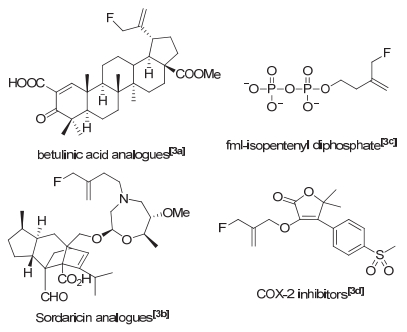
图 1.
有生物活性的含氟烯丙基化合物
Figure 1.
Biologically interesting allylic fluorides

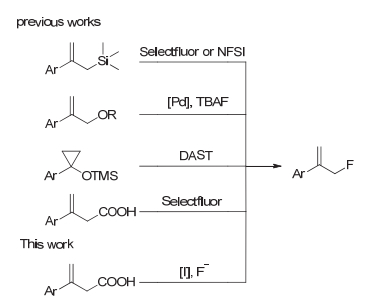
含有烯丙基片段的化合物是一类非常有用的中间体, 在很多的金属催化反应中有很广泛的运用[1].在分子中引入氟原子可以改变分子结构和电性, 这对药物代谢和药效等有很大影响[2].开发一种新的合成含氟烯丙基的方法对合成一些抑制剂[3](图 1)、含氟聚合物[4]等有较高附加值的氟化物很有意义.到目前为止, 合成含氟烯丙基的方法有以下几种(图 2):利用对硝基苯甲酰基[5]或三甲基硅基[6]作为离去基团在亲电氟化试剂的进攻下脱去合成; 环丙烷基三甲基硅醚重排[7]; 羧酸在亲电氟化试剂进攻脱羧氟化[8].
脱羧官能团化是有机合成中常用的合成手段[9].但是对于烯丙基选择性的脱羧氟化的方法并不多见.在过去的研究中, 脱羧氟化的研究取得了长足的进步, 大部分常见的脱羧氟化的方法都涉及XeF2[10], AgF或者AgF2[11], Selectfluor[12], N-氟代双苯磺酰胺(NFSI)[13]等试剂, 这些试剂的使用都会伴随着氧化反应或自由基反应的发生, 有时会对末端烯烃造成破坏[14].
像氟化钾、氟化季铵盐或氢氟酸一类的氟负离子由于其自身较低的亲核性很难作为亲核氟化试剂直接应用于氟化反应, 相反经常作为添加剂来调节反应催化过程, 控制产物比例[15].但是有了过渡金属催化剂[16]或有机催化剂[17]的帮助, 氟负离子也可以在一些烯烃或炔烃参与的反应中用作氟源. Groves等[18]在最近就研究开发了锰催化剂条件下氢氟酸盐作为氟源, 三价碘作为温和氧化剂的脱羧氟化反应.
另一个方面, 高价碘试剂有活化碳碳多重键的能力, 有时可以替代金属催化剂[19]. 2014年Minakata等[20]报道了一种新颖的用碘苯二乙酸做氧化剂的β, γ-不饱和羧酸脱羧官能团化的反应.基于我们[21]之前对开发合成有机氟化物新方法的努力, 以及受前人探索的启发, 我们希望开发一种用便宜的亲核氟化试剂做氟源实现的烯丙基脱羧氟化方法.
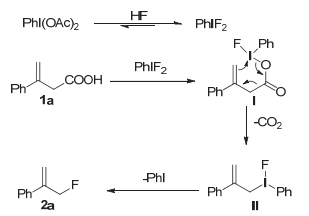
最初筛选条件选用β, γ-不饱和羧酸1a作为模板反应底物进行尝试, 结果如表 1所示.二氟碘苯可以作为亲核氟化试剂和烯烃或脂肪族化合物反应的[22].但是二氟碘苯本身是一种不稳定且有害的物质, 难以保存.碘化学中常见策略之一是利用稳定的高价碘化合物(如碘苯二乙酸、亚碘酰苯等)通过配体交换在体系里促进二氟碘苯的生成, 完成氟化反应[23].还有一种策略是用氧化剂氧化碘苯到高价态在体系中原位生成二氟碘苯参与反应, 反应完毕后高价碘被还原回碘苯, 此时碘试剂作为催化剂参与到催化循环中.然而在实验中(表 1, Entries 1~3)间氯过氧苯甲酸(mCPBA)作为氧化剂会产生副反应, 使得目标产物产率低于5%.最终我们选择了另一种策略, 用不同的三价碘试剂进行了尝试.亚碘酰苯在模板反应中得到了27%的产率(表 1, Entry 4).碘苯二乙酸和三乙胺三氢氟酸盐的组合可以得到较好的反应选择性, 产物基本为2a和2a'(表 1, Entry 5)[24].随后对三乙胺氢氟酸的投料比进行了调整, 在加入5 equiv.时可以获得76%的产率(表 1, Entry 7).然而乙酰氧化的产物2a'依然存在, 提高氢氟酸的浓度最终只能降低2a'到16%(表 1, Entry 8).降低反应温度以及选用其他的高碘化试剂对提高反应产率都没有帮助(表 1, Entries 11~14).最终确定了最优反应条件为1.2 equiv.的碘苯二乙酸, 5 equiv.的三乙胺三氢氟酸盐在二氯甲烷中加热到75 ℃反应12 h (表 1, Entry 7).
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entrya | [I] | TEA•3HF(equiv.) | 2a'(%) | 2a(%) |
| 1b | PhI (0.2 equiv.)+mCPBA (1.5 equiv.) | 5 | ― | <5 |
| 2b | p-Iodotoluene (0.2 equiv.)+mCPBA (1.5 equiv.) | 5 | ― | <5 |
| 3b | p-Iodotoluene (0.2 equiv.)+mCPBA (1.5 equiv.) | py•9HF (5 equiv.) | ― | <5 |
| 4c | PhIO (1.2 equiv.) | 5 | ― | 27 |
| 5 | PhI(OAc)2 (1.2 equiv.) | 1 | 72 | 23 |
| 6 | PhI(OAc)2 (1.2 equiv.) | 2.5 | 43 | 51 |
| 7 | PhI(OAc)2 (1.2 equiv.) | 5 | 17 | 76 |
| 8 | PhI(OAc)2 (1.2 equiv.) | 7.5 | 16 | 77 |
| 9 | PhI(OAc)2 (1.2 equiv.) | 10 | 17 | 75 |
| 10c | PhI(OAc)2 (1.2 equiv.) | 5 | 13 | 49 |
| 11d | PhI(OAc)2 (1.2 equiv.) | 5 | 15 | 57 |
| 12 | PhI(OCOPh)2 (1.2 equiv.) | 5 | ― | 73 |
| 13 | PhI(OCOCF3)2 (1.2 equiv.) | 5 | ― | 57 |
| 14 | PhI(OAc)2 (2.4 equiv.) | 5 | 41 | 43 |
| a Reaction condition: 1a (0.2 mmol), DCM (1 mL), 75 ℃, 12 h, GC yields; b room temperature; c 40 ℃; d 60 ℃. | ||||
有了最优反应条件, 我们对反应的底物范围进行了拓展(表 2).带有不同取代基的芳基β, γ-不饱和羧酸化合物都可以被转化为对应的烯丙基氟化产物, 并取得了中等到较好的收率.含有供电子基团(2b, 2c, 2d, 2g, 2h, 2i)和卤原子(2e, 2f)的底物都可以耐受该反应条件.从三种不同取代位置的甲苯同分异构体(2b, 2c, 2d)的反应结果看, 位阻效应对反应的影响不大.不幸的是, 有吸电基三氟甲基取代的底物2j不能得到目标产物.联苯底物2k得到71%的收率.稠环类底物也都对反应有很好的适用性, α-萘基2l和β-萘基2m分别得到了64%和71%的收率, 9-菲基2n得到了62%的收率.杂环类底物如苯并噻吩环也分别得到了61%和57%的产率(2o, 2p).最后, 两个α, α-二甲基化合物也分别得到了68%和63%的收率(2q, 2r).
在前人工作的基础上[20], 我们提出了一个可能的机理(图 3).首先是碘苯二乙酸通过配体交换产生二氟碘苯, 然后在反应的最初, 由二氟碘苯和1a发生配体交换产生中间体Ⅰ, 然后分子内的自发的重排反应脱去羧酸, 得到中间体Ⅱ.在随后的还原消除反应产生PhI, 与碘相连的氟原子通过SN2取代最终得到了2a.在反应体系中乙酰氧化和氟化是一对竞争反应, 在体系里的离子浓度影响了最终产物的分配.
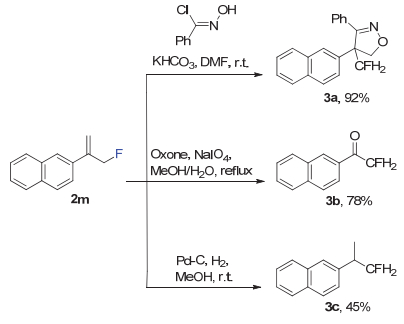
烯丙基氟化物是很好的合成子可以转化为各种含有一氟甲基的化合物.一氟甲基常用作电子等排体在药物设计中替代原有结构[25~27].我们尝试了一些由产物2m转化为可以保留具有一定生物活性的一氟甲基化合物的拓展反应(图 4). 2m与邻氯苯甲醛肟发生环加成反应得到异噁唑产物3a (92%).也可以在硫酸氢钾复合盐和高碘酸钠氧化条件下得到2-氟乙酮产物3b (78%).与氢气在钯碳催化下氢化加成则得到氟代异丙基产物3c (45%).
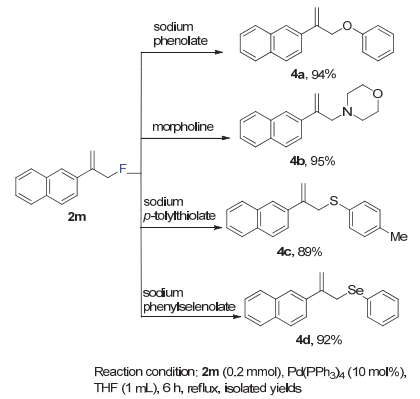
氟原子在化合物中不仅可以改变其理化性质, 在有机合成中也可以作为前驱体转换成其他官能团[28].通过脱氟反应构建C―X键, 有很多新颖的具有原子经济的方法[29].过渡金属催化可以与烯烃发生络合反应, 活化烯丙基位的氟原子, 实现脱卤官能团化[30].在钯催化剂的帮助下, 不同亲核试剂分别与2m反应以高产率得到了4a~4d (89%~95%)(图 5).
我们开发了一种新的使用碘苯二乙酸做氧化剂, 三乙胺三氢氟酸做氟源的脱羧氟化方法.可以将β, γ-不饱和羧酸进行脱羧氟化合成各类含氟烯丙基化合物.文章还进一步开发了一系列利用含氟烯丙基化合物合成一氟甲基化合物的方法, 以及脱氟官能团化的应用.
合成含氟烯丙基2a~2r的通用方法:在干燥的特氟龙管中加入磁力搅拌子, β, γ-不饱和羧酸1 (0.2 mmol), 碘苯二乙酸(0.24 mmol), 二氯甲烷(1 mL), 反应管用氮气吹扫10 min, 将三乙胺三氢氟酸盐(1 mmol)用注射器加入, 封管.反应液在75 ℃下反应12 h.等反应液冷却至室温, 用饱和碳酸氢钠溶液淬灭反应.混合物用乙醚萃取并用盐水清洗有机相.合并有机相用无水硫酸钠干燥.溶剂在低压下被移除后, 用硅胶柱分离得到产物2a~2r.
合成3a的方法:在火焰干燥的耐压管中加入磁力搅拌子, 2m (0.2 mmol), 邻氯苯甲醛肟(0.2 mmol), 二甲基甲酰胺(1 mL), 碳酸氢钾(0.2 mmol).反应液在常温下搅拌12 h.反应液先用一段短硅胶柱用50%洗脱液(乙酸乙酯/石油醚, 1/1, V/V)快速冲下, 在减压下除去溶剂, 然后用20%洗脱液(乙酸乙酯/石油醚, 1/4, V/V)过硅胶柱分离得到产物3a (92%).
合成3b的方法:在火焰干燥的耐压管中加入磁力搅拌子, 2m (0.2 mmol), 过一硫酸氢钾复合盐(0.5 mmol), 高碘酸钠(0.5 mmol), 甲醇/水(1:1, 1 mL).反应液在回流下搅拌12 h.反应液先用一段短硅胶柱用50%洗脱液(乙酸乙酯/石油醚, 1/1, V/V)快速冲下, 在减压下除去溶剂, 然后用10%洗脱液(乙酸乙酯/石油醚, 1/9, V/V)过硅胶柱分离得到产物3b (78%).
合成3c的方法:在火焰干燥的耐压管中加入磁力搅拌子, 2m (0.2 mmol), 钯碳(0.02 mmol), 甲醇(1 mL), 氢气置换(1 atm).反应液在常温下搅拌12 h.反应液先用一段短硅胶柱用50%洗脱液(乙酸乙酯/石油醚, 1/1, V/V)快速冲下, 在减压下除去溶剂, 然后用石油醚过硅胶柱分离得到产物3c (45%).
合成4a~4d的通用方法:在火焰干燥的耐压管中加入磁力搅拌子, 2m (0.2 mmol), 四三苯基膦钯(0.02 mmol), 亲核试剂(0.2 mmol), 四氢呋喃(1 mL).反应液在常温下搅拌12 h.反应液先用一段短硅胶柱快速冲下, 在减压下除去溶剂, 然后用硅胶柱分离得到产物4a~4d.
(a) Singh, O. V.; Han, H. Org. Lett. 2007, 9, 4801; (b) Mizuno, S.; Terasaki, S.; Shinozawa, T.; Kawatsura, M. Org. Lett. 2017, 19, 504. (c) Wu, R. H.; Yang, W.; Cheng, G.; Li, Y.; Yang, D. Q. Chinese J. Org. Chem. 2016, 36, 2368(in Chinese). (仵瑞华, 杨文, 程果, 李玥, 杨定乔, 有机化学, 2016, 36, 2368); (d) Chen, C. H.; Fu, L.; Chen, P. H.; Liu, G. S. Chin. J. Chem. 2017, 35, 1781; (e) Gu, Y.; Lu, C.; Gu, Y.; Shen, Q. Chin. J. Chem. 2018, 36, 55.
(a) Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2004; (b) Hiyama, T. Organofluorine Compounds: Chemistry and Applications, Springer, Berlin, 2000; (c) Uneyama, K. Organofluorine Chemistry, Blackwell, Oxford, U.K., 2006; (d) Richard, C. Fluorine in Organic Chemistry, CRC Press, Boca Raton, FL, 2004.
(a) Honda, T.; Liby, K. T.; Su, X.; Sundararajan, C.; Honda, Y.; Suh, N.; Risingsong, R.; Williams, C. R.; Royce, D. B.; Sporn, M. B.; Gribble, G. W. Bioorg. Med. Chem. Lett. 2006, 16, 6306; (b) Kaneko, S.; Arai, M.; Uchida, T.; Harasaki, T.; Fukuoka, T.; Konosu, T. Bioorg. Med. Chem. Lett. 2002, 12, 1705; (c) Rothman, S. C.; Johnston, J. B.; Lee, S.; Walker, J. R.; Poulter, C. D. J. Am. Chem. Soc. 2008, 130, 4906; (d) Leblanc, Y.; Roy, P.; Leger, S.; Grimm, E.; Wang, Z. WO 9841516, 1998[Chem. Abstr. 1998, 129, 275831].
(a) Walkowiak-Kulikowska, J.; Szwajca, A.; Boschet, F.; Gouverneur, V.; Ameduri, B. Macromolecules 2014, 47, 8634. (b) Kostov, G.; Tredwell, M.; Gouverneur, V.; Ameduri, B. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 3843. (c) Wall, L. A. Fluoropolymers, Wiley, New York, 1972.
Hollingworth, C.; Hazari, A.; Hopkinson, M. N.; Tredwell, M.; Benedetto, E.; Huiban, M.; Gee, A. D.; Brown, J. M.; Gouverneur, V. Angew. Chem., Int. Ed. 2011, 50, 2613. doi: 10.1002/anie.201007307
Walkowiak, J.; Martinez, D. C. T.; Ameduri, B.; Gouverneur, V. Synthesis 2010, 1883. https://www.researchgate.net/publication/320204431_Synthesis_characterization_thermal_and_surface_properties_of_co-_and_terpolymers_based_on_fluorinated_a-methylstyrenes_and_styrene
(a) Kirihara, M.; Kambayashi, T.; Momose, T. Chem. Commun. 1996, 1103; (b) Kirihara, M.; Kakuda, H.; Tsunooka, M.; Shimajiri, A.; Takuwa, T.; Hatano, A. Tetrahedron Lett. 2003, 44, 8513.
Hu, J. B.; He, Z. B. CN 102219638, 2011[Chem. Abstr. 2011, 155, 588760].
(a) He, Z. B.; Ping, T.; Hu, J. B. Org. Lett. 2016, 18, 72; (b) Ma, J. J.; Yi, W. B.; Lu, G. P.; Cai, C. Adv. Synth. Catal. 2015, 357, 3447. (c) Xu, X. L.; Chen, H. H.; He, J. B.; Xu, H. J. Chin. J. Chem. 2017, 35, 1665; (d) Montazerozohori, M.; Nasr-Esfahani, M.; Akhlaghi, P. Chin. J. Chem. 2009, 27, 1007; (e) Zhang, J. J.; Cheng, Y. B.; Duan, X. H. Chin. J. Chem. 2017, 35, 311; (f) Zhao, Y. W.; Feng, Q.; Song, Q. L. Chin. Chem. Lett. 2016, 27, 571.
(a) Chatalova-Sazepin, C.; Binayeva, M.; Epifanov, M.; Zhang, W.; Foth, P.; Amador, C.; Jagdeo, M.; Boswell, B. R.; Sammis, G. M. Org. Lett. 2016, 18, 4570; (b) Patrick, T. B.; Khazaeli, S.; Nadji, S.; Hering-Smith, K.; Reif, D. J. Org. Chem. 1993, 58, 705.
(a) Yin, F.; Wang, Z.; Li, Z.; Li, C. J. Am. Chem. Soc. 2012, 134, 10401; (b) Zhang, X. Comput. Theor. Chem. 2016, 1082, 11; (c) Zhang, Q. W.; Brusoe, A. T.; Mascitti, V.; Hesp, K. D.; Blakemore, D. C.; Kohrt, J. T.; Hartwig, J. F. Angew. Chem., Int. Ed. 2016, 55, 9758; (d) Mizuta, S.; Stenhagen, I. S. R.; O'Duill, M.; Wolstenhulme, J.; Kirjavainen, A. K.; Forsback, S. J.; Tredwell, M.; Sandford, G.; Moore, P. R.; Huiban, M.; Luthra, S. K.; Passchier, J.; Solin, O.; Gouverneur, V. Org. Lett. 2013, 15, 2648.
(a) Leung, J. C. T.; Chatalova-Sazepin, C.; West, J. G.; Rueda-Becerril, M.; Paquin, J. F.; Sammis, G. M. Angew. Chem., Int. Ed. 2012, 51, 10804; (b) Leung, J. C. T.; Sammis, G. M. Eur. J. Org. Chem. 2015, 2197; (c) Ventre, S.; Petronijevic, F. R.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 5654; (d) Wang, D. H.; Yuan, Z. L.; Liu, Q. L.; Chen, P. H. Liu, G. S. Chin. J. Chem. 2018, 36, 507; (e) Dong, Y.; Wang, Z.; Li, C. Nat. Commun. 2017, 8, 277.
Rueda-Becerril, M.; Chatalova Sazepin, C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J. F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026. doi: 10.1021/ja211679v
Yang, Q.; Mao, L. L.; Yang, B.; Yang, S. D. Org. Lett. 2014, 16, 3460. doi: 10.1021/ol501357w
(a) Fagnou, K.; Lautens, M. Angew. Chem. 2002, 114, 26; (b) Yan, N.; Lei, Z. W.; Su, J. K.; Liao, W. L. Hu, X. G. Chin. Chem. Lett. 2017, 28, 467; (c) Wang, L. Y.; Jiang, X. H.; Tang, P. P. Org. Chem. Front. 2017, 4, 1958.
(a) Katcher, M. H.; Sha, A.; Doyle, A. G. J. Am. Chem. Soc. 2011, 133, 15902; (b) Lee, E.; Hooker, J. M.; Ritter, T. J. Am. Chem. Soc. 2012, 134, 17456; (c) Fier, P. S.; Luo, J.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 2552; (d) Fier, P. S.; Hartwig, J. F. J. Am. Chem. Soc. 2012, 134, 10795; (e) Liu, Z.; Chen, H.; Lv, Y.; Tan, X.; Shen, H.; Yu, H.-Z.; Li, C. J. Am. Chem. Soc. 2018, 140, 6169; (f) Ma, J. A.; Li, S. Org. Chem. Front. 2014, 1, 712.
Woerly, E. M.; Banik, S. M.; Jacobsen, E. N. J. Am. Chem. Soc. 2016, 138, 13858. doi: 10.1021/jacs.6b09499
Huang, X.; Liu, W.; Hooker, J. M.; Groves, J. T. Angew. Chem., Int. Ed. 2015, 54, 5241. doi: 10.1002/anie.v54.17
(a) Souto, J. A.; Becker, P.; Iglesias, A.; Muñiz, K. J. Am. Chem. Soc. 2012, 134, 15505; (b) Souto, J. A.; Martínez, C.; Velilla, I.; Muñiz, K. Angew. Chem., Int. Ed. 2013, 52, 324; (c) Röben, C.; Souto, J. A.; Escudero-Adán, E. C.; Muñiz, K. Org. Lett. 2013, 15, 1008; (d) Farid, U.; Malmedy, F.; Claveau, R.; Albers, L.; Wirth, T. Angew. Chem., Int. Ed. 2013, 52, 7018; (e) Wang, Y.; Wang, Y.; Zhang, Q.; Li, D. Org. Chem. Front. 2017, 4, 514; (f) Gao, P.; Fan, M. J.; Bai, Z. J.; Wei, Y. Y. Chin. J. Chem. 2015, 33, 479.
(a) Kiyokawa, K.; Yahata, S.; Kojima, T.; Minakata, S. Org. Lett. 2014, 16, 4646; (b) Kiyokawa, K.; Kojima, T.; Hishikawa, Y.; Minakata, S. Chem. Eur. J. 2015, 21, 15548.
(a) Jiang, L. Q.; Qian, J. L.; Yi, W. B.; Lu, G. P.; Cai, C.; Zhang, W. Angew. Chem., Int. Ed. 2015, 54, 14965; (b) Lin, Y.-M.; Yi, W. B.; Shen, W. Z.; Lu, G. P. Org. Lett. 2016, 18, 592; (c) Song, Z. D.; Yi, W. B. Adv. Synth. Catal. 2016, 358, 2727.
(a) Kitamura, T.; Muta, K.; Kuriki, S. Tetrahedron Lett. 2013, 54, 6118; (b) Carpenter, W. J. Org. Chem. 1966, 31, 2688; (c) Zupan, M.; Pollak, A. J. Fluorine Chem. 1976, 7, 445; (d) Arrica, M. A.; Wirth, T. Eur. J. Org. Chem. 2005, 395; (e) Ye, C.; Twamley, B.; Shreeve, J. M. Org. Lett. 2005, 7, 3961.
Kitamura, T.; Muta, K.; Oyamada, J. J. Org. Chem. 2015, 80, 10431. doi: 10.1021/acs.joc.5b01929
Nash, T. J.; Pattison, G. Eur. J. Org. Chem. 2015, 3779. https://www.onacademic.com/detail/journal_1000037686628610_561d.html
Li, Y.; Ni, C.; Liu, J.; Zhang, L.; Zheng, J.; Zhu, L.; Hu, J. B. Org. Lett. 2006, 8, 1693. doi: 10.1021/ol060322t
(a) Fukuzumi, T.; Shibata, N.; Sugiura, M.; Yasui, H.; Nakamura, S.; Toru, T. Angew. Chem., Int. Ed. 2006, 45, 4973; (b) Furukawa, T.; Shibata, N.; Mizuta, S.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2008, 47, 8051.
Prakash, G. K. S.; Ledneczki, I.; Chacko, S.; Olah, G. A. Org. Lett. 2008, 10, 557. doi: 10.1021/ol702500u
Traff, A. M.; Janjetovic, M.; Ta, L.; Hilmersson, G. Angew. Chem., Int. Ed. 2013, 52, 12073. doi: 10.1002/anie.201306104
(a) Yi, W. B.; Huang, X.; Zhang, Z.; Zhu, D.; Cai, C.; Zhang, W. Green Chem. 2012, 14, 3185; (b) Qian, J. L.; Yi, W. B.; Huang, X.; Miao, Y. B.; Zhang, J. K.; Cai, C.; Zhang, W. Org. Lett. 2015, 17, 1090; (c) Song, Z. D.; Huang, X.; Yi, W. B.; Zhang, W. Org. Lett. 2016, 18, 5640.
Benedetto, E.; Keita, M.; Tredwell, M.; Hollingworth, C.; Brown, J. M.; Gouverneur, V. Organometallics 2012, 31, 1408. https://www.researchgate.net/publication/263950393_Platinum-Catalyzed_Substitution_of_Allylic_Fluorides

图 4 含氟烯丙基化合物到一氟甲基化合物的转化研究
Figure 4 Transformations of allylic fluoride to the monofluoromethyl compounds
表 1 脱羧氟化反应的条件筛选
Table 1. Optimization of the decarboxylative fluorination
| ||||
| Entrya | [I] | TEA•3HF(equiv.) | 2a'(%) | 2a(%) |
| 1b | PhI (0.2 equiv.)+mCPBA (1.5 equiv.) | 5 | ― | <5 |
| 2b | p-Iodotoluene (0.2 equiv.)+mCPBA (1.5 equiv.) | 5 | ― | <5 |
| 3b | p-Iodotoluene (0.2 equiv.)+mCPBA (1.5 equiv.) | py•9HF (5 equiv.) | ― | <5 |
| 4c | PhIO (1.2 equiv.) | 5 | ― | 27 |
| 5 | PhI(OAc)2 (1.2 equiv.) | 1 | 72 | 23 |
| 6 | PhI(OAc)2 (1.2 equiv.) | 2.5 | 43 | 51 |
| 7 | PhI(OAc)2 (1.2 equiv.) | 5 | 17 | 76 |
| 8 | PhI(OAc)2 (1.2 equiv.) | 7.5 | 16 | 77 |
| 9 | PhI(OAc)2 (1.2 equiv.) | 10 | 17 | 75 |
| 10c | PhI(OAc)2 (1.2 equiv.) | 5 | 13 | 49 |
| 11d | PhI(OAc)2 (1.2 equiv.) | 5 | 15 | 57 |
| 12 | PhI(OCOPh)2 (1.2 equiv.) | 5 | ― | 73 |
| 13 | PhI(OCOCF3)2 (1.2 equiv.) | 5 | ― | 57 |
| 14 | PhI(OAc)2 (2.4 equiv.) | 5 | 41 | 43 |
| a Reaction condition: 1a (0.2 mmol), DCM (1 mL), 75 ℃, 12 h, GC yields; b room temperature; c 40 ℃; d 60 ℃. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: