Received Date:
09 April 2018 Available Online:
15 July 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21325208, 21572212, 21732006, 21702041), Ministry of Science and Technology of China (No. 2017YFA0303500), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB20000000), Fundamental Research Funds for the Central Universities, and Program for Changjiang Scholars and Innovative Research Team in University
Abstract:
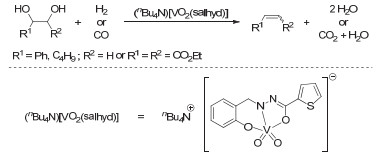
In view of the depletion of fossil fuels, the development and utilization of environment-friendly and sustainable resources widely play an indispensable role in alleviating and resolving problems about resources and environment. Biomass could be utilized as biofuels and renewable platform chemicals. However, biomass-derived molecules are fairly oxygen-rich and hyperfunctionalized. Therefore, new synthetic routes for the regenerative production of chemicals, fuels, and energy from renewable biomass sources are currently investigated especially the routes of transforming high-oxygen-content biomassderived vicinal diols and poly vicinal alcohols into fuels and value-added chemicals. A range of reductive deoxygenation methods consisting of direct deoxygenation, pyrolysis, hydrogenolysis, decarbonylation, decarboxylation, hydrodeoxygenation, and deoxydehydration (DODH) are under investigation. In this review, we detail the recent-evolutionary and efficient strategies of transition metal-catalyzed DODH of vicinal diols into corresponding alkenes, including rhenium, molybdenum, vanadium, and ruthenium catalysts. Rhenium-catalyzed DODH reactions are very selective and active to provide high yields of olefin products, which keep important functionality in place as well as can be readily functionalized. Recent efforts in rhenium-mediated systems include the development of new rhenium catalysts, the application of cheaper and more available reductants, and growing mechanistic understandings owing to both theoretical and experimental studies. A new emerging trend within DODH is the development of heterogeneous rhenium-based catalysts which demonstrates their ability to rival and in some cases surpass their homogeneous counterparts. Furthermore, catalysts based on the transition metals molybdenum, vanadium and ruthenium show great potential as inexpensive alternatives to rhenium catalysts.
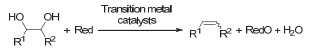
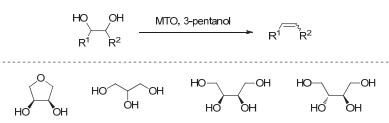
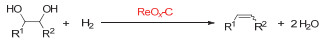
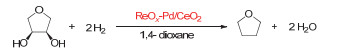
Figure 1.
Transition metal-catalyzed deoxydehydration of vicinal diols into alkenes in the presence of reductants (referred to as "Red"), R1, R2=alkyl, aryl, or H
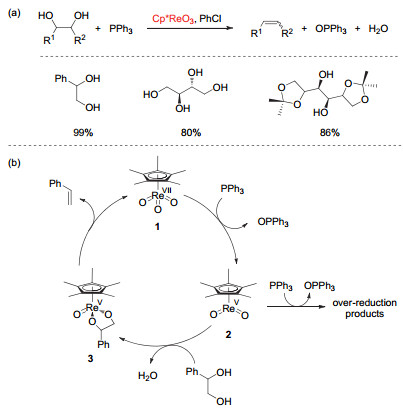
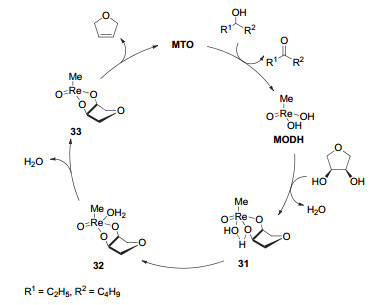
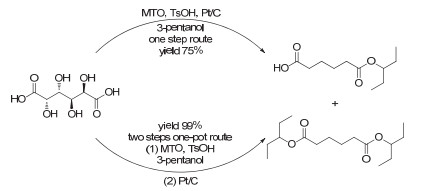
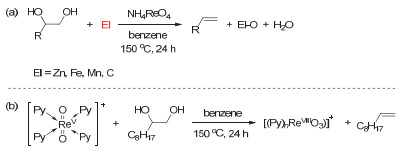
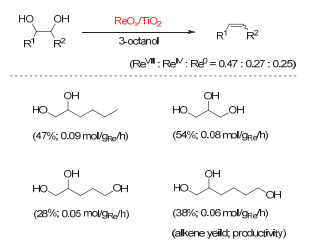
Figure 7.
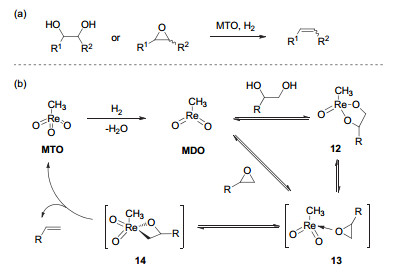
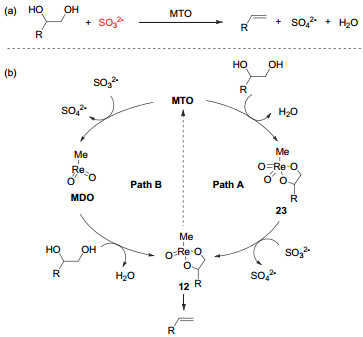
(a) Deoxydehydration of diols and deoxygenation of epoxides catalyzed by methyl trioxorhenium; (b) plausible mechanism of deoxydehydration and deoxygenation
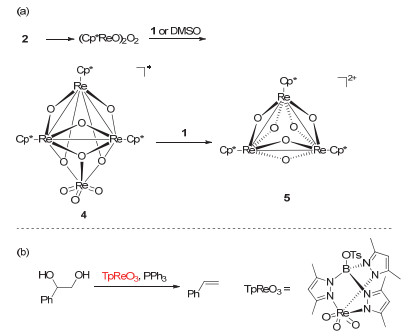
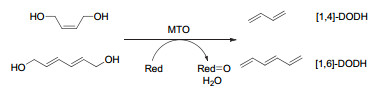
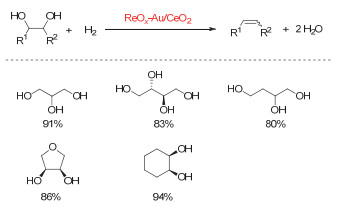
Figure 10.
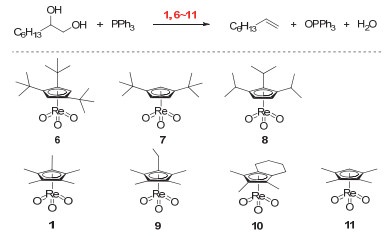
(a) DODH reactions catalyzed by MTO and perrhenates (XReO4) using of sulfites as the reductant; (b) Schematic representation of erythritol reactivity for sulfite-driven DODH
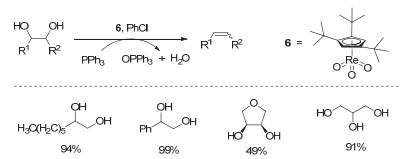
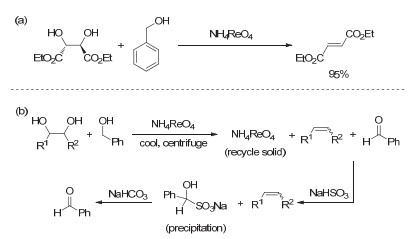
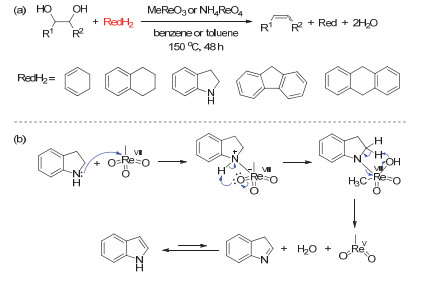
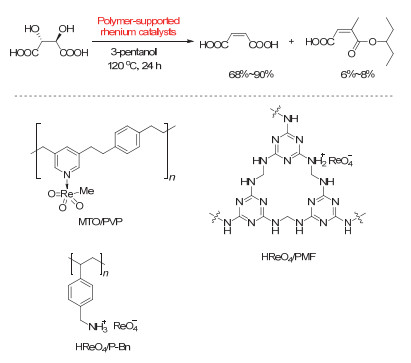
Figure 16.
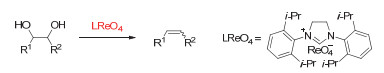
(a) NH4ReO4/benzyl-alcohol-catalyzed DODH of diethyl L-tartrate to diethyl fumarate; (b) process of product separation and benzaldehyde recovery
Huang, Y.; Yang, Z.; Chen, M.; Dai, J.; Guo, Q.; Fu, Y. ChemSusChem 2013, 6, 1348. doi: 10.1002/cssc.201300190
[33]
Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B. C.; Rahimpou, M. R. Energy Environ. Sci. 2014, 7, 103. doi: 10.1039/C3EE43081B
[34]
Chen, M.; Huang, Y.; Pang, H.; Liu, X.; Fu, Y. Green Chem. 2015, 17, 1710. doi: 10.1039/C4GC01992J
Cook, G. K.; Andrews, M. A. J. Am. Chem. Soc. 1996, 118, 9448. doi: 10.1021/ja9620604
[64]
Gable, K. P. ; Ross, B. ACS Symposium Series, Vol. 921, Eds. : Bozell, J. J. ; Patel, M. K. American Chemical Society, Washington, DC, 2006, Chapter 11.
[65]
Bergman, R. G.; Cundari, T. R.; Gillespie, A. M.; Gunnoe, T. B.; Harman, W. D.; Klinckman, T. R.; Temple, M. D.; White, D. P. Organometallics 2003, 22, 2331. doi: 10.1021/om021048j
[66]
Raju, S.; Jastrzebski, J. T. B. H.; Lutz, M.; Klein Gebbink, R. J. M. ChemSusChem 2013, 6, 1673. doi: 10.1002/cssc.201300364
[67]
Raju, S.; Jastrzebski, J. T. B. H.; Lutz, M.; Witteman, L.; Dethlefsen, J. R.; Fristrup, P.; Moret, M. E.; Klein Gebbink, R. J. M. Inorg. Chem. 2015, 54, 11031. doi: 10.1021/acs.inorgchem.5b02366
[68]
Raju, S.; van Slagmaat, C. A. M. R.; Li, J.; Lutz, M.; Jastrzebski, J. T. B. H.; Moret, M. E.; Klein Gebbink, R. J. M. Organometallics 2016, 35, 2178. doi: 10.1021/acs.organomet.6b00120
[69]
Yanagi, T.; Suzuki, H.; Oishi, M. Chem. Lett. 2013, 42, 1403. doi: 10.1246/cl.130699
Hillea, C.; Kühn, F. E. Dalton Trans. 2016, 45, 15. doi: 10.1039/C5DT03641K
[73]
Ziegler, J. E.; Zdilla, M. J.; Evans, A. J.; Abu-Omar, M. M. Inorg. Chem. 2009, 48, 9998. doi: 10.1021/ic901792b
[74]
Bi, S. W.; Wang, J. Y.; Liu, L. J.; Li, P.; Lin, Z. Y. Organometallics 2012, 31, 6139. doi: 10.1021/om300485w
[75]
Larson, R. T.; Samant, A.; Chen, J.; Lee, W.; Bohn, M. A.; Ohlmann, D. M.; Zuend, S. J.; Toste, F. D. J. Am. Chem. Soc. 2017, 139, 14001. doi: 10.1021/jacs.7b07801
[76]
Ahmad, I.; Chapman, G.; Nicholas, K. M. Inorg. Chem. 2010, 49, 4744. doi: 10.1021/ic100467p
[77]
Vkuturi, S.; Chapman, G.; Ahmad, I.; Nicholas, K. M. Organometallics 2011, 30, 2810. doi: 10.1021/om2001662
[78]
Arceo, E.; Ellman, J. A.; Bergman, R. G. J. Am. Chem. Soc. 2010, 132, 11408. doi: 10.1021/ja103436v
Kwok, K. M.; Choong, C. K. S.; Ong, D. S. W.; Ng, J. C. Q.; Gwie, C. G.; Chen, L.; Borgna, A. ChemCatChem 2017, 9, 2443. doi: 10.1002/cctc.v9.13
[115]
Stanowski, S.; Nicholas, K. M.; Srivastava, R. S. Organometallics 2012, 31, 515. doi: 10.1021/om200447z
图 1
过渡金属催化的邻二醇的脱氧脱水反应, Red为还原剂, R1, R2=烷基、芳基或氢
Figure 1
Transition metal-catalyzed deoxydehydration of vicinal diols into alkenes in the presence of reductants (referred to as "Red"), R1, R2=alkyl, aryl, or H
Figure 7
(a) Deoxydehydration of diols and deoxygenation of epoxides catalyzed by methyl trioxorhenium; (b) plausible mechanism of deoxydehydration and deoxygenation
Figure 10
(a) DODH reactions catalyzed by MTO and perrhenates (XReO4) using of sulfites as the reductant; (b) Schematic representation of erythritol reactivity for sulfite-driven DODH
Figure 16
(a) NH4ReO4/benzyl-alcohol-catalyzed DODH of diethyl L-tartrate to diethyl fumarate; (b) process of product separation and benzaldehyde recovery

 下载:
下载:

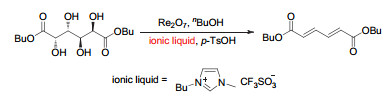

 下载:
下载:










































 下载:
下载:
