表 1
常见含氟基团的Hansch疏水性参数
Table 1.
The Hansch's hydrophobicity parameter of fluorine-containing groups

近二十年, 含氟化合物由于其特殊的“氟效应”受到越来越多药物化学家的重视.在有机分子内引入含氟基团可以显著地影响化合物的物理化学性质以及生物学特性.因此合成化学家们利用含氟取代基对分子进行修饰来调控分子的药理性质, 比如脂溶性、代谢稳定性以及生物利用率等[1].
在众多的的含氟基团中, 三氟甲硫基(SCF3)由于具有较高脂溶性与较强的吸电子能力而备受青睐.从Hansch疏水性参数[2]中可以看出, 三氟甲硫基在常见的含氟基团中具有最高的π系数(Table 1), 将其引入药物分子可以极大地提高分子的脂溶性, 增加药物对细胞膜的穿透性, 因此也提高了药物的生物利用率.此外, 三氟甲硫基具有较强的吸电子能力, 这就使得被三氟甲硫基修饰后的分子具有较高的代谢稳定性.
 下载:
导出CSV
下载:
导出CSV
| X | SCF3 | SF5 | OCF3 | CF3 | F |
| πx | 1.44 | 1.23 | 1.04 | 0.88 | 0.14 |
基于以上提及的三氟甲硫基的特点, 发展将SCF3引入有机化合物的有效方法是合成化学家们感兴趣的一个领域.目前, 已经有很多构建C—SCF3键的方法学, 它们为合成各种各样含三氟甲硫基的有机化合物提供了很好的工具[3].但是不对称三氟甲硫基化在有机合成中仍然是一个挑战, 目前相关报道较少[3i, 4~21].而在药物开发中, 药物分子的手性对其药性影响巨大, 沙利度胺(Thalidomide)事件清楚地说明了不同立体构型的药物分子具有相差悬殊的药理作用.因此, 有机分子的不对称三氟甲硫基化在药物开发中具有重要的意义.尽管目前为止相关文献报道较少, 主要集中于过去五年期间, 我们还是希望能对该领域做一个总结与展望, 以引起更多化学家的重视和促进该研究方向的发展.
目前构建含C—SCF3键的立体中心主要有两种策略, 一种是利用亲电SCF3试剂[4~19], 将三氟甲硫基“转移”到亲核性的分子中(Scheme 1a); 另一种是利用含有SCF3的合成砌块通过不同的反应历程得到三氟甲硫基化的分子[20, 21] (Scheme 1b).本文将对该领域的研究进展进行总结与展望.
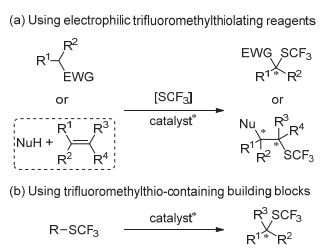
直接使用亲电或者亲核的SCF3试剂是构建C—SCF3键的常用方法.目前只实现了将亲电的SCF3试剂应用于有机化合物的不对称三氟甲硫基化反应, 而亲核型或者自由基型的该类不对称转化还未见报道.
羰基化合物一直是有机合成化学中的一类重要原料.在羰基的α位置引入一个三氟甲硫基将有可能合成具有生物活性的药物分子.尤其是在羰基α位置构建一个含C—SCF3键的立体中心将在药物合成中具有重要意义.
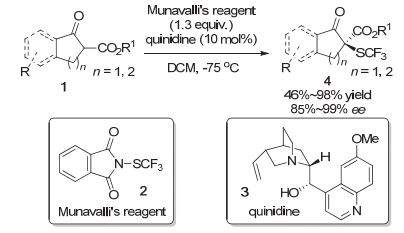
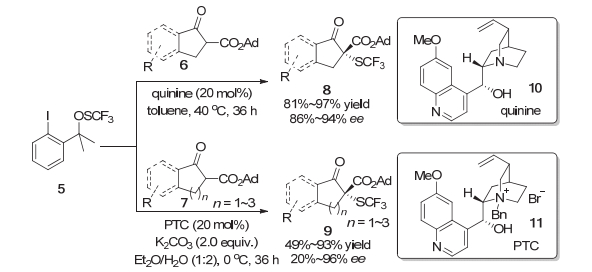
2013年, Rueping等[4]与沈其龙等[5]同时首次独立报道了β-酮酸酯的不对称三氟甲硫基化反应.在Rueping等的研究中, 他们使用了Munavalli等首次合成的N-三氟甲硫基邻苯二甲酰胺2作为亲电的三氟甲硫基试剂, 在有机碱奎尼丁3的催化下, 高对映选择性地构建了含有C—SCF3键的四级碳立体中心(Scheme 2).该方法对于苯环骨架上带有给电子或者吸电子基团的1-茚酮衍生的β-酮酸酯底物都可以获得优秀的产率以及优秀的对映选择性.而对于苯并六元环的β-酮酸酯以及环戊烯酮衍生的β-酮酸酯底物, 反应的产率会有所下降.与此同时, 沈其龙等使用了他们在2013年开发的亲电三氟甲硫基试剂5, 在奎宁或者奎宁衍生的相转移催化剂11催化下, 实现了苯并五元、六元以及七元环酮或者普通的五元、六元环酮衍生的β-酮酸酯的不对称三氟甲硫基化.当反应体系的催化剂是奎宁时, 1-茚酮衍生的各种吸电子基或者给电子基取代的β-酮酸酯6均可以获得较高的产率以及优秀的对映选择性.环戊酮衍生的β-酮酸酯也可以以优秀的产率(95%)与优秀的对映选择性(94%)在该体系中转化为对应的产物.而对于苯并环己酮与苯并环庚酮衍生的β-酮酸酯7则不能在该条件下很好地转化为三氟甲硫基化的产物, 转化率都小于5%.对于这些在奎宁催化下不能很好转化的底物, 他们又通过设计奎宁衍生的手性相转移催化剂(PTC)11来完成相应转化(Scheme 3).对于苯并环己酮衍生的β-酮酸酯, 在新条件下可以以较高的产率以及中等到良好的对映选择性转化为对应的三氟甲硫基化的产物.对于苯并环庚酮, 反应获得了较高的产率以及优秀的对映选择性.而环己酮衍生的β-酮酸酯只能以中等的对映选择性完成转化.此外, 苯并环戊酮在该新条件下的反应具有较低的对映选择性.
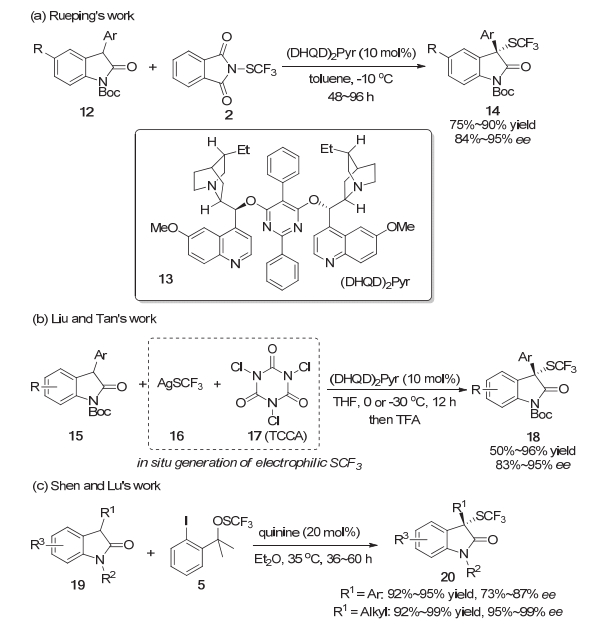
2014年, Rueping等[6]再次使用N-三氟甲硫基邻苯二甲酰胺2作为亲电的三氟甲硫基试剂, 实现了金鸡纳碱(DHQD)2Pyr催化的2-吲哚酮底物12的不对称三氟甲硫基化反应.在该反应体系中, C3位取代基是各种不同电子性质及不同取代模式的芳环时, 反应都能以良好的产率以及良好到优秀的对映选择性得到三氟甲硫基化的产物.底物中C5位无论是吸电子还是给电子的取代基都能以中等到良好的产率以及良好到优秀的对映选择性完成相应转化(Scheme 4a).同年, 谭斌与刘心元等[7]以及吕龙与沈其龙等[8]也分别报道了2-吲哚酮的不对称三氟甲硫基化反应.在谭斌等的工作中, 采用了通过TCCA(三氯异氰尿酸)和AgSCF3原位制备亲电三氟甲硫基活性物种的策略完成了相应转化.该原位制备活性亲电物种的策略, 不需要对三氟甲硫基物种进行分离, 简化了反应操作; 同时, 该反应也具有中等到优秀的收率以及良好到优秀的对映选择性(Scheme 4b), 具有一定的实用价值.在他们对该反应机理的初步研究中, 发现该体系中CF3S-SCF3是亲电的三氟甲硫基活性物种之一, 该体系中还存在其他的尚未确定的三氟甲硫基活性物种.在Rueping等和谭斌与刘心元等发展的2-吲哚酮的不对称三氟甲硫基化反应中, 底物主要局限于3-芳基- 2-吲哚酮.而随后吕龙与沈其龙等发展的方法中, 3-芳基-2-吲哚酮与3-烷基-2-吲哚酮都能很好地实现不对称三氟甲硫基化, 尤其是3-烷基-2-吲哚酮, 可以获得优秀的产率以及优秀的对映选择性(Scheme 4c).
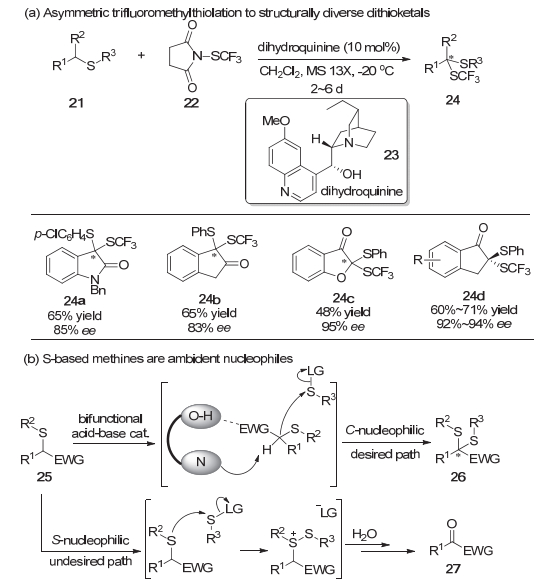
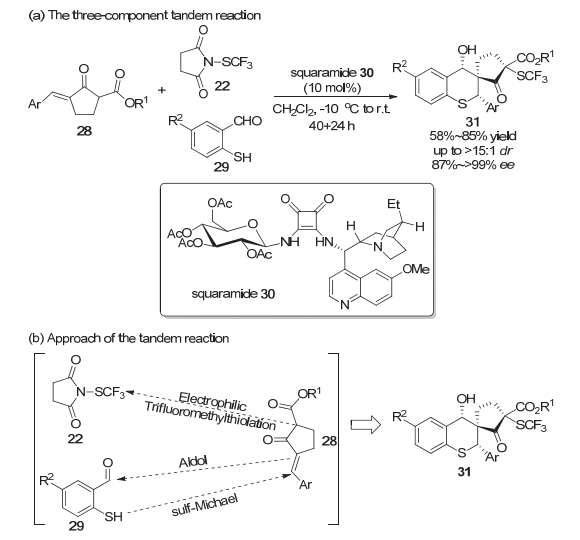
2015年, 周锋等[9]利用N-三氟甲硫基丁二酰亚胺22在二氢奎宁23催化下实现了α-硫代羰基化合物21的不对称三氟甲硫基化反应, 高对映选择性地合成了各种二硫缩酮化合物24 (Scheme 5a).研究发现, 二氢奎宁这类双功能的酸-碱催化剂在该反应体系中起着至关重要的作用.实验结果显示α-硫代羰基化合物能以硫或者次甲基的碳作为亲核位点与亲电试剂反应.他们认为二氢奎宁中羟基作为氢键给体与底物之间形成氢键, 有利于二氢奎宁中含氮部分作为Brønsted碱进一步完成α-硫代羰基化合物的去质子过程, 从而使得碳作为亲核位点与亲电试剂反应的途径更加有利(Scheme 5b).杜大明等[10]同样采用N-三氟甲硫基丁二酰亚胺试剂22, 于2017年报道了金鸡纳碱衍生的方酰胺30催化的不对称三氟甲硫基化/Michael加成/aldol串联反应.该体系一锅构建了同时具有三个连续手性中心和一个连接SCF3的全碳立体中心的复杂螺环骨架31, 并且得到了中等到良好的收率以及优秀的立体选择性(Scheme 6a).该串联反应得以实现的关键是底物分子中同时具有活化的亲核位点以及亲电位点, 使得亲电取代反应与亲核进攻可以连续地进行(Scheme 6b).显然, 手性的方酰胺诱导产生了反应较高的立体选择性.
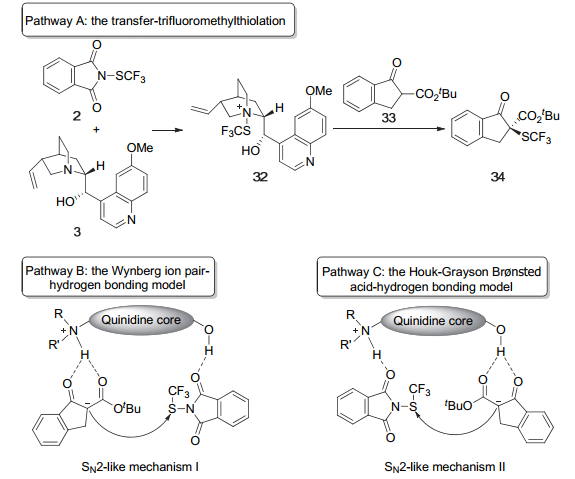
2017年, 薛小松和程津培等[11]就Rueping等在2013年报道的金鸡纳碱催化下β-酮酸酯的不对称三氟甲硫基化反应的机理和对映选择性的起源进行了详细的DFT研究.首先他们评估了三种可能的机理: (A)三氟甲硫基转移机理; (B) Wynberg离子对氢键模型; (C) Houk-Grayson双功能Brønsted酸氢键模型(Scheme 7).根据他们的计算结果, (A)反应历程的活化能至少比(B)历程的活化能高14.6 kcal/mol, 因此可以首先排除(A)历程.接着他们通过寻找C—SCF3键形成步骤中能量最低的过渡态结构, 分别确定了(B)、(C)两种历程中生成主要构型产物与次要构型产物的过渡态结构.其中(B)历程中产生主要构型产物的过渡态能量比产生次要构型产物的过渡态能量低4.3 kcal/mol, 这将导致(B)历程具有优秀的对映选择性(ee>99%), 这一计算结果与实验结果非常吻合.而(C)历程对应的两个过渡态的能量只相差0.4 kcal/mol, 理论上(C)历程会具有较差的对映选择性.并且(C)历程中两个过渡态分别比(B)历程中能量较低的过渡态在能量上高出8.0 kcal/mol和8.4 kcal/mol.这两点都说明该反应更有可能经历(B)历程.即Wynberg离子对氢键模型是该反应的最优催化模式, 同时SCF3转移通过类SN2机理进行.最后, 他们对该反应对映选择性的起源的研究表明反应的对映选择性由过渡态中多重弱相互作用协同诱导产生, 这些弱相互作用包括: C—H…O, C—H…S, C—H…π以及π…π相互作用等.该机理研究为不对称三氟甲硫基化领域中新反应和新催化剂的设计与开发提供了一定的理论依据.
从以上金鸡纳碱及其衍生物催化的不对称三氟甲硫基化反应以及相关的机理研究中我们可以发现, 双功能的手性金鸡纳碱催化剂是该类催化反应得以进行的关键.该类生物碱的使用, 不仅使得有机化合物的不对称三氟甲硫基化得到突破, 也为后续不对称三氟甲硫基化反应催化剂的设计与选择提供了一种参考.
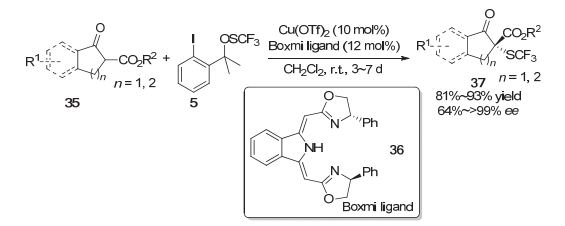
除了前面提到的生物碱之外, 铜催化剂也可以催化羰基化合物的不对称三氟甲硫基化反应. 2014年, Gade等[12]采用亲电三氟甲硫基试剂5, 使用他们自己发展的Boxmi配体36, 在铜催化下实现了环状β-酮酸酯底物35的不对称三氟甲硫基化.对于五元和六元环的β-酮酸酯底物都能以良好到优秀的收率完成转化, 并且绝大多数产物都具有优秀的对映选择性.在该反应体系中手性铜催化剂主要是作为Lewis酸来稳定底物的烯醇结构并在过渡态中诱导该反应手性的产生(Scheme 8).
之前发展的羰基α位不对称三氟硫甲基化的方法中, 底物一般局限于环状的β-酮酸酯与吲哚酮类底物. 2018年, 南方科技大学的汪君等[13]利用烷基锌与N-三氟甲硫基-4-硝基邻苯二甲酰亚胺39通过铜催化实现了α, β-不饱和酮的1, 4-加成/三氟甲硫基化串联反应.该方法具有产率优良、对映选择性良好等特点, 为合成α位含C—SCF3键立体中心的非环状酮提供了一种有效的途径(Scheme 9).
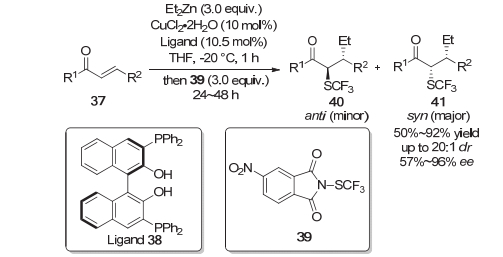
对于羰基类化合物的不对称三氟甲硫基化反应, 除了使用手性催化剂, 也可以使用光学纯的手性三氟甲硫基试剂或者使用手性辅基来完成手性的传递.
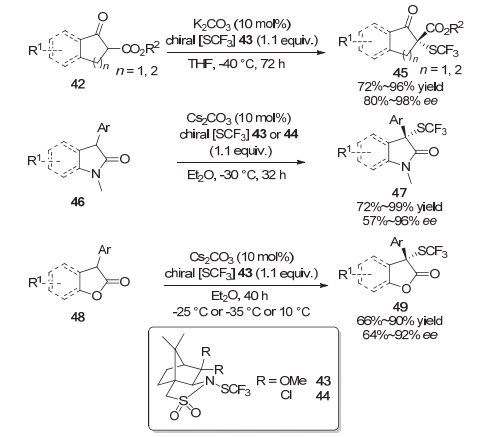
2017年, 沈其龙等[14]首次报道了手性的亲电三氟甲硫基试剂43和44, 并利用光学纯的该试剂在催化量的碱作用下, 实现了β-酮酸酯42、2-吲哚酮46以及苯并呋喃酮48的不对称三氟甲硫基化反应, 获得了优良的对映选择性(Scheme 10).
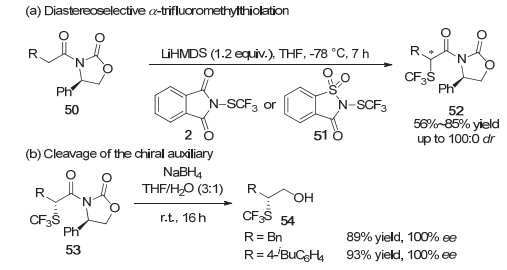
随后在2018年, Cahard等[15]报道了Evans手性辅基控制的羰基化合物α位非对映选择性的三氟甲硫基化反应, 获得了具有中等到优良非对映选择性的产物(Scheme 11a).这些非对映异构体经过柱层析分离、脱掉手性辅基便可得到光学纯的化合物(Scheme 11b).
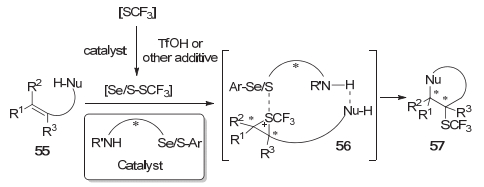
与前面提及的羰基化合物的不对称三氟甲硫基化反应相对应, 赵晓丹等[16~19]在烯烃的双官能团化/不对称三氟甲硫基化领域开展了工作.在该反应设计中首先是硫醚或者硒醚与亲电的三氟甲硫基试剂反应生成活性的[S-SCF3]或[Se-SCF3]中间体, 该活性中间体被体系中的烯烃捕获得到硫鎓离子中间体, 硫鎓离子中间体再进一步被体系中的亲核组分进攻得到烯烃双官能团化的产物.从该类反应的机理可知, 该体系的对映选择性的控制主要受两个因素干扰:其一, 手性的硫鎓离子可以在烯烃间发生转移而消旋化; 其二, 亲电的三氟甲硫基试剂或者手性的硫鎓离子可能被其他的亲核试剂捕获而形成[Nu-SCF3]物种, 再被烯烃捕获形成消旋的硫鎓离子.他们认为, 引入双功能的手性硫醚和手性硒醚催化剂可以很好的解决这两个问题, 其中催化剂中的硫/硒醚部分作为Lewis碱, 催化剂的NH部分作为氢键给体与底物中的亲核部分相互作用可以很好地稳定手性的硫鎓离子中间体[16].这一策略主要适用于分子内的反应(Scheme 12).
2016年, 赵晓丹等[16]首次报道了手性硫醚60催化的反式烯烃的不对称三氟甲硫基化内酯化反应.该体系获得了良好到优秀的对映选择性、高非对映选择性以及中等到优秀的产率(Scheme 13a).高对映选择性仅仅局限于4-芳基烯酸底物, 而4-烷基烯酸底物只能获得中等的对映选择性.该工作为他们之后对烯烃不对称三氟甲硫基化反应的进一步研究奠定了基础. 2017年, 该课题组[17]又报道了手性硒醚63催化的烯烃的不对称三氟甲硫基化分子内胺化反应(Scheme 13b).对于反式烯烃, 该反应体系可以以优良的产率以及立体选择性获得饱和的氮杂环产物64或65; 对于顺式烯烃, 没有目标产物产生; 而对于末端烯烃, 反应的产率和对映选择性会有所降低.
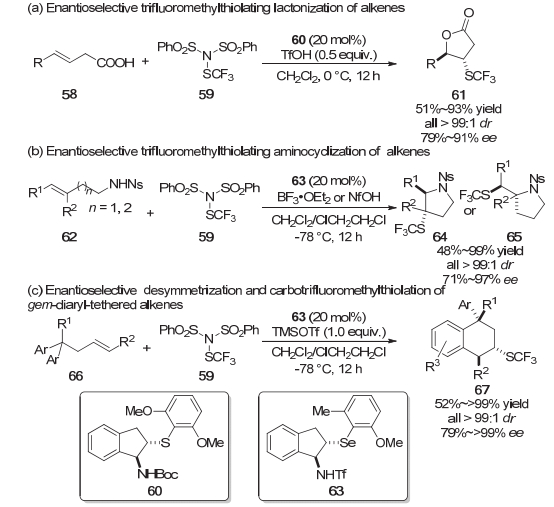
2018年, 该课题组[18]又发展了结构中包含偕二芳基的烯烃底物66的去对称化、不对称三氟甲硫基化芳基化反应.他们同样是采用了双功能的手性硒醚63作为催化剂, 以优秀的立体选择性和较高的产率构建了不对称三氟甲硫基化的四氢化萘骨架67, 该骨架中还包含一个全碳立体中心(Scheme 13c).该反应体系还具有底物普适性好, 可以克级制备等特点.此外, 他们还对该反应体系进行了理论计算研究, 研究结果表明, 在该类反应体系中硒醚是比硫醚更好的活化试剂, 硒醚更容易与亲电的三氟甲硫基试剂形成[Se-SCF3]活性物种.最后, 理论计算还表明, 三氟乙酸根负离子与催化剂之间的氢键相互作用是该反应具有较高立体选择性的关键.
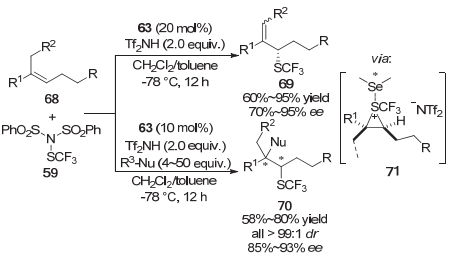
在上面提到的方法中, 为了解决硫鎓离子或者硒鎓离子的外消旋化问题, 亲核基团需要预先引入到底物中, 这就使得反应只能以分子内的形式进行.为此赵晓丹等[19]又发展了手性硒醚63催化的烯丙位不对称三氟甲硫基化和烯烃的分子间双官能团化/不对称三氟甲硫基化的方法.他们认为采用比较富电子的三取代烯烃68, 可以使得到的硫鎓离子中间体71具有一定的稳定性, 从而解决硫鎓离子中间体易发生外消旋化的问题.当体系中不加入亲核试剂时, 反应以烯丙位官能团化的形式进行, 以中等到较高的产率以及优良的对映选择性得到了烯丙位不对称三氟甲硫基化的产物69.当体系加入亲核试剂时, 反应以烯烃双官能团化的形式进行.尽管体系中加入了过量甚至大大过量的亲核试剂, 反应只能以中等的收率得到烯烃双官能团化的产物70, 但是反应仍然获得了优良的对映选择性和非对映选择性(Scheme 14).这一工作以不同的反应模式实现了烯烃到包含一个连接SCF3手性中心的化合物的转化, 可能会促进烯烃的其他不对称三氟甲硫基化方法的发展.
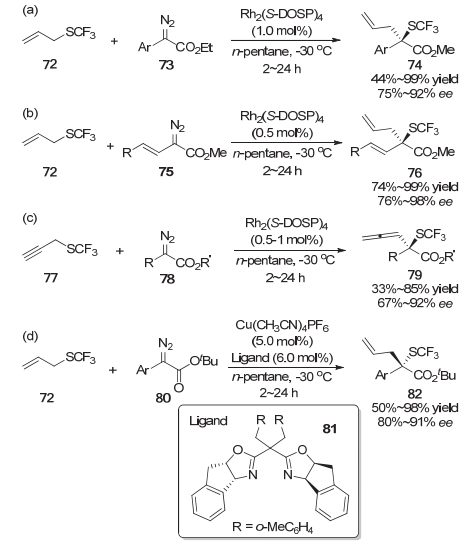
目前, 对于采用亲电或亲核的三氟甲硫基试剂在有机分子中引入三氟甲硫基的报道已经相对较多.而直接从含有SCF3的合成砌块出发合成三氟甲硫基化的有机分子是另一种可供选择的策略.然而, 利用含SCF3的合成砌块对映选择性地构建C—SCF3键的相关报道却相对较少[20, 21].基于金属卡宾的高效催化反应, 王剑波等[20]使用烯丙基三氟甲基硫醚72经Doyle-Kirmse反应实现了一种独特的不对称三氟甲硫基化反应.该反应是首例利用含SCF3的合成砌块来构建一系列连接三氟甲硫基的全取代碳立体中心的报道(Scheme 15)[22, 23].在该项研究中, 采用Rh2(S-DOSP)4作为催化剂, 烯丙基三氟甲基硫醚72能与各种取代的芳基重氮乙酸乙酯73反应给出优良的产率和对映选择性(Scheme 15a).对于烯基重氮乙酸乙酯75, 当催化剂Rh2(S-DOSP)4的载量降低到0.5 mol%, 同样获得了优异的产率和对映选择性(Scheme 15b).此外, 炔丙基三氟甲基硫醚77也可以顺利地以中等到优秀的收率和对映选择性转化为联烯类化合物79 (Scheme 15c).值得一提的是, 他们用廉价的一价铜催化剂代替二价铑催化剂, 在体系中加入手性双噁唑啉配体81时, 对于烯丙基三氟甲基硫醚72和芳基重氮乙酸乙酯80参与的该不对称三氟甲硫基化的反应, 同样得到了优良的产率以及对映选择性(Scheme 15d).但是在一价铜催化的体系中, 烯基重氮乙酸乙酯的反应给出了较低的对映选择性, 而炔丙基三氟甲基硫醚的反应活性较低.
该反应的机理也是人们比较感兴趣的一个方面.其中比较有趣的一个问题是, [2, 3]-σ重排是经历自由硫叶立德的重排过程还是经历一个金属相连的硫叶立德的重排过程.为了确定重排到底经历哪种模式, 他们首先探索了不同结构的催化剂对烯丙基硫醚83和重氮化合物84反应结果的影响.
由于此时重排后的产物存在非对映选择性, 假如反应经历的是自由叶立德的重排过程, 那么非对映选择性应当是由底物决定的, 几乎不受催化剂的影响; 假如反应经历金属相连的叶立德的重排过程, 那么催化剂的结构应当会对非对映选择性产生影响.他们选取了几种结构相差较大的铑催化剂来进行反应, 结果显示反应的非对映选择性随着催化剂的改变几乎保持不变(Scheme 16a), 说明该体系中[2, 3]-σ重排更可能是自由叶立德的重排过程.接着他们使用对称的双烯丙基硫醚86进一步对反应进行了机理探究实验.假如反应经历的是自由叶立德的重排过程, 由于此时硫叶立德中间体没有手性, 得到的产物也不会有对映选择性; 反之, 金属相连的硫叶立德中间体是手性的, 这会诱导产物出现一定的对映选择性.他们使用不同的催化剂以及不同的重氮化合物与双烯丙基硫醚反应, 最终给出的对映选择性几乎都接近0%(Scheme 16b), 该实验结果也说明在该体系中[2, 3]-σ重排更可能是自由叶立德的重排过程.在铜催化的体系中, 相关机理实验也得到了相同的结论.
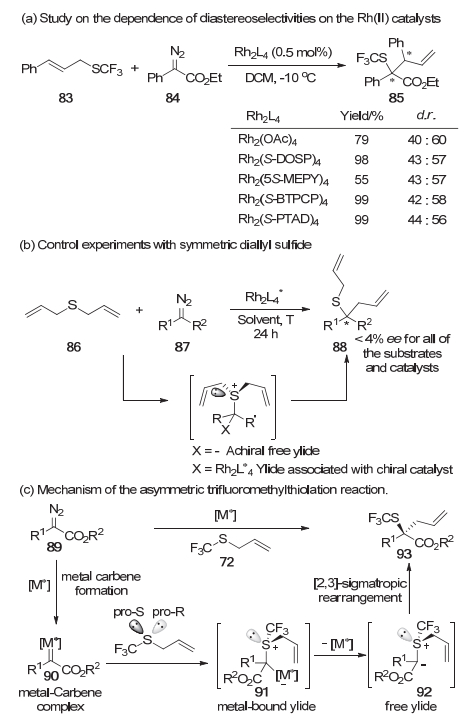
基于以上实验事实, 他们认为该反应具有如下机理:首先是手性金属催化剂分解重氮化合物89得到手性的金属卡宾中间体90, 随后其会高对映选择性地识别硫原子上的孤对电子得到金属相连的硫叶立德中间体91, 该中间体进一步发生金属的解离得到自由的硫叶立德中间体92(该自由的手性硫叶立德中间体具有足够稳定性), 然后发生协同的[2, 3]-σ重排使得手性从硫中心转移到碳中心(Scheme 16c).最后他们还提出了不对称诱导模型, 他们认为在手性金属卡宾识别硫醚中两对对映异伦的孤对电子的过渡态中三氟甲基与配体之间的电子排斥作用诱导了硫叶立德手性的产生, 从而控制了整个反应的对映选择性.
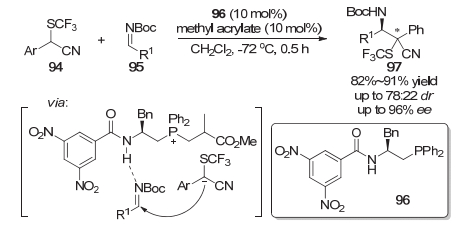
在2017年赵刚等[21]也报道了基于含SCF3合成砌块构建含C—SCF3键立体中心的工作.在该项工作中, 采用了有机膦96与甲基丙烯酸甲酯双试剂催化的策略, 经历Mannich类型的反应, 获得了较高的收率以及中等到优秀的对映选择性, 但是反应的非对映选择性较差(Scheme 17).
从以上总结中我们可以发现, 自2013年首次报道以来, 不对称三氟甲硫基化反应在过去的五年间得到了一些发展.目前主要有使用亲电的三氟甲硫基试剂和使用含三氟甲硫基的合成砌块这两种策略, 前者主要应用于β-酮酸酯、苯并或普通的(杂)环酮以及烯烃的不对称三氟甲硫基化反应中, 一般都是经历SN2类型的机理; 后者目前只有2017年的两例报道, 其中由烯丙基三氟甲基硫醚作为合成砌块经历Doyle-Kirmse反应的相关策略为不对称三氟甲硫基化反应开启了新的模式.此外, 使用含三氟甲硫基合成砌块这一策略理论上可以通过多种多样的反应模式来构建含C—SCF3键的立体中心.
尽管过去五年中, 不对称三氟甲硫基化反应取得了重要的进展, 但是相对而言, 该领域还处于发展的起步阶段, 已有的报道中反应模式还比较少, 所采取的策略也比较单一.同时, 该领域中还存在着较多的挑战:例如利用亲电的或其它三氟甲硫基试剂发展普通羰基α位的不对称三氟甲硫基化反应; 用亲核的或其它三氟甲硫基试剂通过金属催化的偶联反应或者其它非金属催化的反应途径实现有机化合物的不对称三氟甲硫基化; 将含三氟甲硫基的简单合成砌块通过更多的有机反应途径合成各种各样含有C—SCF3键的立体中心的复杂有机化合物等.总而言之, 有机化合物的不对称三氟甲硫基化反应还存在着较大的发展空间.
(a) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359. (b) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320. (c) Manteau, B.; Pazenok, S.; Vors, J.-P.; Leroux, F. R. J. Fluorine Chem. 2010, 131, 140. (d) Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315.
(a) Leo, A.; Hansch, C.; Elkins, D. Chem. Rev. 1971, 71, 525. (b) Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207.
For recent reviews on trifluoromethylthiolation, see: (a) Toulgoat, F.; Alazet, S.; Billard, T. Eur. J. Org. Chem. 2014, 2014, 2415. (b) Shao, X.; Xu, C.; Lu, L.; Shen, Q. Acc. Chem. Res. 2015, 48, 1227. (c) Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731. (d) Zhang, K.; Xu, X.; Qing, F. Chin. J. Org. Chem. 2015, 35, 556(in Chinese). (张柯, 徐修华, 卿凤翎, 有机化学, 2015, 35, 556.) (e) Xu, J.; Chen, P.; Ye, J.; Liu, G. Acta Chim. Sinica 2015, 73, 1294(in Chinese). (徐佳斌, 陈品红, 叶金星, 刘国生, 化学学报, 2015, 73, 1294.) (f) Zheng, H.; Huang, Y.; Weng, Z. Tetrahedron Lett. 2016, 57, 1397. (g) Li, G.; Sun, D. Chin. J. Org. Chem. 2016, 36, 1715(in Chinese). (李恭铭, 孙德群, 有机化学, 2016, 36, 1715). (h) Barata-Vallejo, S.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 7150. (i) Guo, Y.; Huang, M.-W.; Fu, X.-L.; Liu, C.; Chen, Q.-Y.; Zhao, Z.-G.; Zeng, B.-Z.; Chen, J. Chin. Chem. Lett. 2017, 28, 719. (j) Zhang, P.; Lu, L.; Shen, Q. Acta Chim. Sinica 2017, 75, 744(in Chinese). (张盼盼, 吕龙, 沈其龙, 化学学报, 2017, 75, 744.)(k)Zhao, X.; Li, T.; Tian, M.; Su, Z.; Wei, A.; Lu, K. Chin. J. Org. Chem. 2018, 38, 677(in Chinese). (赵霞, 李天骄, 田苗苗, 苏志扬, 魏奥琪, 芦逵, 有机化学, 2018, 38, 677.)
Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. Angew. Chem., Int. Ed. 2013, 52, 12856. doi: 10.1002/anie.201304957
Wang, X.; Yang, T.; Cheng, X.; Shen, Q. Angew. Chem., Int. Ed. 2013, 52, 12860. doi: 10.1002/anie.201305075
Rueping, M.; Liu, X.; Bootwicha, T.; Pluta, R.; Merkens, C. Chem. Commun. 2014, 50, 2508. doi: 10.1039/c3cc49877h
Zhu, X.-L.; Xu, J.-H.; Cheng, D.-J.; Zhao, L.-J.; Liu, X.-Y.; Tan, B. Org. Lett. 2014, 16, 2192. doi: 10.1021/ol5006888
Yang, T.; Shen, Q.; Lu, L. Chin. J. Chem. 2014, 32, 678. doi: 10.1002/cjoc.201400392
Liao, K.; Zhou, F.; Yu, J.-S.; Gao, W.-M.; Zhou, J. Chem. Commun. 2015, 51, 16255. doi: 10.1039/C5CC07010D
Zhao, B.-L.; Du, D.-M. Org. Lett. 2017, 19, 1036. doi: 10.1021/acs.orglett.6b03846
Li, M.; Xue, X.-S.; Cheng, J.-P. ACS Catal. 2017, 7, 7977. doi: 10.1021/acscatal.7b03007
Deng, Q.-H.; Rettenmeier, C.; Wadepohl, H.; Gade, L. H. Chem.-Eur. J. 2014, 20, 93. doi: 10.1002/chem.201303641
Jin, M. Y.; Li, J.; Huang, R.; Zhou, Y.; Chung, L. W.; Wang, J. Chem. Commun. 2018, 54, 4581. doi: 10.1039/C8CC02097C
Zhang, H.; Leng, X.; Wan, X.; Shen, Q. Org. Chem. Front. 2017, 4, 1051. doi: 10.1039/C7QO00042A
Chachignon, H.; Kondrashov, E. V.; Cahard, D. Adv. Synth. Catal. 2018, 360, 965. doi: 10.1002/adsc.v360.5
Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X. Angew. Chem., Int. Ed. 2016, 55, 5846. doi: 10.1002/anie.201601713
Luo, J.; Liu, Y.; Zhao, X. Org. Lett. 2017, 19, 3434. doi: 10.1021/acs.orglett.7b01392
Luo, J.; Cao, Q.; Cao, X.; Zhao, X. Nat. Commun. 2018, 9, 527. doi: 10.1038/s41467-018-02955-0
Liu, X.; Liang, Y.; Ji, J.; Luo, J.; Zhao, X. J. Am. Chem. Soc. 2018, 140, 4782. doi: 10.1021/jacs.8b01513
Zhang, Z.; Sheng, Z.; Yu, W.; Wu, G.; Zhang, R.; Chu, W.-D.; Zhang, Y.; Wang, J. Nat. Chem. 2017, 9, 970. doi: 10.1038/nchem.2789
Xu, L.; Wang, H.; Zheng, C.; Zhao, G. Adv. Synth. Catal. 2017, 359, 2942. doi: 10.1002/adsc.v359.17
Hock, K. J.; Koenigs, R. M. Angew. Chem., Int. Ed. 2017, 56, 13566. doi: 10.1002/anie.201707092
Mao, G.; Chen, L.; Wang, C. Sci. China Chem. 2017, 60, 1565. doi: 10.1007/s11426-017-9113-5

图式 1 构建含C—SCF3键立体中心的两种策略
Scheme 1 Two strategies for the construction of trifluoromethylthio-containing stereocentres

图式 2 奎尼丁催化的β-酮酸酯底物的不对称三氟甲硫基化反应
Scheme 2 Quinidine-catalyzed asymmetric trifluoromethylthiolation of β-ketoesters

图式 3 奎宁催化的β-酮酸酯底物的不对称三氟甲硫基化反应
Scheme 3 Quinine-catalyzed asymmetric trifluoromethylthiolation of β-ketoesters

图式 5 α-硫代羰基化合物的不对称三氟甲硫基化反应
Scheme 5 Asymmetric trifluoromethylthiolation of S-based active methine compounds

图式 6 不对称三氟甲硫基化/Michael加成/aldol串联反应
Scheme 6 Asymmetric trifluoromethylthiolation/sulfur-Michael/aldol cascade reaction

图式 7 奎尼丁催化的不对称三氟甲硫基化反应机理
Scheme 7 Mechanism of quinidine-catalyzed asymmetric trifluoromethylthiolation reaction

图式 8 铜催化的环状β-酮酸酯底物的不对称三氟甲硫基化反应
Scheme 8 Copper-catalyzed asymmetric trifluoromethylthiolation of cyclic β-ketoesters

图式 9 铜催化的烯酮化合物的1, 4-加成/三氟甲硫基化串联反应
Scheme 9 Copper-catalyzed tandem 1, 4-addition/trifluoromethylthiolation of acyclic enones

图式 10 使用光学纯的手性试剂完成的不对称三氟甲硫基化反应
Scheme 10 Asymmetric trifluoromethylthiolation using optically pure chiral trifluoromethylthiolation reagents

图式 11 使用Evans手性噁唑啉辅基完成的不对称三氟甲硫基化反应
Scheme 11 Asymmetric trifluoromethylthiolation using Evans' chiral oxazolidinone auxiliary

图式 12 双功能手性硫/硒醚催化剂的催化途径
Scheme 12 Approach of the bifunctional chiral sulfide/selenide catalyst

图式 13 手性硫/硒醚催化的烯烃的分子内双官能团化/不对称三氟甲硫基化反应
Scheme 13 Chiral sulfide/selenide catalyzed intramolecular difunctionalization/asymmetric trifluoromethylthiolation of alkenes

图式 14 手性硒醚催化的烯丙位三氟甲硫基化反应以及烯烃的分子间双官能团化/不对称三氟甲硫基化反应
Scheme 14 Chiral selenide catalyzed allylic asymmetric trifluoromethylthiolation and intermolecular difunctionalization/asymmetric trifluoromethylthiolation of alkenes


图式 16 控制实验进一步理解不对称三氟甲硫基化反应
Scheme 16 Control experiments to gain insights into the mechanism of the asymmetric trifluoromethylthiolation reaction

图式 17 经双试剂催化实现的不对称三氟甲硫基化
Scheme 17 Asymmetric trifluoromethylthiolation via dual-reagent catalysis
表 1 常见含氟基团的Hansch疏水性参数
Table 1. The Hansch's hydrophobicity parameter of fluorine-containing groups
| X | SCF3 | SF5 | OCF3 | CF3 | F |
| πx | 1.44 | 1.23 | 1.04 | 0.88 | 0.14 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: