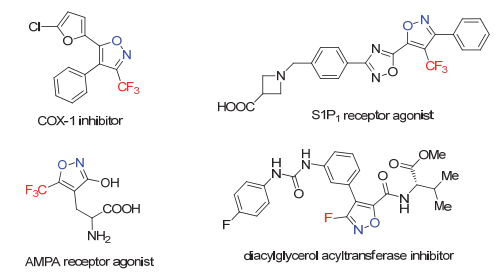
图 1.
几种具有生物活性的含氟异噁唑分子
Figure 1.
Bio-active fluorine-containing isoxazoles

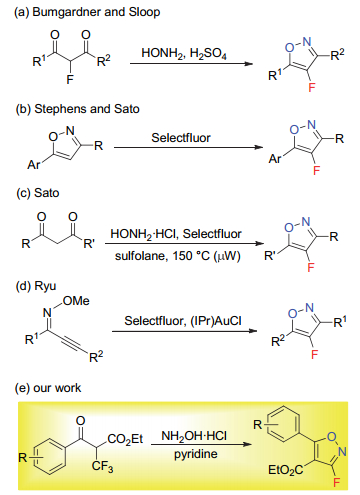
含氟杂环是药物、农药及功能材料分子中广泛存在的重要结构单元[1], 其中五元含氟芳香杂环如含氟呋喃、吡咯、吡唑、噻吩等在合成及药物化学领域已被广泛研究[2], 而有关含氟异噁唑化合物的报道则相对较少[3].异噁唑化合物大多具有丰富的生物特性[4], 氟原子的引入往往可以进一步提高其亲脂性及代谢稳定性等[5], 因而含氟异噁唑逐渐成为药物及农药开发的重要筛选砌块, 如图 1所示是几种具有生物活性的含氟异噁唑分子.然而, 迄今为止, 含氟异噁唑特别是单氟代异噁唑化合物的合成方法仍然非常有限[6], 其中代表性的例子如Scheme 1所示. 1992年, Bumgardner及Sloop等[7]报道了含氟1, 3-二酮类化合物在几种含氟杂环合成中的应用, 其中2-氟代1, 3-二酮可在盐酸羟胺存在条件下以较好的收率得到4-氟代异噁唑产物(Scheme 1, a), 然而文中仅展示了两例合成.近年来, 随着碳氢键直接官能团化反应的兴起, Stephens及Sato研究小组[8]相继发展了异噁唑化合物的直接C—H氟化反应(Scheme 1, b), 不足之处是收率偏低, 且异噁唑杂环的预先合成也限制了其在合成中的广泛应用.值得一提的是在Sato小组[8b]的工作中, 他们也成功发展了1, 3-二酮类化合物与盐酸羟胺及亲电氟化试剂Selectfluor的一锅反应直接得到4-氟代异噁唑产物(Scheme 1, c), 但反应条件较为苛刻, 底物类型局限, 且会生成难分离的区域异构体. 2014年, Ryu小组[9]发展了金催化下炔基酮肟的串联环化-氟化反应, 该方法可在温和条件下以较高的收率得到一系列4-氟代异噁唑产物(Scheme 1, d).从以上的文献调研可以看出, 氟代异噁唑化合物的合成近年来逐渐取得了很好的发展, 但仍面临诸多不足, 特别是3-氟代异噁唑化合物至今未见其通用合成路线[10], 极大限制了此类分子在合成及药物化学等领域的进一步发展与应用.基于此, 我们从易得的α-三氟甲基-β-羰基酯出发, 在盐酸羟胺及吡啶存在条件下, 发展了一锅高效合成氟代异噁唑化合物的新方法, 该反应条件温和, 操作简便, 官能团兼容性良好, 可以中等到较好的收率得到一系列3-氟-5-芳基异噁唑(Scheme 1, e), 且所得产物可进一步转化为1, 2, 4-氧杂二氮唑取代的氟代异噁唑衍生物及氟代异噁唑的酰胺类衍生物, 这两类分子均为生物活性分子的类似物.在空白对照实验基础上, 推测该反应为碱促进的双重碳氟键断裂历程[11], 中间体可能涉及偕二氟烯烃的反应路径.
α-三氟甲基-β-羰基酯(1a)可由β-羰基酯经三氟甲基化反应快速大量制备[12], 当在吡啶存在条件下其与盐酸羟胺进行反应时, 我们预期会得到4-三氟甲基异噁唑酮化合物[13], 但反应结果却是意外得到了3-氟代异噁唑化合物.推测该反应可能经历了双重碳氟键断裂历程, 且其成功实现可为氟代异噁唑化合物的合成提供一条新的合成路线, 因此我们接下来对该反应进行了一系列研究.如表 1所示, 当用吡啶作为碱时, 在乙醇中80 ℃反应12 h, 产物2a的收率为37% (Entry 1), 而其他各种有机碱(Entries 2~5)及无机碱(Entry 6)均未能取得更好的结果.接下来我们选定吡啶为碱, 对多种反应溶剂进行了考察, 遗憾的是, 在甲苯、N, N-二甲基甲酰胺(DMF)及1, 4-二氧六环(dioxane)等溶剂中(Entries 7~10), 反应未能检测到目标产物, 不过当选用乙腈作为溶剂时, 3-氟代异噁唑2a收率可达38% (Entry 11), 且反应体系较纯, 因此接下来选定乙腈作为溶剂对该反应进行进一步优化.升高反应温度会造成更多副产物的产生, 而降低反应温度会造成原料转化率降低, 因此确定最优反应温度为75 ℃ (Entries 12~14).随后对盐酸羟胺及吡啶的当量进行了优化(Entries 15~18), 当使用3.3 equiv.盐酸羟胺及4.4 equiv.吡啶时, 反应可取得最好的效果, 产物2a收率可达70% (Entry 17).
 下载:
导出CSV
下载:
导出CSV
 | |||||
| Entry | Base | x/y | Solvent | Temp./℃ | Yieldb/% |
| 1 | Pyridine | 2.2/2.2 | EtOH | 80 | 37 |
| 2 | Et3N | 2.2/2.2 | EtOH | 80 | nd |
| 3 | DBU | 2.2/2.2 | EtOH | 80 | nd |
| 4 | DABCO | 2.2/2.2 | EtOH | 80 | 19 |
| 5 | DMAP | 2.2/2.2 | EtOH | 80 | nd |
| 6 | Cs2CO3 | 2.2/3.0 | EtOH | 80 | nd |
| 7 | Pyridine | 2.2/2.2 | Toluene | 80 | nd |
| 8 | Pyridine | 2.2/2.2 | DMF | 80 | nd |
| 9 | Pyridine | 2.2/2.2 | Dioxane | 80 | nd |
| 10 | Pyridine | 2.2/2.2 | iPrOH | 80 | nd |
| 11 | Pyridine | 2.2/2.2 | CH3CN | 80 | 38 |
| 12 | Pyridine | 2.2/2.2 | CH3CN | 85 | 28 |
| 13 | Pyridine | 2.2/2.2 | CH3CN | 75 | 44 |
| 14 | Pyridine | 2.2/2.2 | CH3CN | 65 | 30 |
| 15 | Pyridine | 1.1/1.1 | CH3CN | 75 | 31 |
| 16 | Pyridine | 3.3/3.9 | CH3CN | 75 | 46 |
| 17 | Pyridine | 3.3/4.4 | CH3CN | 75 | 70 |
| 18 | Pyridine | 3.3/4.9 | CH3CN | 75 | 41 |
| 1a Unless noted otherwise, reactions were performed with substrate 1a (0.1 mmol), HONH2·HCl (x equiv.), base (y equiv.) in solvent (2.0 mL) at indicated temperature for 12 h. b Isolated yield. | |||||
在确定最优反应条件后, 我们对该反应的底物普适性进行了考察.如表 2所示, 从一系列α-三氟甲基-β-羰基酯出发, 在盐酸羟胺与吡啶条件下, 可顺利得到14个3-氟代异噁唑产物, 收率最高可达83%.其中当芳基取代基为卤素时, 反应效果普遍较好(2b~2g), 但当卤素取代基在芳基邻位时, 可能由于位阻原因造成目标产物收率明显下降(2d).为进一步确认所得化合物的分子结构, 特别是排除所得产物是5-氟-3-芳基异噁唑的可能性, 选择产物2f重结晶并成功对其进行了X射线单晶衍射分析[14], 结果显示该化合物确为3-氟-5芳基异噁唑分子.当反应底物羰基酯中芳环上为吸电子取代基时, 该反应可顺利进行并得到相应目标产物(2h~2i).该反应对供电子性基团取代的底物也可很好的兼容, 如不同位置取代的甲基及甲氧基类α-三氟甲基-β-羰基酯均可生成相应的目标产物(2j~2m).此外, 萘基衍生的α-三氟甲基-β-羰基酯也可进行此反应, 并以中等的收率得到3-氟代异噁唑2n.遗憾的是, 当尝试脂肪基取代的α-三氟甲基-β-羰基酯时, 未能顺利得到目标产物.
下载:
导出CSV

|
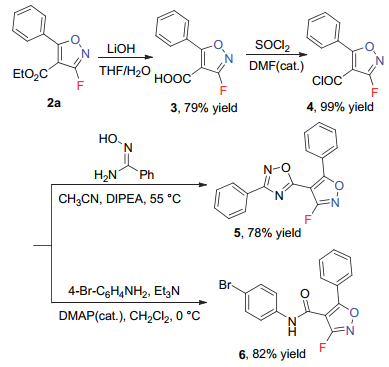
为进一步展示该反应的潜在合成应用价值, 我们选择产物2a进行了合成转化.如Scheme 2所示, 经过水解及氯化反应, 可以良好的收率顺利得到氟代异噁唑酰氯4, 随后分别进行环化反应及胺化反应, 可以78%及82%的收率分别得到1, 2, 4-氧杂二氮唑取代的氟代异噁唑衍生物5及氟代异噁唑的酰胺类衍生物6, 其中化合物5为具有S1P1受体拮抗剂活性的三氟甲基异噁唑的氟代类似物[15], 为此类分子的深入研究提供了一定的合成基础.
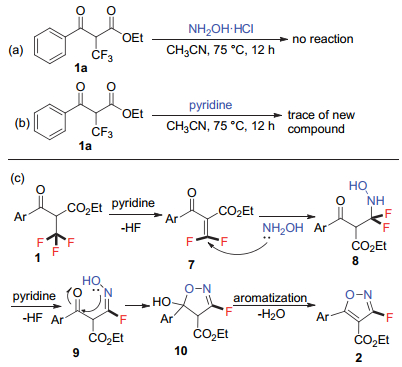
为研究该反应的反应历程, 我们进行了一系列控制实验.如Scheme 3所示, 原料1a与盐酸羟胺在无吡啶的反应条件下未检测到任何转化(Scheme 3, a), 这说明α-三氟甲基-β-羰基酯未与盐酸羟胺反应生成肟类中间体; 而原料1a与吡啶在无盐酸羟胺的反应条件下则检测到可生成新的化合物(Scheme 3, b), 氟谱检测显示其裂分形式为两组双重峰, 我们推测其可能为化合物1a脱除一分子氟化氢后生成的偕二氟烯烃中间体, 遗憾的是转化率太低, 未能分离得到该化合物.根据所得实验结果及文献信息[16], 推测该反应的机理如Scheme 3 (c)所示, α-三氟甲基-β-羰基酯在吡啶存在条件下脱除一分子氟化氢生成偕二氟烯烃中间体7, 随后羟胺的氮原子进攻亲电性的α, β-不饱和烯烃7得到中间体8, 这一二氟烷基衍生物在吡啶存在条件下可进一步脱除氟化氢生成氟代肟类中间体9, 紧接着肟骨架中的氧原子进攻羰基生成环化产物10, 进一步脱水芳构化即生成最终产物3-氟代异噁唑2.
α-三氟甲基-β-羰基酯类化合物在盐酸羟胺及吡啶存在条件下, 可以中等到较好的收率一锅反应生成一系列3-氟代异噁唑化合物, 该反应条件温和, 操作简便, 官能团兼容性良好, 且所得产物可进一步合成转化为1, 2, 4-氧杂二氮唑取代的氟代异噁唑衍生物及氟代异噁唑的酰胺类衍生物, 为此类生物活性分子的合成提供了新的思路.根据初步机理研究推测该反应为碱促进的双重碳氟键断裂历程, 中间体可能涉及偕二氟烯烃的反应路径, 详细的机理实验及该方法的合成拓展应用正在进行之中.
在一烘干的反应管中依次加入α-三氟甲基-β-羰基酯1 (0.2 mmol, 1.0 equiv.), 盐酸羟胺(46 mg, 0.66 mmol), 及吡啶(71 μL, 0.88 mmol).随后加入3 mL乙腈并将反应管置于75 ℃条件下反应12 h.薄层色谱板(TLC)检测显示原料已反应完全, 冷却至室温后, 加入10 mL水, 用乙酸乙酯萃取3次, 合并有机相用饱和食盐水洗涤后用无水硫酸钠干燥, 过滤后真空脱除溶剂, 粗产品用硅胶色谱柱分离提纯(石油醚/乙酸乙酯, V:V=30:1)得到产物3-氟-5-取代苯基异噁唑-4-甲酸乙酯2.
(a) Schlosser, M. Angew. Chem., Int. Ed. 2006, 45, 5432. (b) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881. (c) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359. (d) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320. (e) Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1. (f) Fluorinated Hetreocycles, ACS Symp. Ser. No. 1003, Eds.: Gakh, A. A.; Kirk, K. L. ACS, Washington, DC, 2009, pp. 3~20. (g) Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470. (h) Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496. (i) Li, S.; Ma, J.-A. Chem. Soc. Rev. 2015, 44, 7439. (j) Jeschke, P. Pest Manage. Sci. 2017, 73, 10536. (k) Chen, C.; Fu, L.; Chen, P.; Liu, G. Chin. J. Chem. 2017, 35, 1781. (l) Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338.
(a) Fluorine in Heterocyclic Chemistry, Vol. 1, Eds.: Nenajdenko, V., Springer International Publishing, 2014. (b) Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.-P.; Leroux, F. R. J. Fluorine Chem. 2013, 152, 2.
(a) Cordero, F. M.; Giomi, D.; Lascialfari, L. In Progress in Heterocyclic Chemistry, Vol. 27, Eds.: Gribble, G. W.; Joule, J. A. Elsevier Ltd., 2015, p. 321. (b) Agrawal, N.; Mishra, P. Med. Chem. Res. 2018, 27, 1309.
(a) Pinho, M.; Teresa, M. V. D. Curr. Org. Chem. 2005, 9, 925. (b) Kaur, K.; Kumar, V.; Sharma, A. K.; Gupta, G. K. Eur. J. Med. Chem. 2014, 77, 121. (c) Hu, F.; Szostak, M. Adv. Synth. Catal. 2015, 357, 2583. (d) Morita, T.; Yugandar, S.; Fuse, S.; Nakamura, H. Tetrahedron Lett. 2018, 59, 1159.
(a) Fluorine in Medicinal Chemistry and Chemical Biology, Eds.: Ojima, I., Wiley-Blackwell, Chichester, UK, 2009. (b) Isanbora, C.; O'Hagan, D. J. Fluorine Chem. 2006, 127, 303.
(a) Peng, W.-M.; Zhu, S.-Z. Acta Chim. Sinica 2003, 61, 455. (彭卫民, 朱仕正, 化学学报, 2003, 61, 455.) (b) Kumar, V.; Kaur, K. J. Fluorine Chem. 2015, 180, 55.
(a) Bumgardner, C. L.; Sloop, J. C. J. Fluorine Chem. 1992, 56, 141. (b) Sloop, J. C.; Bumgardner, C. L.; Loehle, W. D. J. Fluorine Chem. 2002, 118, 135.
(a) Stephens, C. E.; Blake, J. A. J. Fluorine Chem. 2004, 125, 1939. (b) Sato, K.; Sandford, G.; Shimizu, K.; Akiyama, S.; Lancashire, M. J.; Yufit, D. S.; Tarui, A.; Omote, M.; Kumadaki, I.; Harusawa, S.; Ando, A. Tetrahedron 2016, 72, 1690.
Jeong, Y.; Kim, B.-I.; Lee, J. K.; Ryu, J.-S. J. Org. Chem. 2014, 79, 6444. doi: 10.1021/jo5008702
(a) Dighe, S. U.; Mukhopadhyay, S.; Kolle, S.; Kanojiya, S.; Batra, S. Angew. Chem., Int. Ed. 2015, 54, 10926. (b) Yuan, X.; Yao, J.-F.; Tang, Z.-Y. Org. Lett. 2017, 19, 1410.
(a) Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119. (b) Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Chem. Rev. 2015, 115, 931. (c) Shen, Q.; Huang, Y.-G.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y. J. Fluorine Chem. 2015, 179, 14.
Ohtsuka, Y.; Uraguchi, D.; Yamamoto, K.; Tokuhisa, K.; Yamakawa, T. J. Fluorine Chem. 2016, 181, 1. doi: 10.1016/j.jfluchem.2015.10.013
Okamoto, K.; Nanya, A.; Eguchi, A.; Ohe, K. Angew. Chem., Int. Ed. 2018, 57, 1039. doi: 10.1002/anie.201710920
CCDC 1843944 contains the supplementary crystallographic data for this compound 2f, these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
(a) Watterson, S. H.; Guo, J.; Spergel, S. H.; Langevine, C. M.; Moquin, R. V.; Shen, D. R.; Yarde, M.; Cvijic, M. E.; Banas, D.; Liu, R.; Suchard, S. J.; Gillooly, K.; Taylor, T.; Rex-Rabe, S.; Shuster, D. J.; McIntyre, K. W.; Cornelius, G.; D'Arienzo, C.; Marino, A.; Balimane, P.; Warrack, B.; Salter-Cid, L.; McKinnon, M.; Barrish, J. C.; Carter, P. H.; Pitts, W. J.; Xie, J.; Dyckman, A. J. J. Med. Chem. 2016, 59, 2820. (b) Hou, X.; Zhang, H.; Chen, B.-C.; Guo, Z.; Singh, A.; Goswami, A.; Gilmore, J. L.; Sheppeck, J. E.; Dyckman, A. J.; Carter, P. H.; Mathur, A. Org. Process Res. Dev. 2017, 21, 200.
(a) Menozzi, G.; Schenone, P.; Mosti, L. J. Heterocycl. Chem. 1983, 20, 645. (b) Strah, S.; Golobič, A.; Golobič, L.; Stanovnik, B. J. Heterocycl. Chem. 1997, 34, 1511. (c) Tang, X.-H.; Hu, C.-M. J. Fluorine Chem. 1995, 74, 9. (d) Ohtsuka, Y.; Uraguchi, D.; Yamamoto, K.; Tokuhisa, K.; Yamakawa, T. Tetrahedron 2012, 68, 2636. (e) Xu, L.; Zhang, Q.; Xie, Q.; Huang, B.; Dai, J.-J.; Xu, J.; Xu, H.-J. Chem. Commun. 2018, 54, 4406.

图式 2 3-氟代异噁唑产物的合成转化
Scheme 2 Synthetic transformations of obtained 3-F-substituted isoxazoles
表 1 盐酸羟胺作用下碳氟键断裂构建氟代异噁唑的条件筛选a
Table 1. Screening studies of consecutive double C—F bond cleavage to access fluorine-substituted isoxazoles
| |||||
| Entry | Base | x/y | Solvent | Temp./℃ | Yieldb/% |
| 1 | Pyridine | 2.2/2.2 | EtOH | 80 | 37 |
| 2 | Et3N | 2.2/2.2 | EtOH | 80 | nd |
| 3 | DBU | 2.2/2.2 | EtOH | 80 | nd |
| 4 | DABCO | 2.2/2.2 | EtOH | 80 | 19 |
| 5 | DMAP | 2.2/2.2 | EtOH | 80 | nd |
| 6 | Cs2CO3 | 2.2/3.0 | EtOH | 80 | nd |
| 7 | Pyridine | 2.2/2.2 | Toluene | 80 | nd |
| 8 | Pyridine | 2.2/2.2 | DMF | 80 | nd |
| 9 | Pyridine | 2.2/2.2 | Dioxane | 80 | nd |
| 10 | Pyridine | 2.2/2.2 | iPrOH | 80 | nd |
| 11 | Pyridine | 2.2/2.2 | CH3CN | 80 | 38 |
| 12 | Pyridine | 2.2/2.2 | CH3CN | 85 | 28 |
| 13 | Pyridine | 2.2/2.2 | CH3CN | 75 | 44 |
| 14 | Pyridine | 2.2/2.2 | CH3CN | 65 | 30 |
| 15 | Pyridine | 1.1/1.1 | CH3CN | 75 | 31 |
| 16 | Pyridine | 3.3/3.9 | CH3CN | 75 | 46 |
| 17 | Pyridine | 3.3/4.4 | CH3CN | 75 | 70 |
| 18 | Pyridine | 3.3/4.9 | CH3CN | 75 | 41 |
| 1a Unless noted otherwise, reactions were performed with substrate 1a (0.1 mmol), HONH2·HCl (x equiv.), base (y equiv.) in solvent (2.0 mL) at indicated temperature for 12 h. b Isolated yield. | |||||
 下载: 导出CSV
下载: 导出CSV
表 2 盐酸羟胺作用下碳氟键断裂构建3-氟代异噁唑底物拓展a
Table 2. Substrate scope of 3-F-substituted isoxazoles via consecutive double C—F bond cleavage
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: