磺酰基结构的甲基丙烯酸酯类单体(图 4).这类甲基丙烯酸酯单体可以2, 2-偶氮二异丁腈为引发剂, 进行自由基聚合; 也可以2-溴丙酸甲酯为引发剂, CuBr/PMDETA为催化体系, 进行原子转移自由基聚合(ATRP)来完成共聚.由此合成的聚甲基丙烯酸酯类聚合物具有优异的热稳定性, 分解温度随相对分子质量的增加大幅上升.由于全氟环丁基芳基醚结构位于侧链上, 对双键没有影响, 不会影响其聚合, 得到结构单一的均聚物, 不会产生相容性的问题, 而且全氟环丁基芳基醚结构的存在在有效提高聚甲基丙烯酸酯类聚合物的玻璃化转变温度的同时, 其透光率等性能不会受到影响.
图 4

最近, 梁永民小组[77]成功开发出一种通过钯催化体系来构建含有共轭氟代烷基的1, 3-烯炔和1, 4-烯-3-酮的方法, 并成功合成了具有优异耐化学性、耐高温和优异疏水性的新型功能性含氟聚合物材料.该材料在温度达到400 ℃时重量损失仅有10%, 热稳定性十分出色, 未来在航空航天领域具有广阔的应用前景.
4.2 高性能绝缘材料
随着电子信息技术的突飞猛进, 电子产品正朝着轻量薄型化、高性能化和多功能化的方向发展.进入21世纪以来, 特别是近几年, 超大规模集成电路(ULSI: Ultra Large Scale Integrated Circuit)器件的集成度越来越高, 比如Intel公司所生产的酷睿2双核处理器的特征尺寸已经达到65 nm.当器件的特征尺寸逐渐减小时即集成度不断提高时, 会引起电阻-电容延迟上升, 从而出现信号传输延时、噪声干扰增强和功率损耗增大等一系列问题, 这将极大限制器件的高速性能.降低电阻-电容延迟和功率损耗有两个途径, 一是降低导线电阻也就是用铜取代传统的铝来制备导线, 另外一个同时也是更重要的是降低介质层带来的寄生电容.由于电容正比于介电常数, 所以就需要开发新型、低成本以及具有良好性能的绝缘材料(介电常数小于3)来代替传统的SiO2(介电常数约为4.0)作为介质层.而对于应用于金属间的绝缘材料, 除了满足绝缘性能的要求外, 还必须具有较高的热稳定性, 因为在器件的制造过程中需经历较高的加工温度, 例如金属互联线的成型就需要在400~450 ℃的高温条件下进行.
含氟高分子由于其突出的耐热性、耐化学腐蚀性、耐候性和电绝缘性等性能, 广泛应用于绝缘材料领域, 目前也有大量关于含氟聚合物绝缘材料的报道, 主要包括以下几类聚合物: (1)含氟聚酰亚胺: Watanabe等[78]用芳香二酐与含有亚苯基醚和全氟联苯结构的芳香二胺合成了含氟聚酰亚胺, 介电常数为2.65~2.68, 5%热失重温度为450 ℃左右. (2)含氟聚芳基醚:性能最优异的为Ailhd Signal公司的FLARE系列产品[79], 具有低出气、高热稳定和优异的力学性能, FLARE2.0的介电常数为2.8, 玻璃化转变温度为400 ℃.该公司还为抛光FLARE膜开发了专用泥浆. Tsuchiya等[80]通过氧化偶联聚合的方法将4, 4-双(1-萘氧基)-2, 2-双(三氟甲基)联苯聚合成一种含有氟原子和萘结构的芳基醚聚合物, 该聚合物的介电常数在1~20 GHz范围内有一个小的单调递减(2.70~2.65), 他们认为这样低的介电常数是由于三氟甲基和大体积联萘结构的存在而导致聚合物自由体积的增加所引起的. (3)含氟聚芳基醚酮: Niu等[81]在聚芳基醚酮分子骨架上引入了体积较大的含氟基团支链, 支链的引入增加了聚合物的自由体积, 这既增加了聚合物在有机溶剂中的溶解度, 同时降低了聚合物的介电常数.该聚合物的介电常数为2.67, 同时在N, N-二甲基甲酰胺等溶剂中有较好的溶解性. (4)含氟苯并噁唑聚合物: Dang等[82]通过在主链结构上结合全氟异丙基来降低介电常数, 这是由几种机理同时起作用的, 包括疏水性、增加自由体积和减小电极化率, 氟化也能够提高聚合物的热稳定性, 得到的大部分苯并噁唑聚合物的热分解温度达533 ℃, 而介电常数在2.1~2.5之间. Fukukawa等[83]则通过引入金刚烷结构合成了一种含氟苯并噁唑聚合物, 该结构的引入既能降低介电常数, 又能增加热稳定性.其制得的含氟苯并噁唑聚合物的介电常数为2.55, 5%热失重温度为518 ℃. (5)含氟苯并噁嗪聚合物: Su等[84]合成一种含氟的B-α型苯并噁嗪, 并用含氟和无氟的B-α型苯并噁嗪共混以弥补含氟苯并噁嗪聚合物力学性能较低的问题, 其得到的最低的介电常数为2.36, 两者的配比为1:1, Tg为283 ℃, 5%热失重为368 ℃. (6)非晶聚四氟乙烯:四氟乙烯本身具有低介电常数(1.9~2.1), 较高的玻璃化转变温度和很好的力学性能, 但由于很难与金属进行粘合, 在微电子电路方面的应用主要利用化学气相沉积(CVD)工艺.美国Clemson大学Sharangpani等[85]设计了CAV设备工艺, 得到了性质更好的Teflon AF材料(介电常数1.9); Rosenmeyer等[86]从水乳液中得到了介电常数1.9~2.1的聚四氟乙烯薄膜. (7)聚全氟环丁烷:多(三氟乙烯基)单体在300~350 ℃活化1 min, 再在250 ℃固化1 h, 即制得介电常数2.35, 玻璃化转变温度400 ℃的聚全氟环丁烷.材料拉伸模量2.37 GPa, 最大吸水量0.2%.虽然目前含氟聚合物绝缘材料取得了很大的进展, 然而仍存在着种类单一, 生产成本高等缺点, 因此开发新种类的含氟聚合物具有重要的意义.另外, 鲜有关于含氟聚合物纳米材料的报道, 开发含氟聚合物纳米材料, 将纳米空穴引入到含氟聚合物中降低介电常数也是一个新的研究方向.
上海有机化学研究所房强小组[87]开展了一系列含氟耐高频低介电常数材料方面的研究.三氟乙烯基醚加热成环可得到全氟环丁基芳基醚聚合物, 他们设计了多官能度大分子单体, 从而得到了能向不同方向进行链增长的高分子量聚合物.该方法有效改善了含氟聚合物的加工性, 同时又能兼顾耐热性、透明性、绝缘性等优点.该反应通过一个带有季碳中心和三氟乙烯基醚基团为侧链的功能性单体实现, 该单体具有四面体骨架结构, 易于进行热交联, 得到了微孔(平均尺寸为8 Å)含氟聚合物.由于微孔的存在, 这种聚合物表现出绝佳的绝缘性能, 其介电常数在10 MHz以下低于2.29, 在高频5 GHz下的介电常数和介电损耗分别为2.36和1.29× 10-3, 优于传统的商用低介电材料, 同时聚合物具有良好的疏水性(99 ℃, 72 h吸水量小于0.08%)和透光率(400~1100 nm光源透过率为93%).
倍半硅氧烷(POSS)是一种具有独特笼型结构的纳米材料, 引入聚合物中可改善力学和介电等性能, 所以被广泛应用.近年来, 研究发现将POSS氟化能使材料拥有更好的疏水性、热稳定性、机械强度和抗氧化性等, 所以如何将POSS进行氟官能化成为了重要研究方向.该小组[88]通过一步Pt催化硅氢加成反应, 将氟引入到POSS中, 从而实现了POSS的氟烷基化和功能化(图 5).这种新型有机-无机杂交聚合物表现出良好的热稳定性(436 ℃下仅损失5 wt%重量)和透光率(1.5 mm厚度的材料能透过92%波长为400~1100 nm的光线). 40 Hz到30 MHz甚至5 GHz频率下, 仍能保持低介电常数(小于2.56)和低介电损耗(小于3.1×10-3), 甚至在潮湿环境中放置数日, 仍能保持稳定性能.这种良好的绝缘材料在微电子、高频通讯等领域有广阔的应用前景.
图 5

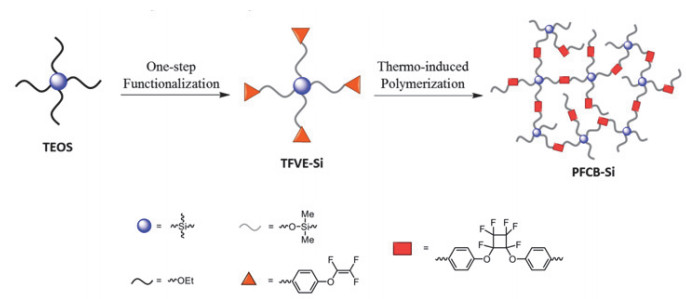
该小组[89]还发现, 有机硅氧烷(TEOS)参与的反应中产生的Si-OH和-Si-CH2-CH2-基团在高频下会影响聚硅氧烷的介电性能和热稳定性.于是, 他们研发了一种由TEOS进行Piers-Rubinsztajn反应而得到的, 含有四个官能团的氟化大分子单体, 可有效解决这个问题(图 6).该大分子单体热聚合后得到高透光率、低介电常数(2.50)、低介电损耗(4.0×10-3)的透明薄膜, 可应用于微电子领域, 尤其是高频通信方向.
图 6

4.3 有机含氟高分子纳米材料的生物毒性
含氟材料在前面章节所叙述的所有极端条件下的应用中都免不了和生物体发生接触.除此之外, 含氟材料在许多其他方面也有着重要的生物学应用.浙江理工大学的研究人员开发了一组由2-全氟辛基甲基丙烯酸酯(FMA)和甲基丙烯酸2-羟乙酯(HEMA)单体组成的氟化两亲共聚物, 并研究了它们的表面性质和防污性能, 证明了氟化两亲性共聚物表面具有优异的蛋白质抗性, 能在防污应用中起到重要作用[90].华东师范大学的程义云研究员[91]发表了专论, 详细论述了含氟高分子在基因载体中的研究, 其独特的理化性质能帮助改善传统的阳离子高分子作为基因载体时不稳定、有毒性等缺陷, 使其成为基因治疗、基因编辑的理想材料.
由于含氟材料具有广泛的生物应用, 使得氟的潜在毒性成为了一个重要研究方向.研究发现, 长期暴露在氟原子和含氟化合物环境中会损坏动物的大脑、骨骼、肾脏和牙齿等部位[92].例如, 全氟辛烷磺酸盐(PFOS)对哺乳动物有持续并且蓄积的毒性, 动物实验发现PFOS会导致癌症、内分泌紊乱、新生儿死亡等结果[93~95].对于有机含氟纳米材料而言, 有关它的生物毒性研究还不成熟, 但是近年来受到了越来越广泛的关注.
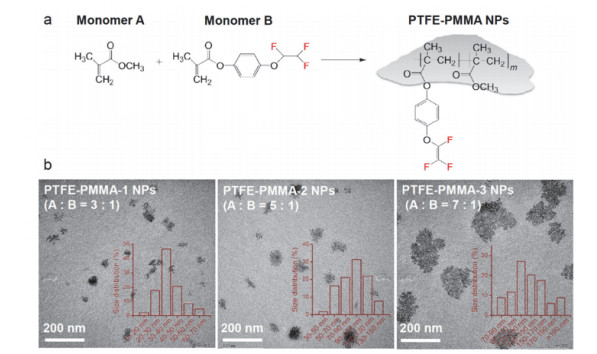
为了研究有机含氟纳米材料的生物毒性, 中国科学院生态环境研究中心杜宇国小组合成制备了三种不同的有机含氟纳米颗粒(PTFE-PMMA-1 NPs, PTFE- PMMA-2 NPs和PTFE-PMMA-3 NPs), 它们的氟含量分别为12.0 wt%, 6.1 wt%和5.0 wt% (图 7)[96~98].
图 7
 图 7. (a) 有机含氟纳米材料的合成反应; (b) PTFE-PMMA-1 NPs, PTFE-PMMA-2 NPs和PTFE-PMMA-3 NPs的电镜图和粒径分布[98]
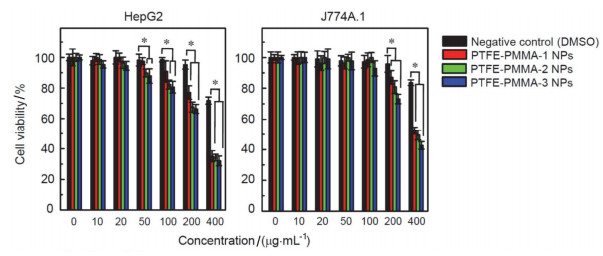
图 7. (a) 有机含氟纳米材料的合成反应; (b) PTFE-PMMA-1 NPs, PTFE-PMMA-2 NPs和PTFE-PMMA-3 NPs的电镜图和粒径分布[98]他们选取了人肝脏肿瘤细胞HepG2和小鼠巨噬细胞J774A.1为体外细胞模型, 研究了三种有机含氟纳米颗粒的细胞暴露的“剂量-效应关系”.实验结果显示在暴露剂量范围内(10~400 μg/mL), 当有机含氟纳米颗粒的浓度超过50 μg/mL时, 会显著影响两种细胞的活力(图 8).与阴性对照组(DMSO)相比, 细胞活力降低与暴露浓度呈现正相关的关系, 且PTFE-PMMA-3 NPs引起的细胞活力降低要强于PTFE-PMMA-1 NPs和PTFE-PMMA-2 NPs.以上结果说明, 与典型的人工纳米材料(如纳米银和氧化石墨烯)相比, 有机含氟纳米颗粒的细胞毒性相对较低, 因为当纳米银和氧化石墨烯暴露浓度超过10 μg/mL时, 即可引起细胞毒性.此外, 有机含氟纳米颗粒毒性的大小可能与氟含量有关, 因为含氟侧链的引入可以改变纳米颗粒的亲疏水性及其他理化性质, 进而影响其与细胞的作用[98].
图 8

由此可见, 在生物应用方面, 有机含氟纳米材料相比于其他典型的人工纳米材料有着更广阔的应用空间和更好的安全性, 而官能团的含氟量影响材料细胞毒性的具体生物机制还有待科学家们的进一步深入研究.
5. 结语
含氟材料由于其特有的化学结构而具有特别优异的性能, 因此含氟材料的发展可谓蒸蒸日上, 通过在有机材料中引入氟原子(或含氟基团)来提高有机材料的耐极端条件的能力, 是一个无论在理论上还是实践上都被证实的关键策略.此外, 含氟材料的高成本是目前制约其推广应用的主要障碍.因此, 发展温和安全的方法高效制备有机含氟纳米材料及其前体, 从而研究加工和规模化放大制备耐极端条件的有机含氟纳米材料, 进一步通过在模拟极端条件下评价有机含氟纳米材料, 研究有机含氟纳米材料结构与性能间的内在规律, 将对提升我国在耐极端条件的有机纳米材料的科技实力以及工程化能力起到极其重要的推动作用, 进而在工业、国防、航空、电讯电缆及其它领域得到应用.
-
-
[1]
Bai, C. L.; Liu, M. H. Angew. Chem., Int. Ed. 2013, 52, 2678. doi: 10.1002/anie.201210058
-
[2]
Hiyama, T. ; Kanie, K. ; Kusumoto, T. ; Morizawa, Y. ; Shimizu, M. Organofluorine Compounds: Chemistry and Applications, Springer, New York, 2000, pp. 1~272.
-
[3]
Uneyama, K. Organofluorine Chemistry, Blackwell, Oxford, 2006, pp. 1~337.
-
[4]
Sharpless, K. B. Efficient Preparations of Fluorine Compounds, Wiley, Hoboken, 2013, pp. I~XI.
-
[5]
Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, 65.
-
[6]
Hintermann, L.; Togni, A. Angew. Chem., Int. Ed. 2000, 39, 4359. doi: 10.1002/(ISSN)1521-3773
-
[7]
Chen, Q. Y.; Duan, J. X. Tetrahedron Lett. 1993, 34, 4241. doi: 10.1016/S0040-4039(00)60538-5
-
[8]
Su, D. B.; Duan, J. X.; Chen, Q. Y. Tetrahedron Lett. 1991, 32, 7689. doi: 10.1016/0040-4039(91)80566-O
-
[9]
Grushin, V. V. Acc. Chem. Res. 2010, 43, 160. doi: 10.1021/ar9001763
-
[10]
Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679. doi: 10.1126/science.1190524
-
[11]
Wang, X. S.; Mei, T. S.; Yu, J. Q. J. Am. Chem. Soc. 2009, 131, 7520. doi: 10.1021/ja901352k
-
[12]
Hull, K. L.; Anani, W. Q.; Sanford, M. S. J. Am. Chem. Soc. 2006, 128, 7134. doi: 10.1021/ja061943k
-
[13]
Furuya, T.; Kuttruff, C. A.; Ritter, T. Curr. Opin. Drug Discov. Devel. 2008, 11, 803.
-
[14]
Tang, P.; Furuya, T.; Ritter, T. J. Am. Chem. Soc. 2010, 132, 12150. doi: 10.1021/ja105834t
-
[15]
Sun, H.; Dimagno, S. G. Angew. Chem., Int. Ed. 2006, 45, 2720. doi: 10.1002/(ISSN)1521-3773
-
[16]
Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2010, 49, 2219. doi: 10.1002/anie.v49:12
-
[17]
Barker, T. J.; Boger, D. L. J. Am. Chem. Soc. 2012, 134, 13588. doi: 10.1021/ja3063716
-
[18]
Liu, W.; Huang, X. Y.; Cheng, M. J.; Nielsen, R. J.; William, A. G. I.; Groves, J. T. Science 2012, 337, 1322. doi: 10.1126/science.1222327
-
[19]
Yin, F.; Wang, Z.; Li, Z., Li, C. J. Am. Chem. Soc. 2012, 134, 10401. doi: 10.1021/ja3048255
-
[20]
Chen, H.; Wang, X.; Guo, M.; Zhao, W.; Tang, X.; Wang, G. Org. Chem. Front. 2017, 4, 2403. doi: 10.1039/C7QO00611J
-
[21]
周能能, 胥攀, 李伟鹏, 成义祥, 朱成建, 化学学报, 2017, 75, 60. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345727.shtmlZhou, N.; Xu, P.; Li, W.; Cheng, Y.; Zhu, C. Acta Chim. Sinica 2017, 75, 60(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345727.shtml
-
[22]
Amii, H.; Uneyana, K. Chem. Rev. 2009, 109, 2119. doi: 10.1021/cr800388c
-
[23]
Aizenberg, M.; Milstein, D. Science 1994, 265, 359. doi: 10.1126/science.265.5170.359
-
[24]
Peterson, A. A.; McNeill, K. Organometallics 2006, 25, 4938. doi: 10.1021/om0607292
-
[25]
Ishii, Y.; Chatani, N.; Yorimitsu, S.; Murai, S. Chem. Lett. 1998, 27, 157. doi: 10.1246/cl.1998.157
-
[26]
Bohm, V. P. W.; Gstottmayr, C. W. K.; Weskamp, T.; Herrmann, W. A. Angew. Chem., Int. Ed. 2001, 40, 3387. doi: 10.1002/(ISSN)1521-3773
-
[27]
Braun, T.; Perutz, R. N.; Sladek, M. I. Chem. Commun. 2001, 21, 2254.
-
[28]
Mongin, F.; Mojovic, L.; Guillamet, B.; Trecourt, C. F.; Queguiner, G. J. Org. Chem. 2002, 67, 8991. doi: 10.1021/jo026136s
-
[29]
Schaub, T.; Backes, M.; Radius, U. J. Am. Chem. Soc. 2006, 128, 15964. doi: 10.1021/ja064068b
-
[30]
Widdowson, D. A.; Wilhelm, R. Chem. Commun. 2000, 1, 2211.
-
[31]
Kim, Y.; Yu, S. J. Am. Chem. Soc. 2003, 125, 1696. doi: 10.1021/ja028966t
-
[32]
Cargill, M. R.; Sanford, G.; Tadeusiak, A. J.; Yufit, D. S.; Howard, J. A.; Kilickiran, P.; Nelles, G. J. Org. Chem. 2010, 75, 5860. doi: 10.1021/jo100877j
-
[33]
Mikami, K.; Miyamoto, T.; Hatano, M. Chem. Commun. 2004, 2082.
-
[34]
Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188. doi: 10.1126/science.1159979
-
[35]
Grushin, V. V.; Marshall, W. J. J. Am. Chem. Soc. 2006, 128, 12644. doi: 10.1021/ja064935c
-
[36]
Grushin, V. V.; Marshall, W. J. J. Am. Chem. Soc. 2006, 128, 19880.
-
[37]
Watson, D. A.; Su, M.; Teverovskiy, G.; Zhang, Y.; García-Fortanet, J.; Kinzel, T.; Buchwald, S. L. Science 2009, 325, 1661. doi: 10.1126/science.1178239
-
[38]
Chu, L. L.; Qing, F. L. J. Am. Chem. Soc. 2010, 132, 7262. doi: 10.1021/ja102175w
-
[39]
Chu, L. L.; Qing, F. L. Org. Lett. 2010, 12, 5060. doi: 10.1021/ol1023135
-
[40]
Liu, T. F.; Shen, Q. L. Org. Lett. 2011, 13, 2342. doi: 10.1021/ol2005903
-
[41]
Liu, T. F.; Shao, X. X.; Wu, Y. M.; Shen, Q. L. Angew. Chem., Int. Ed. 2012, 51, 540. doi: 10.1002/anie.201106673
-
[42]
Prakash, G. K. S.; Hu, J. B. Acc. Chem. Res. 2007, 40, 921. doi: 10.1021/ar700149s
-
[43]
Zhao, Y. C.; Huang, W. Z.; Zhu, L. G.; Hu, J. B. Org. Lett. 2010, 12, 1444. doi: 10.1021/ol100090r
-
[44]
Zhou, Q. H.; Ruffoni, A.; Gianatassio, R.; Fujiwara, Y.; Sella, E.; Shabat, D.; Baran, P. S. Angew. Chem., Int. Ed. 2013, 52, 3949. doi: 10.1002/anie.v52.14
-
[45]
荣健, 倪传法, 王云泽, 匡翠文, 顾玉诚, 胡金波, 化学学报, 2017, 75, 105. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345728.shtmlRong, J.; Ni, C.; Wang, Y.; Kuang, C.; Gu, Y.; Hu, J. Acta Chim. Sinica 2017, 75, 105(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345728.shtml
-
[46]
Lafrance, M.; Rowley, C. N.; Woo, T. K.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 8754. doi: 10.1021/ja062509l
-
[47]
Do, H. Q.; Daugulis, O. J. Am. Chem. Soc. 2008, 130, 1128. doi: 10.1021/ja077862l
-
[48]
He, C. Y.; Fan, S. L.; Zhang, X. G. J. Am. Chem. Soc. 2010, 132, 12850. doi: 10.1021/ja106046p
-
[49]
Zhang, X. G.; Fan, S. L.; He, C. Y.; Wan, X.; Min, Q. Q.; Yang, J.; Jiang, Z. X. J. Am. Chem. Soc. 2010, 132, 4506. doi: 10.1021/ja908434e
-
[50]
Zeng, Y. W.; Zhang, L. J.; Zhao, Y. C.; Ni, C. F.; Zhao, J. W.; Hu, J. B. J. Am. Chem. Soc. 2013, 135, 2955. doi: 10.1021/ja312711c
-
[51]
Wang, X.; Xu, Y.; Mo, F.; Ji, G. J.; Qiu, D.; Feng, J. J.; Ye, Y. X.; Zhang, S. N.; Zhang, Y.; Wang, J. B. J. Am. Chem. Soc. 2013, 135, 10330. doi: 10.1021/ja4056239
-
[52]
Dai, J. J.; Fang, C.; Xiao, B.; Yi, J.; Xu, J.; Liu, Z. J.; Lu, X.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 8436. doi: 10.1021/ja404217t
-
[53]
Lewis, G. N.; Macdonald, R. T. J. Am. Chem. Soc. 1936, 58, 2519. doi: 10.1021/ja01303a045
-
[54]
Jeon, Y. S.; Jang, N. H.; Kang, B. M.; Jeon, Y. S.; Kim, C. S.; Chio, K. Y.; Ryu, H. Bull. Korean Chem. Soc. 2007, 28, 451. doi: 10.5012/bkcs.2007.28.3.451
-
[55]
Kim, D. W. J. Radioanal. Nucl. Chem. 2002, 252, 559. doi: 10.1023/A:1015815123149
-
[56]
Takahashi, H.; Zhang, Y. H.; Miyajima, T.; Oi, T. J. Mater. Chem. 2006, 16, 1462. doi: 10.1039/B514857J
-
[57]
Kim, D. W.; Kang, B. M.; Jeon, B. K.; Joen, Y. S. J. Radioanal. Nucl. Chem. 2003, 256, 81. doi: 10.1023/A:1023352126261
-
[58]
Araki, H.; Umeda, M.; Enokida, Y.; Yamamoto, I. Fusion. Eng. Des. 1998, 39, 1009.
-
[59]
Otake, K.; Suzuki, T.; Kim, H. J.; Nomura, M.; Fujii, Y. J. Nucl. Sci. Technol. 2006, 43, 419. doi: 10.1080/18811248.2006.9711115
-
[60]
Black, J. R.; Umeda, G.; Dunn, B.; Mcdonough, W. F.; Kavner, A. J. Am. Chem. Soc. 2009, 131, 9904. doi: 10.1021/ja903926x
-
[61]
Saleem, M.; Hussain, S.; Rafiq, M.; Baig, M. A. J. Appl. Phys. 2006, 100, 053111-1.
-
[62]
Olivares, I. E.; Duarte, A. E.; Saravia, E. A.; Duarte, F. J. Appl. Opt. 2002, 41, 2973. doi: 10.1364/AO.41.002973
-
[63]
中国科学院上海有机化学研究所, ZL2012104371552, 2012.Shanghai Institute of Organic Chemistry, CAS. ZL2012104371552, 2012.
-
[64]
胡金波, 张伟, 郑卫琴, 陈光华, 施啸, 徐永昌, 吕红贵, 袁承业, ZL201310239535X, 2013.Hu, J. B. ; Zhang, W. ; Zheng, W. Q. ; Chen, G. H. ; Shi, X. ; Xu, Y. C. ; Lv, H. G. ; Yuan, C. Y. ZL201310239535X, 2013.
-
[65]
胡金波, 张伟, 郑卫琴, 施啸, 徐永昌, 吕红贵, 袁承业, ZL2013102395326, 2013.Hu, J. B. ; Zhang, W. ; Zheng, W. Q. ; Shi, X. ; Xu, Y. C. ; Lv, H. G. ; Yuan, C. Y. ZL2013102395326, 2013.
-
[66]
Shanghai Institute of Organic Chemistry, CAS. PCT/CN2013/075340, 2013.
-
[67]
Hu, J. B. ; Zhang, W. ; Zheng, W. Q. ; Chen, G. H. ; Shi, X. ; Xu, Y. C. ; Lv, H. G. ; Yuan, C. Y. PCT/CN2014/074304, 2014.
-
[68]
胡金波, 张伟, 徐永昌, 顾洪熙, CN2017103408158, 2017.Hu, J. B. ; Zhang, W. ; Xu, Y. C. ; Gu, H. X. CN2017103408158, 2017.
-
[69]
胡金波, 张伟, 张丽君. CN2017103398796, 2017.Hu, J. B. ; Zhang, W. ; Zhang, L. J. CN2017103398796, 2017.
-
[70]
胡金波, 张伟, 顾洪熙, 郑卫琴, CN2017103398917, 2017.Hu, J. B. ; Zhang, W. ; Gu, H. X. Zheng, W. Q. CN2017103398917, 2017.
-
[71]
胡金波, 张伟, 郑卫琴, 徐永昌, CN2017103398936, 2017.Hu, J. B. ; Zhang, W. ; Zheng, W. Q. Xu, Y. C. CN2017103398936, 2017.
-
[72]
Mahl, A.; Lim, A.; Latta, J.; Yemam, H. A.; Greife, U.; Sellinger, A. Nucl. Instrum. Methods Phys. Res. A 2018, 884, 113. doi: 10.1016/j.nima.2017.11.091
-
[73]
Babb, D. A.; Ezzell, B. R.; Clement, K. S.; Richey, W. F.; Kennedy, A. P. J. Polym. Sci. Polym. Chem. 1993, 31, 3465. doi: 10.1002/pola.1993.080311336
-
[74]
Zhu, Y. Q.; Huang, Y. G.; Meng, W. D.; Li, H.; Qing, F. L. Polymer 2006, 47, 6272. doi: 10.1016/j.polymer.2006.06.066
-
[75]
Yao, R. X.; Kong, L.; Yin, Z. S.; Qing, F. L. J. Fluorine Chem. 2008, 129, 1003. doi: 10.1016/j.jfluchem.2008.04.012
-
[76]
Li, Y. J.; Chen, S.; Zhang, S.; Li, Q. N.; Lu, G. L.; Li, W. X.; Liu, H.; Huang, X. Y. Polymer 2009, 50, 5192. doi: 10.1016/j.polymer.2009.09.018
-
[77]
Wang, Q.; Yu, X.; Jin, J.; Wu, Y.; Liang, Y. Chin. J. Chem. 2018, 36, 223. doi: 10.1002/cjoc.v36.3
-
[78]
Watanabe, Y.; Shibasaki, Y.; Ando, S.; Ueda, M. Polym J. 2006, 38, 79. doi: 10.1295/polymj.38.79
-
[79]
Towery, D.; Fury, M. A. J. Electron. Mater. 1998, 27, 1088. doi: 10.1007/s11664-998-0142-z
-
[80]
Tsuchiya, K.; Shibasaki, Y.; Aoyagi, M.; Ueda, M. Macromolecules 2006, 39, 3964. doi: 10.1021/ma0521607
-
[81]
Niu, Y. M.; Zhu, X. L.; Liu, L. Z.; Zhang, Y.; Wang, G. B.; Jiang, Z. H. React. Funct. Polym. 2006, 66, 559. doi: 10.1016/j.reactfunctpolym.2005.10.009
-
[82]
Dang, T. D.; Mather, P. T.; Alexander, M. D.; Grayson, C. J.; Houtz, M. D.; Spry, R. J.; Arnold, F. E. J. Polym. Sci. Polym. Chem. 2000, 38, 1991. doi: 10.1002/(ISSN)1099-0518
-
[83]
Fukukawa, K.; Shibasakl, Y.; Ueda, M. Macromolecules 2004, 37, 8256. doi: 10.1021/ma049063i
-
[84]
Su, Y. C.; Chang, F. C. Polymer 2003, 44, 7989. doi: 10.1016/j.polymer.2003.10.026
-
[85]
Sharangpani, R.; Singh, R. Rev. Sci. Instrum. 1997, 68, 1564. doi: 10.1063/1.1147926
-
[86]
Rosenmeyer, C. T.; Wu, H. MRS Proceedings 1996, 427, 463. doi: 10.1557/PROC-427-463
-
[87]
Luo, Y. J.; Jin, K. K.; He, C. Q.; Wang, J. J.; Sun, J.; He, F. K.; Zhou, J. F.; Wang, Y. Q.; Fang, Q. Macromolecules 2016, 49, 7314. doi: 10.1021/acs.macromol.6b01678
-
[88]
Wang, J. J.; Sun, J.; Zhou, J. F.; Jin, K. K.; Fang, Q. ACS Appl. Mater. Interfaces 2017, 9, 12782. doi: 10.1021/acsami.7b01415
-
[89]
Wang, J. J.; Zhou, J. F.; Jin, K. K.; Wang, L.; Sun, J.; Fang, Q. Macromolecules 2017, 50, 9394. doi: 10.1021/acs.macromol.7b02000
-
[90]
Zhao, Z.; Ni, H.; Han, Z.; Jiang, T.; Xu, Y.; Lu, X.; Ye, P. ACS Appl. Mater. Interfaces 2013, 5, 7808. doi: 10.1021/am401568b
-
[91]
Cheng, Y. Acta Polym. Sinica 2017, 8, 1234.
-
[92]
Jha, S. K.; Mishra, V. K.; Sharma, D. K.; Damodaran, T. Rev. Environ. Contam. Toxicol. 2011, 211, 121.
-
[93]
Lau, C.; Butenhoff, J. L.; Rogers, J. M. Toxicol. Appl. Pharm. 2004, 198, 231. doi: 10.1016/j.taap.2003.11.031
-
[94]
Dewitt, J. C.; Shnyra, A.; Badr, M. Z.; Loveless, S. E.; Hoban, D.; Frame, S. R.; Cunard, R.; Anderson, S. E.; Meade, B. J.; Peden-Adams, M. M.; Luebke, R. W.; Luster, M. I. Crit. Rev. Toxicol. 2009, 39, 76. doi: 10.1080/10408440802209804
-
[95]
Younglai, E. V.; Wu, Y. J.; Foster, W. G. Curr. Pharm. Design 2007, 13, 3005. doi: 10.2174/138161207782110499
-
[96]
Yao, W. Q.; Li, Y. J.; Huang, X. Y. Polymer 2014, 55, 6197. doi: 10.1016/j.polymer.2014.09.036
-
[97]
Tong, L.; Shen, Z.; Zhang, S.; Li, Y. J.; Lu, G. L.; Huang, X. Y. Polymer 2008, 49, 4534. doi: 10.1016/j.polymer.2008.08.033
-
[98]
Wang, X.; Cheng, W. G.; Yang, Q. Y.; Niu, H. Y.; Liu, Q.; Liu, Y.; Gao, M.; Xu, M.; Xu, A.; Liu, S. J.; Huang, X. Y.; Du, Y. G. J. Environ. Sci. 2018, 69, 217. doi: 10.1016/j.jes.2017.10.014
-
[1]
-

图 3 含氟萃取剂材料在锂同位素分离中的应用
Figure 3 Application of fluorine-containing extractant materials in lithium isotope separation
图 7 (a) 有机含氟纳米材料的合成反应; (b) PTFE-PMMA-1 NPs, PTFE-PMMA-2 NPs和PTFE-PMMA-3 NPs的电镜图和粒径分布[98]
Figure 7 (a) Synthesis of organic fluorine-containing nanomaterials, (b) Electron micrograph and particle size distribution of PTFE-PMMA-1 NPs, PTFE-PMMA-2 NPs and PTFE-PMMA-3 NPs[98]. Reprinted with permission from ref. [98]. Copyright (2018) Elsevier
-

 下载:
下载:

 下载:
下载:







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 34
- 文章访问数: 2335
- HTML全文浏览量: 549
 下载:
下载:
