表 1
Na与Li的性质对比[1]
引用本文:
王玲, 杨国锐, 王嘉楠, 王思岚, 彭生杰, 延卫. 静电纺丝在钠离子电池中的应用研究进展[J]. 化学学报,
2018, 76(9): 666-680.
doi:
10.6023/A18040129

Citation: Wang Ling, Yang Guorui, Wang Jianan, Wang Silan, Peng Shengjie, Yan Wei. Research Progress on Electrospun Materials for Sodium-Ion Batteries[J]. Acta Chimica Sinica, 2018, 76(9): 666-680. doi: 10.6023/A18040129
Citation: Wang Ling, Yang Guorui, Wang Jianan, Wang Silan, Peng Shengjie, Yan Wei. Research Progress on Electrospun Materials for Sodium-Ion Batteries[J]. Acta Chimica Sinica, 2018, 76(9): 666-680. doi: 10.6023/A18040129
静电纺丝在钠离子电池中的应用研究进展
摘要:
钠离子电池具有与锂离子电池相似工作机理,因其原料资源丰富,是一种极具应用前景的新一代储能设备.然而,钠离子电池面临着电极材料体积膨胀过大、钠离子传输动力学较慢和能量密度偏低等问题,阻碍了其实用化.静电纺丝技术合成的一维钠离子电池电极材料,可通过形貌调控或碳复合方式有效缓冲储钠过程中电极的体积膨胀,而且具有连续的电子传递和较短的离子传输路径,从而改善钠离子传输动力学,以提高电池倍率性能.通过电纺还可简便地制备直接用于钠离子电池的柔性纤维膜来提高电池的能量密度.综述了静电纺丝技术制备钠离子电池材料的研究进展,主要包括正极和负极材料,对今后静电纺丝在钠离子电池中的发展进行了展望.
English
Research Progress on Electrospun Materials for Sodium-Ion Batteries
Abstract:
The scarce lithium resources would ultimately fail to satisfy the ever-growing industrial demand, especially for the large-scale stationary energy storage. Sodium-ion batteries (SIBs) are considered as promising next-generation power sources because sodium is widely available and exhibits similar chemistry to that of lithium-ion batteries (LIBs). Although sodium share similar physical and chemical properties to lithium, the lager ionic radius, heavier molar mass and less negative redox potential of Na+/Na of the sodium jointly lead to some issues beset the SIBs, such as sluggish sodiation kinetics, larger volume expansion and lower energy density, which need to be tackled to promote the practical applications of the SIBs. Therefore, developing appropriate electrode materials is crucial to achieve SIBs with long lifespan and high energy density. One-dimensional nanostructures can provide orientated electronic (ionic) transport and strong tolerance to volume change, thus enhancing the electrochemical performance of electrode materials. Electrospinning technique is a low cost and versatile method to fabricate continuous one-dimensional functional materials with various morphology and targeted components that has been widely applied in SIBs. The volume change could be buffered efficiently by facilely modifying the morphology of electrospun materials or in-situ compositing with carbon materials. Benefiting from the ultra-high aspect ratio, electrospun one-dimensional electrodes can reduce the ionic transport distance, while provide continuous transport way for electron along the longitudinal direction, which is helpful to improve the sluggish sodiation kinetics. It is also worth noting that free-standing or flexible fibers could be easily obtained via the electrospinning technique, which can be used as binder-free electrode to enhance the energy density of the batteries. The research progress on electrospun materials for sodium-ion batteries is summarized in this review, including cathode materials and anode materials. Their electrochemical performance in sodium storage is discussed in detail. The advantages and challenges of these materials were pointed out, and the future development of electrospun materials for sodium ion batteries was also prospected.
-
Key words:
- sodium-ion batteries
- / electrospinning
- / cathode materials
- / anode materials
- / nanofiber
- / binder-free
-
1. 引言
近年来, 能源危机和化石能源消费引起的环境问题备受世界关注, 侵袭我国多地的雾霾形成主因也是煤炭燃烧和石油大量使用.可再生能源和电动汽车的推广是缓解能源危机和应对当前环境问题的有效途径.可再生能源如:风能、太阳能和潮汐能等, 具有时间或空间分布不均、连续性不强、不稳定等缺陷, 从而需求有效的储能设备和系统来克服, 为构建稳定的绿色智能电网提供保障.绿色环保的电动汽车的发展, 也离不开动力储能设备的支持.
锂离子电池由于具有能量密度和功率密度高、循环寿命长、安全性好等优点, 在便携式电子设备及电动车上得到了广泛的应用.但锂元素在地壳中丰度只有0.002%, 且主要分布于南美和澳大利亚地区, 根据相关预测若锂消耗量以每年5%递增, 则全球锂元素存储量将在65年内耗尽.因此, 开发新的电池体系来替代锂离子电池迫在眉睫.考虑到金属钠与锂处于同一主族, 具有相似的物理化学性质, 且在自然界中储量丰富、成本低廉(如表 1[1]所示), 因此钠离子电池在大规模储能方面再次引起关注.虽然钠离子半径比锂离子大, 导致钠离子电池的能量密度比锂离子电池低, 但是钠的标准电极电位为-2.7 V, 比锂高约0.3 V, 具有更稳定的电化学性能和安全性, 且钠与铝不会形成合金, 所以可用成本更低廉的铝箔替代铜箔作为集流体, 因此虽然在电子产品上钠离子电池很难与锂离子电池竞争, 但是在大规模储能上钠离子电池则具有巨大的潜力[2].
表 1

 下载:
导出CSV
下载:
导出CSV
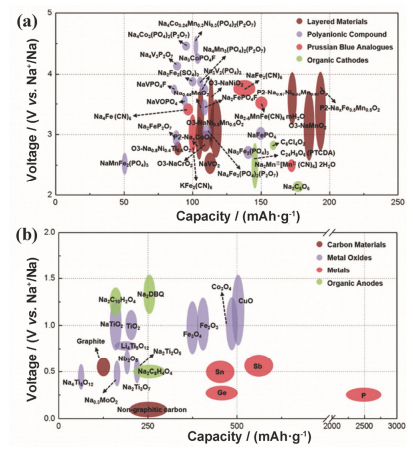
Na Li Cation radius 97 pm 68 pm Atomic weight 23.0 g•mol-1 6.9 g•mol-1 E0 vs. SHE -2.7 V -3.04 V A-O Coordination Octahedral Octahedral or tetrahedral Melting point 97.7 ℃ 180.5 ℃ Abundance 23.6×103 mg•kg-1 20 mg•kg-1 Distribution Everywhere 70% in South America Price, Carbonates ca. 2 RMB per kg ca. 40 RMB per kg 与锂离子电池相似, 钠离子电池的结构主要由正负极材料、电解液、隔膜和封装组件等组成.正负极由活性物质、导电剂炭黑、粘结剂和集流体构成.隔膜以聚乙烯、聚丙烯等聚合物以及玻璃纤维为主, 起到传输离子隔绝电子的作用.电解液为有机电解液, 通过将NaClO4或NaPF6溶解在碳酸丙烯酯(PC)、碳酸乙烯酯(EC)、碳酸二甲酯(DMC)、碳酸二乙酯(DEC)等有机溶剂中得到. 图 1[3]为钠离子电池正负极的理论容量与电压关系图.从图中可以看出, 钠离子电池正极材料主要为具有层状结构的氧化物如NaMO2(M=Fe, Co, Ni, Cr, Mn等)[4~7]、普鲁士蓝、聚阴离子化合物如Na2FeP2O7[8], Na3V2(PO4)3[9, 10], Na3V2(PO4)2F3[11]以及它们的改性材料.负极材料主要集中在碳材料、钛基材料、金属化合物及其合金、磷化物等方面.
图 1

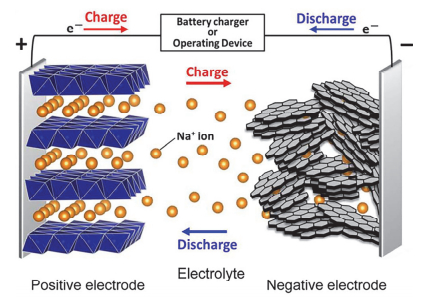
钠离子电池的工作原理如图 2[12]所示.放电过程中, Na+从负极材料脱出经电解液进入正极, 同时电子经过外电路由负极到达正极, 以达到系统电荷的平衡, 充电过程则刚好相反, 是一种“摇椅式电池”.但是由于Na+半径和质量均比Li+大, 导致其迁移动力学缓慢, 且较大的Na+也会造成嵌钠过程中更大的体积膨胀, 严重限制了钠离子电池的寿命和快速充放电性能.因此, 开发能够稳定储钠的电极材料成为当前的研究热点[13].
图 2
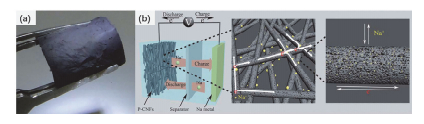
纳米材料通常具有较大的比表面积和较小的粒径, 可以缩短电荷传输距离, 被证明可以提高电极材料的活性[14, 15].在这些纳米材料中, 一维纳米材料可以促进电子在纵向方向上的快速传递, 是能源存储领域的理想选择之一[16~18].一维材料的制备方法有很多种, 包括自组装[19, 20]、水热或溶剂热[21~23]、静电纺丝[24~26]等.其中, 静电纺丝是一种简单有效且易量产的制备一维纳米纤维的技术, 通过调整电纺过程的参数可以灵活调整纳米纤维的直径、长度以及表面形貌等[27, 28].并且通过将静电纺丝和其它方法联用可以制备出各种形貌和特殊形貌的材料.对于钠离子电池来说静电纺丝材料具有很多优势.首先, 静电纺丝是一种工业化的技术, 可以实现大规模的制备, 为材料的工业化应用奠定基础.其次, 通过静电纺丝可以获得柔性的纤维膜(图 3(a)[29]), 既免去了电池制备过程中搅浆的过程, 节省了成本, 提高了电池的能量密度, 又可以减少离子扩散距离, 提高电池的功率密度和循环稳定性.而且, 通过电纺制备的材料多为一维纤维, 可以为电子的传递提供连续的一维通道(图 3(b)[29]), 有利于材料电化学性能的发挥.因此, 近年来利用静电纺丝制备钠离子电池材料的研究越来越多.
图 3

2. 静电纺丝技术原理
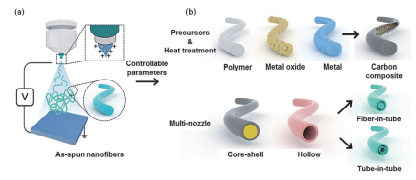
静电纺丝(简称电纺)主要由三个部分组成, 即高压电源、喷丝头以及接收装置(如图 4[28]所示).电纺过程中, 前驱体溶液在注射泵的推动下挤到喷丝头形成带电液滴, 带电液滴在电场和表面张力的共同作用下形成Taylor锥.当电场力大到可以克服表面张力时, 液滴就会发生非稳定性弯曲进而被拉伸, 分裂成更细的射流到达接收极, 同时溶剂快速挥发, 最终在接收极上形成纺丝薄膜.通过调整前驱体溶液种类和配比、电纺电压、接收距离以及温度和湿度等参数, 可以得到不同的一维纤维.而且, 通过控制后期煅烧过程也可以方便地获得各种形貌的纤维.由于静电纺丝的诸多优势, 近年来被广泛用作制备钠离子电池材料.
图 4

3. 静电纺丝正极材料
正极材料是钠离子电池的关键组成之一, 用来提供钠离子和高电位氧化还原电对, 对电池的工作电压和比容量有非常重要的影响[30].因此, 近年来研究者们在正极材料性能的改善和提升, 以及新型正极材料的开发上投入了大量的工作.
聚阴离子化合物具有高的工作电压良好的循环稳定性, 且结构多样、性能稳定, 是非常有前景的钠离子电池正极材料.其中钠快离子导体NASICON (Na super ionic conductor)型正极具有三维网状结构, Na+能在其晶体结构所含的所有三维通道中迁移, 具有迁移速度快、结构稳定等优点, 成为正极材料的研究重点之一, 但此类材料导电性较差, 通常需要与碳复合提高导电性. Li等[31]将五氧化二磷、乙酸钠和钛酸异丙酯进行电纺然后碳化或在空气中煅烧制备了NaTi2(PO4)3/C复合纤维和NaTi2(PO4)3纤维.钠离子电池充放电测试表明, NaTi2(PO4)3/C复合纤维无论在倍率性能还是循环寿命上均优于NaTi2(PO4)3纤维, 前者在5C下循环500次容量为97 mAh•g-1, 容量保持率为93%.余彦课题组[32]通过静电纺丝将20~30 nm的Na3V2(PO4)3纳米颗粒均匀分布在电纺纤维中, 在2C, 5C, 10C和20C下分别得到了77, 58, 39和20 mAh•g-1的容量.吴川课题组[33]制备了带芽孢的柳条状的Na3V2(PO4)3/C纳米纤维, 在0.2C下首次放电容量为106.8 mAh•g-1, 循环125次后仍有107.2 mAh•g-1, 良好的循环稳定性主要来自于其独特的结构以及(113)晶面的优先生长.然后他们又用电纺得到Na3V2(PO4)3/C纳米棒[34], 在0.5C下首次放电容量为105.3 mAh•g-1, 且循环50次后容量仍有92.6%. Zhu等[35]也制备了Na3V2(PO4)3/C纤维并同时用作正负极进行测试, 在0.01~4.00 V的电压区间和100 mA•g-1下得到了106.2 mAh•g-1的全电池容量. Kajiyama等[36]制备了以Na3V2(PO4)3为核碳层为壳的核壳结构Na3V2(PO4)3/C纳米纤维, 由于碳和聚阴离子通过化学键稳定从而可以防止副反应的反生, 这种材料在1C下首次放电容量为94 mAh•g-1且循环50次其容量保持率为74%. NASICON型材料一般含有无毒且储量丰富的元素, 如果一些氧化还原电对(如Fe3+/Fe2+, Ni4+/Ni2+等)能在钠离子电池中实现可逆的氧化还原反应, 则此类材料将会发挥更大的潜力[13].
氟磷酸钠盐NaVPO4F也是典型的聚阴离子型正极材料, 具有高的理论容量(143 mAh•g-1)、高的电压平台以及结构稳定性.早在2003年, Barker等[37]发现NaVPO4F与硬碳材料制成钠离子电池的工作电压与锂离子电池一致.其首次放电容量为78 mAh•g-1, 首次库伦效率大于95%, 在30次循环后容量保持率为50%.而且他们发现Na+在NaVPO4F中的嵌入电位过低, 可逆脱嵌性能良好.焦丽芳课题组[38]利用电纺制备了NaVPO4F/C复合材料, 表现出优异的电化学性能:高的容量(1C下126.3 mAh•g-1), 极佳的倍率(50C下61.2 mAh•g-1)以及超长的循环性能(2C下循环1000次容量保持率96.5%).其性能表现来自于一维纳米结构组成的含碳网络既增加了导电性和结构稳定性, 又可阻止超小的NaVPO4F颗粒在脱嵌过程中的团聚和粉化.此外, 柔性薄膜可以直接用来装配电池, 充分发挥材料的性能并可提高电池的能量密度.
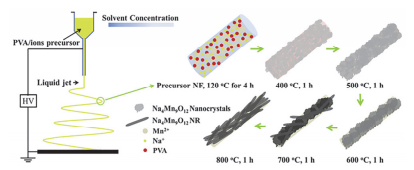
层状过渡金属氧化物具有可逆的离子脱嵌能力, 被广泛用于二次电池电极材料.在钠离子电池中, 钠基层状过渡金属氧化物是正极材料的研究热点.中国科学院物理研究所胡勇胜等[39]首次发现了NaxCux/2Ti1-x/2O2化合物能够实现Na+的可逆脱嵌, 为钠离子电池正极材料的设计提供了一些新思路.由于Na+比Li+大, 使得Na+在层状结构间的脱嵌相对困难, 因此, 改善材料结构有利于提高钠离子电池的电化学性能. Na0.44MnO2晶体结构具有两种由MnO6八面体和MnO5正方金字塔形成的大的通道, 便于Na+的传输, 但由于嵌钠动力学缓慢以及结构的破坏造成倍率性能不理想. Fu等[40]通过调控电纺过程和改变煅烧条件制备了Na0.44MnO2纤维和纳米棒(如图 5), 其中纤维在10C下可逆容量69.5 mAh•g-1, 倍率性能较好, 而具有单晶的S型隧道结构的纳米棒在循环140次容量仍有120 mAh•g-1, 循环性能也比较优异.郭再萍课题组分别制备了P2型Na2/3(Fe1/2Mn1/2)O2[41]和Li1+x(Mn1/3Ni1/3Fe1/3)O2[42]分级纳米纤维, 均具有较小的电荷转移阻抗、较高的容量和良好的循环稳定性.麦立强课题组[43]采用不同分子量的PVA进行梯度电纺合成了Na0.7Fe0.7Mn0.3O2纳米管.这种纳米管在100 mA•g-1下循环1000次时容量保持率为90%, 且在500 mA•g-1下循环5000次时容量保持率为80%, 具有极佳的循环稳定性.
图 5

焦磷酸盐易于合成, 其稳定的结构可以提供良好的Na+迁移性和热稳定性, 强P—O键的存在使得电化学性能比较稳定. Na6.24Fe4.88(P2O7)4是其中的一种, 其基于Fe2+/Fe3+还原电对的理论容量为117.4 mAh•g-1, 适合用作钠离子电池正极, 但导电性较差. Niu等[44]首次合成了基于石墨烯的Na6.24Fe4.88(P2O7)4纳米纤维(NFPO@C@rGO), 其导电性明显提高, 在40 mA•g-1下循环320次可逆容量约99 mAh•g-1, 循环性能优异, 且在1280 mA•g-1的大电流下容量约53.9 mAh•g-1, 倍率性能也远高于之前报道的NFPO材料.虽然焦磷酸盐作为钠离子电池正极材料有一定的新颖性, 但由于含有较大的P2O7离子, 导致分子量较大, 理论容量偏低.
硫酸盐基材料因有很强的电负性和高的工作电位引起了关注, 铁基硫酸盐表现出最为优异的电化学性能, 其中包括符合化学计量比的Na2Fe2(SO4)3及其非计量比的衍生物. Yu等[45]首次制备了柔性Na2+2xFe2-x(SO4)3@多孔碳纤维膜, 在40C和5C的倍率下交替循环500次后容量仍保持95%.铁基硫酸盐材料具有较高的工作电压和合适的容量, 而且价格低廉、环境适应性好, 通过构造合适的导电网络可以获得优异的电化学性能, 具有良好的潜在应用前景.
除以上材料外, 金属氟化物也是近年来研究的比较有应用前景的新型正极材料. Gocheva等[46]首次用机械化学的方法制备了NaMF3(M=Fe、Mn、Ni)并证明其有储钠容量.普鲁士蓝类化合物为过渡金属离子与CN-配位形成的配合物, 具有开放的3D结构, 有利于Na+的传输, 也被广泛用于钠离子电池正极材料. Goodenough等[47]研究了普鲁士蓝作为钠离子电池正极材料的性能, 得到约100 mAh•g-1的可逆容量且循环30次没有容量损失.武汉大学杨汉西等对苯胺-硝基苯胺共聚物[48]和聚吡咯有机物[49]作为钠离子电池正极进行了研究, 发现有机物作为正极材料具有循环稳定性好和容量高的优点.因为有机物价格低廉、容易获得, 也被认为是很有潜力的钠离子电池正极材料.但是目前为止, 并未见利用静电纺丝法制备这些材料用于钠离子电池正极的报道.
目前静电纺丝制备钠离子电池正极材料已经取得一些进展, 但仍存在储钠容量较低的问题, 限制了高容量钠离子电池的开发.因此开发稳定性好的高容量正极材料是将来研究的重要方向. 表 2总结了静电纺丝法所制备的钠离子电池正极材料的电化学性能.
4. 静电纺丝负极材料
负极材料是钠离子电池中的一个重要部分, 也是当前研究的热点.由于金属钠的化学活性极高, 直接用作钠离子电池负极时, 反复充放电过程中会造成钠在电极表面的不均匀沉积, 产生枝晶刺穿隔膜, 引发电池短路.此外, 商业化锂离子电池石墨负极材料无法与Na+形成热力学稳定的插层化合物, 从而很难表现出储钠性能.而且, 大部分在锂离子电池中表现优异的负极材料, 储钠动力学过程缓慢, 限制了其在钠离子电池中的应用.因此, 开发合适的储钠负极材料至关重要.
表 2
表 2 静电纺丝钠离子电池正极材料Table 2. Electrospun cathode materials for sodium-ion batteries下载:
导出CSV
Material Initial discharge/(mAh•g-1) Rate Cycle Capacity retention Cut-off voltage/V Reference NaTi2(PO4)3/C nanofiber 105 5C 500 93% 1.5~3.3 [31] Na3V2(PO4)3/C nanofiber 101 0.1C — — 2.7~3.8 [32] Na3V2(PO4)3/C nanofiber 106.8 0.2C 125 100% 2.5~4.0 [33] Na3V2(PO4)3/C nanorods 105.3 0.5C 50 92.6% 2.5~4.0 [34] Na3V2(PO4)3/C nanofiber 106.2 100 mA•g-1 300 34.8% 0.01~4 [35] Na3V2(PO4)3/C core-shell NWs 94 1C 50 74% 2.5~3.8 [36] NaVPO4F/C nanofiber — 2C 1000 96.5% 2.6~4.4 [38] Na0.44MnO2 NFs and NRs 120 0.42C 140 ca. 100% 1.5~4.0 [40] P2-type Na2/3(Fe1/2Mn1/2)O2 NFs ca. 195 0.1C 80 85.6% 1.5~4.2 [41] Li1+x(Mn1/3Ni1/3Fe1/3)O2 NFs ca. 88 0.1C 100 — 2~4.5 [42] Na0.7Fe0.7Mn0.3O2 nanotubes 82 500 mA•g-1 5000 70% 3.0~4.5 [43] Na6.24Fe4.88(P2O7)4@C@rGO ca. 99 40 mA•g-1 320 — 1.5~4.0 [44] Na2+2xFe2-x(SO4)3@porous CFs — 1C 300 95% — [45] 4.1 碳材料
在钠离子电池负极材料中, 碳材料是研究最多的一类材料.碳材料不仅具有较低的嵌钠平台、较高的容量和优异的循环稳定性, 而且具有资源丰富、制备简单等优点, 是最有希望推动钠离子电池产业化的负极材料[50].目前研究的碳材料主要分为石墨碳、无定形碳、多孔碳以及硬碳类.石墨是商业化锂离子电池的负极材料, 理论比容量为372 mAh•g-1.但由于Na+的半径比Li+大, 而石墨的层间距只有3.4 Å, 导致Na+在其间很难稳定存在, 且Na+的嵌入会破坏石墨的层状结构, 因此储钠性能较差. Fouletier等[51]早在1988年就对石墨进行了嵌钠性能研究, 最终只得到了30 mAh•g-1的比容量.无定形碳材料已被证明可以可逆地储存Li+, 其结构中较大的层间距和较多的缺陷也为Na+提供了存储位点, 是钠离子电池负极材料最先开展的研究方向[52].近年来利用电纺制备一维碳纳米纤维、纳米管用作钠离子电池负极材料研究的比较多.用作负极材料时, 碳纳米纤维具有多种功能, 既是活性物质, 又是导电添加剂, 同时还可作为支撑活泼金属的基底[53]. Chen等[54]首次在600 ℃的低温下制备电纺碳纤维用于钠离子电池负极, 在50和2000 mA•g-1的电流密度下循环200次容量分别为233和82 mAh•g-1, 容量保持率为97.7%.王成扬课题组[55]在800~1500 ℃的条件下制备了一系列碳纤维并研究其在钠离子电池中的性能, 发现碳化温度会影响碳层结构和储钠容量.随后他们又分别将木质素[56]、富里酸[57]和腐殖酸[58]引入聚丙烯腈(PAN)中进行电纺得到碳纳米纤维, 并在不同的温度下碳化, 均获得了较好的钠电性能.因此合理控制碳纤维的合成条件可以使碳材料具有一定的储钠性能, 但是要想进一步提高其储钠性能, 必须对碳材料进行优化.研究表明, 氮掺杂可以促进更多的电子进入碳的π共轭体系, 提高碳材料的导电性, 而且吡啶氮和吡咯氮可以制造一些缺陷位点, 为Na+提供更多的扩散通道和活性位点, 从而提高电极性能[59]. Zhu等[60]制备了氮掺杂碳纤维(N-CNFs), 在1000 mA•g-1的大电流下循环200次后可逆容量仍然可以达到150 mAh•g-1.研究表明, 多孔或空心结构可以促进电解液与活性物质的接触同时缩短Na+的传输路径, 因此也可以提高碳材料的电极性能.余彦等[29]在PAN的电纺溶液里加入F127作为造孔剂制备了柔性多孔碳纳米纤维, 在0.2C倍率下循环100次容量维持在266 mAh•g-1, 在2C倍率下循环1000次后容量仍达到约140 mAh•g-1, 容量保持率为71.5%. Qi等[61]也做了相似的工作, 其得到的多孔碳纤维在500 mA•g-1下循环1000次后可逆容量为210 mAh•g-1. Chen等[62]将电纺得到的MoO2@C核壳结构经过硝酸处理得到多孔碳纳米管, 其比表面积高达502.9 m2•g-1, 并且表面含有很多含氧官能团.作为钠离子电池负极时, 具有优异的循环稳定性(5 A•g-1下循环1200次后容量为110 mAh•g-1).楼雄文等[63]利用静电纺丝制备了大面积的柔性氮掺杂多孔纳米碳纤维膜直接作为钠离子电池负极使用, 在100 mA•g-1的电流密度下循环100次后容量仍然稳定在377 mAh•g-1, 即使在15 A•g-1的大电流密度下也能达到154 mAh•g-1的可逆容量.而且值得一提的是, 在5 A•g-1的电流密度下循环7000次可逆容量仍然稳定在210 mAh•g-1, 展现出极其优异的循环稳定性和倍率性能.为了减小电荷在纤维之间传递的阻力, Guo等[64]利用Cu(NO3)2作为交联剂制备了相互交叉连接的碳纳米纤维.相比没有交联的碳纳米纤维, 交连纤维具有更高的可逆容量, 更优的倍率性能和更长的循环寿命.
二维材料石墨烯具有较大的比表面积、优异的导电性、较好的柔性和化学稳定性, 在能源存储领域展现出独特的优势.近年来, 由于具有丰富的活性位点, 石墨烯更是在钠离子电池上表现出高的可逆容量[65].为了充分发挥石墨烯的高容量及多孔碳纤维的电化学性能, Liu等[66]将高度剥离的单层或双层石墨烯通过电纺的方法均匀分散到多孔碳纳米纤维中, 在钠离子电池中表现出高的可逆容量(100 mA•g-1电流下容量432.2 mAh•g-1)、优异的倍率性能(10 A•g-1大电流下261.1 mAh•g-1的可逆容量)以及循环稳定性(循环1000次后容量保持率91%).这是高度剥离的石墨烯和多孔碳纤维共同作用的结果, 因为它既可以提供大量的储钠活性位点, 保证与电解液的充分接触, 为离子传输和电子传导提供通道, 同时又可以防止在充放电过程中石墨烯的团聚和纤维的破碎, 因而具有优异的钠电性能, 为高性能碳基钠离子电池负极材料的制备提供了一个新思路.
4.2 钛基材料
钛基材料通过嵌入脱出机理存储钠离子, 主要包括钛基氧化物(主要为TiO2)、钛酸盐(Na2Ti6O13、Na2Ti3O7等).这类材料虽然导电性差、比容量低, 但是具有结构稳定、价格低廉、工作电压低、循环稳定性好等优点, 因此也是研究较多的一类材料.
4.2.1 二氧化钛
TiO2为零应变材料, 在锂离子电池中表现出非常好的循环稳定性, 因此近年来人们对TiO2材料的储钠性能进行了研究. Xiong等[67]首次报道了TiO2材料的储钠性能.目前, 不同晶型的TiO2被用于钠离子电池负极材料, 比如无定型态[68]、锐钛矿[69~71]、金红石[72]、TiO2(B)[16, 73]、TiO2(H)[74]等.这些材料在储钠性能上有很大的差异. Su等[75]的研究发现锐钛矿TiO2的储钠性能要优于金红石、无定型以及混晶TiO2. Yu等[76]利用静电纺丝法先得到二氧化钛电纺纤维, 再将得到的电纺纤维先在空气中煅烧得到锐钛矿和金红石混晶的二氧化钛纤维(a-TiO2/r-TiO2), 然后再在真空中煅烧得到具有氧空位的混晶二氧化钛(a-TiO2-x/r-TiO2-x).这种二氧化钛具有一维结构、多孔通道和高晶界密度.大量氧空位的引入可以提高材料的导电性和Na+的扩散速度, 两种晶形间高密度的晶界的存在也可以促进Na+的扩散.当把此种二氧化钛纤维用作钠离子电池负极时, 具有非常好的循环稳定性(10C下循环4500次容量为134 mAh•g-1)和优异的倍率性能(20C下循环4500次可逆容量为93 mAh•g-1).这种简单有效的方法也为其他金属氧化物的制备提供了借鉴.不仅是引入氧空位可以提高二氧化钛的钠电性能, 近年来的研究发现, 原子掺杂(F, S, Nb, N等)[77]或氢还原也可以提高二氧化钛的导电性进而提高电池性能. Wu等[78]在电纺中引入F127先制备出多孔二氧化钛纤维, 再通过在NH3/Ar混合气中煅烧的方法掺杂氮.氮掺杂不仅可以增加导电性, 而且可以引入Ti3+和氧空位来提高二氧化钛的界面稳定性, 减小了电子和离子传输的阻力, 因而得到了较好的倍率性能(10C下可逆容量为110 mAh•g-1)和循环稳定性(10C下循环500次仅有6%的容量损失). Shen等[79]直接将电纺纤维在N2条件下碳化得到柔性氮掺杂C/TiO2纳米纤维, 在0.1、0.2和10C下分别得到246、185和75 mAh•g-1的可逆容量, 且循环100次容量非常稳定.以上结果说明氮掺杂确实可以提高二氧化钛的钠电性能.
此外, 碳复合也是提高二氧化钛导电性的有效方法. Udomsanti等[80]利用两个针头同时电纺的方法, 将碳纤维和二氧化钛纤维接收到同一个滚筒收集器上, 得到钛碳互穿复合纤维, 虽然得到了较好的循环稳定性, 但储钠容量并不高. Xiong等[81]将电纺得到的TiO2纤维在Ar下煅烧到550 ℃得到TiO2/C复合纤维并将其用于钠离子电池负极.这种将纳米TiO2包裹在碳纳米纤维中的结构既缩短了Na+的传输路径又提高了材料的导电性, 同时提供了足量的表面反应位点, 因而有利于Na+的脱嵌反应, 在200 mA•g-1下循环1000次后容量高达237.1 mAh•g-1, 容量保持率接近100%. Ge等[82]也做了类似的工作, 并考察了碳化温度和二氧化钛晶型对储钠性能的影响.韩国Kim课题组[83]将还原氧化石墨烯包裹在电纺TiO2纤维表面, 并引入PAH使TiO2纤维表面带正电荷以增强TiO2纤维与氧化石墨烯的结合.相比于TiO2纳米纤维, 这种包裹石墨烯的TiO2纳米纤维负极材料可以将首次放电容量提高到217 mAh•g-1, 0.2C下循环200次容量保持率可高达85%, 即使在5C下库仑效率仍高达99.7%.最近, 该课题组又通过将电纺纤维在H2/Ar混合气下低温还原的方式得到具有氧缺陷的无定形二氧化钛与碳复合的纤维(TiO2-x/CNF).由于无定形二氧化钛可以为Na+的嵌入提供空间, 碳复合和Ti3+的引入可以提高材料导电性以及其一维结构的协同作用(如图 6), 这种纤维在第二次放电时容量就高达208 mAh•g-1.
图 6

4.2.2 钛酸盐
钛酸盐化学稳定性好, 安全性高且无毒, 被应用于钠离子电池负极材料研究中.然而其低的电导率和Na+扩散速率限制了其大规模应用. Zou等[85]制备了不同直径的Na2Ti3O7/C复合纤维, 其中直径约120 nm的纤维在4C下储钠容量为101 mAh•g-1并且在1C下循环100次后容量保持在99 mAh•g-1.这样优异的电化学性能得益于纳米颗粒短的离子电子传输路径以及碳材料连续的导电网络.尖晶石Li4Ti5O12热稳定性好, 且与电解液接触没有SEI膜, 使得电池可以满足在混合动力汽车上使用较高的能量和耐极限条件的需求.余彦等[86]将小于10 nm的Li4Ti5O12颗粒通过电纺包埋进碳纤维中, 在0.2C下循环100次容量稳定在162.5 mAh•g-1.
磷酸钛钠(NaTi2(PO4)3)为NASION型材料, 其特有的三维骨架结构有利于Na+的脱嵌, 具有较高的理论比容量(133 mAh•g-1)和优异的结构稳定性, 是理想的钠离子电池电极材料. NaTi2(PO4)3的电位为2.1 V vs. Na+/Na, 根据对电极的性质, 可以作为钠离子电池的正极和负极使用, 但更多的研究是将其用作钠离子电池负极[87].其作为钠离子电池负极时, 仍然面临着电导率低导致电化学性能降低的问题, 因此研究者们提出了将其与各种碳材料复合的方法, 比如多孔碳[88, 89]、石墨烯[87, 90, 91]等, 作为其电子传输网络, 使其具有比较好的倍率性能和较长的循环寿命. Liu等[92]首次利用静电纺丝的方法制备了一维NaTi2(PO4)3/C多孔纳米纤维, 在5C和10C下分别得到了104.7和93.4 mAh•g-1的可逆容量, 为提高NaTi2(PO4)3的导电性提出了一种新的方法.
钛基负极材料具有结构稳定、循环性好、安全性高等优点, 然而与作为锂离子电池负极类似, 其作为钠离子电池负极时也面临着导电率较低以及储钠容量低的问题, 因此目前基本上利用改性后的二氧化钛作为钠离子电池负极.另外由于Na+更大的离子半径, 导致TiO2材料的嵌钠动力学速度更慢, 扩散势垒更高, 因此对材料的设计提出了更高的要求.由于目前对静电纺丝法制备TiO2用于钠离子电池负极的研究比较少, 因此对其储钠机理的深入研究和电化学性能的优化也是提高TiO2材料储钠性能的一个方向.
4.3 过渡金属氧化物
过渡金属氧化物来源广泛, 价格低廉, 并且理论容量高, 是很有前景的负极材料.过渡金属氧化物通过转换反应储钠, 其反应方程式可表示为:
$ {\rm{M}}{{\rm{O}}_y} + 2y{\rm{N}}{{\rm{a}}^ + } + 2y{\rm{e }} \leftrightarrow {\rm{ M}} + y{\rm{N}}{{\rm{a}}_2}{\rm{O}} $
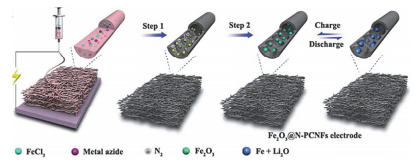
(1) 但此类材料在循环过程中通常会发生巨大的体积膨胀, 导致活性材料粉化脱落, 而且导电性差导致活性物质的利用率低、倍率性能差.近期研究表明, 过渡金属氧化物的电化学性能与其结构和颗粒大小密切相关.纳米电极材料由于粒径小比表面积大, 可以减小电荷传输距离, 提高嵌钠动力学, 因而被证明可以提高电化学性能[93].而中空、核壳等结构较大的空间可以有效缓冲脱嵌过程中的体积膨胀.另一种常用的改性方法是与导电材料复合, 在提高导电性的同时还可以缓冲充放电过程中的体积膨胀. Mao等[94]将Co3O4纳米颗粒包裹进碳纤维中, 在50 mA•g-1下循环100次得到约300 mAh•g-1的容量(Co3O4的理论容量为889 mAh•g-1).相比Co3O4纳米线, 碳复合Co3O4纳米线在500 mA•g-1下循环500次后容量保持率可达84.3%. Fu等[95]也做了类似的工作, 但其颗粒较小(约6.27 nm), 且储钠性能相对更好(0.5C下循环700次后可逆容量为400 mAh•g-1). CuO的理论容量为674 mAh•g-1, 且环境友好, 储量丰富, 价格低廉, 但仍需克服体积膨胀的问题.焦丽芳课题组[96]将约2 nm的CuO量子点通过电纺嵌入碳纳米纤维得到柔性薄膜并直接用于钠离子电池负极, 得到了优异的循环稳定性(500 mA•g-1下循环500次的容量为401 mAh•g-1)和倍率性能(100 mA•g-1下容量528 mAh•g-1和5000 mA•g-1下容量250 mAh•g-1).其优异的电化学性能主要是由于CuO量子点使得转化反应变得高度可逆, 提高了CuO的利用率.作为地球上储量第四丰富的元素, 铁的氧化物Fe2O3的理论储钠容量为1007 mAh•g-1, 是一种理想的钠电负极材料.余学斌等[97]利用叠氮化锂作为造孔剂和氮源, 得到了如图 7所示豌豆状Fe2O3纳米颗粒包裹于氮掺杂多孔碳纳米纤维的复合材料(Fe2O3@N-PCNFs).这种豌豆状结构不仅有足够的空间容纳Fe2O3纳米颗粒的体积膨胀, 同时也为电子传递和Na+的传输提供多孔的连续的快速通道.因此在200 mA•g-1下循环100次后得到了806 mAh•g-1的超高储钠容量, 并且在2000 mA•g-1的大倍率下循环1500次可逆容量仍有396 mAh•g-1, 这在所有报道的Fe2O3储钠材料中循环稳定性最好, 为其他高性能纳米储能材料的制备提供借鉴. Xu等[98]将原子级的无定型FeOx颗粒均匀分散到碳纳米纤维中, 并且发现煅烧温度、时间以及碳纤维的限制作用是形成原子级无定型FeOx纳米颗粒的原因.用于钠离子电池负极时, 在0.5 A•g-1下循环500次后容量为277 mAh•g-1, 容量保持率接近100%, 并且循环后FeOx颗粒仍能维持其大小和无定型状态, 有力地证明了电极良好的稳定性.周震等[99]在碳纤维中包裹T-Nb2O5纳米颗粒, 在1 A•g-1下循环5000次得到150 mAh•g-1的可逆储钠容量, 且在8 A•g-1的大倍率下容量仍有97 mAh•g-1. T-Nb2O5为典型的赝电容材料, 其容量主要来自于两部分:一部分由相扩散控制, 另一部分由电容过程控制.他们通过测试不同扫描速度下的循环伏安曲线, 计算了两部分分别对容量的贡献.其中在0.1 mV•s-1下, 总容量的51.4%是由电容过程贡献, 并且随着扫描速度提高, 电容过程贡献的容量比会逐渐提高到94%, 说明快速充放电过程主要发生在活性物质的表面, 这也是T-Nb2O5/CNFs材料倍率性能好的原因.
图 7

二元过渡金属氧化物具有比单金属氧化物更好的导电性和电化学活性, 但目前为止在钠离子电池负极中研究较少[100, 101].陈军等[59]利用静电纺丝将约3.3 nm的尖晶石MnFe2O4均匀分布在氮掺杂多孔碳纤维中, 直接用于钠离子电池负极, 具有良好的倍率性能(10000 mA•g-1下容量305 mAh•g-1以及100 mA•g-1下容量504 mAh•g-1)和超长的寿命(2000 mA•g-1下循环4200次后容量保持率90%).通过研究其储钠机理, 发现在首次放电后MnFe2O4转化成金属Mn和Fe, 紧接着在充电过程中转化成MnO和Fe2O3, 由于MnO/Fe的还原/氧化电位比Fe2O3/Mn的低, 接下来会再通过还原反应转化成Mn和Fe, 在充放电过程中MnO和Fe2O3之间可以相互缓冲体积膨胀, 再加上碳材料的缓冲作用, 因此循环寿命较长.阎兴斌等[102]制备了Ni掺杂MnCo2O4纳米管, 在0.1 mA•g-1下循环700次得到238.6 mAh•g-1的可逆储钠容量, 且在1 A•g-1的大电流下循环11000次后可逆容量仍有109 mAh•g-1, 容量保持率高达81%, 展示出极佳的循环稳定性.这主要是由于中空多孔的结构可以缓冲体积膨胀的应力, 防止颗粒团聚, 并且其赝电容行为也促进了电化学反应动力学.通过XRD、TEM等测试表明在首次放电后Ni掺杂MnCo2O4转化为Mn, Co和Ni, 再在后续充电过程中转化为MnO, CoO和NiO, 反应机理与MnFe2O4类似.
4.4 过渡金属硫(硒)化物
金属硫化物和硒化物通常具有层状结构, 每层原子之间通过共价键的作用稳定存在, 而层间仅依靠较弱的范德华力形成, 因此一些极性分子可以通过悬挂、吸附或嵌入方式破坏范德华力进入层间而不破坏其层状结构.跟其对应的金属和氧化物相比, 金属硫化物作为钠离子电池负极有几点优势[103]: (1)金属硫化物由于转换反应储钠机理具有较高的理论容量; (2)金属硫化物因为体积膨胀相对较小具有更好的机械稳定性且还原反应的高度可逆使得首次库仑效率更高; (3)金属硫化物的放电产物Na2S的导电性比Na2O更好; (4)金属硫化物的M—S键比金属氧化物的M—O键弱, 可以促进转换反应动力学.因此, 金属硫化物引起了越来越多的关注. Kitajou等[104]证明Na+可以嵌入FeS2的层间并在0.2 mA•g-1的电流下获得了758 mAh•g-1的容量. Qu等[105]报道了SnS2/石墨烯复合材料的初始放电容量可达到630 mAh•g-1并且循环400次后仍有500 mAh•g-1.南开大学陈军课题组[106]利用水热和超声剥离法可控制备了堆积密度不同的铜钱状VS2, 在100 mA•g-1下循环300次得到了410 mAh•g-1的可逆容量且在2000 mA•g-1下可逆容量仍有333 mAh•g-1.这表明金属硫化物在钠离子电池负极材料开发方面是很有潜力的.在金属硫化物中, 研究比较多的是层状MoS2.在锂离子电池中, 平均1 mol MoS2可以嵌入4 mol Li+, 理论容量约为670 mAh•g-1. MoS2的层间距约为0.62 nm, 因此也为较大的Na+的嵌入提供了足够的空间.之前的研究表明, MoS2也存在体积膨胀和导电性差的问题, 导致容量衰减较快.为了解决这些问题, 研究者们提出了将MoS2与比较稳定的材料复合的方法, 如碳材料和二氧化钛. Kim课题组[107]通过一步静电纺丝技术制备了MoS2纤维, 然后在其表面负载一层TiO2层以增加其循环稳定性, 并通过TEM和XRD研究了反应机理.相比块状MoS2, MoS2纤维有更大的比表面积和更多的反应位点, 在第二次循环中得到了840 mAh•g-1的放电容量, 并在5C下表现出更优异的倍率性能. Chen等[108]利用化学气相沉积法将MoS2纳米片沉积到电纺碳纤维的表面, 循环50次后储钠容量仍高达380 mAh•g-1, 并且在1 A•g-1的大电流下循环500次后可逆容量达198 mAh•g-1, 说明此复合材料的循环稳定性和倍率性能良好.余彦等[109]首次将单层MoS2纳米片引入电纺纤维中, 在0.1 A•g-1下得到了854 mAh•g-1的可逆储钠容量, 且在10 A•g-1下容量仍然有253 mAh•g-1, 其循环稳定性和倍率性能在所有MoS2的报道中最优.出色的电化学性能主要是由于单层结构可以使离子在硫化物中各个方向的传输距离最小化, 且将脱嵌和转换反应机理结合起来. Xiong等[110]也用一步电纺法将MoS2纳米片嵌入碳纳米纤维中制备了MoS2/CNFs, 在100 mA•g-1电流密度下循环600次后容量可达到283.9 mAh•g-1, 循环稳定性比较理想.
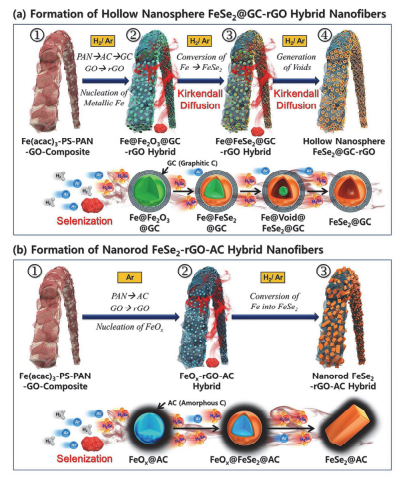
相对硫化物来说, 硒化物研究的非常少, 比如核壳结构MoSe2微球[112]和FeSe2微球[113]被相继合成出来用于钠离子电池负极, 也取得了不错的结果.韩国Kang课题组[114]首次用电纺的方法合成NiSe2并与还原氧化石墨烯以及无定型碳复合(NiSe2-rGO-C), 利用石墨的机械稳定性和导电性以及无定形碳对体积膨胀的缓冲作用, 将复合电极的性能有效的发挥出来, 并为其它金属或非金属硒化物的合成提供了一个很好的方法.他们又充分应用纳米材料的柯肯达尔效应将铁转变成空心结构, 再经过硒化过程分别制备了如图 8所示FeSe2中空纳米球@GC-rGO纳米线和FeSe2纳米棒-rGO-AC复合纤维[111]. FeSe2中空纳米球@GC-rGO纳米线在1 A•g-1下循环150次后容量高达412 mAh•g-1, 容量保持率为82%, 并且在10 A•g-1的大倍率下仍可获得352 mAh•g-1的可逆容量.这种复合材料优异的倍率性能和循环稳定性得益于FeSe2的中空结构可以有效地缓冲嵌钠过程中的体积膨胀以及rGO良好导电性等的协同作用.
图 8

4.5 金属单质及其合金
合金类材料的储钠机理为合金化反应, 其反应第一步为上述的转换反应, 第二步为合金反应, 其方程式为:
$ {\rm{M}} + z{\rm{Na}} \leftrightarrow {\rm{N}}{{\rm{a}}_z}{\rm{M}} $
(2) 这类材料通常具有较大的理论比容量和合适的嵌钠电位[115], 是理想的负极材料.然而有合金化反应的材料并不多, 目前只有Pb、Sn、Sb、Bi几种金属被证实能与Na+发生合金化反应. Pb属于对环境有污染的重金属, 因此没有被广泛研究. Sn、Sb及其氧化物和合金等得到了广泛的关注.锡基钠离子电池负极材料主要包括Sn单质、化合物(SnO2、SnS、SnS2、Sn4P3)以及合金(SnSb、SnGe、SnNi、SnCu、SnGeSb)等. Sn的理论储钠容量为847 mAh•g-1, 远高于各种碳材料[116, 117]. 2011年, Chevrier等[118]通过密度泛函理论计算发现, 金属Sn在放电过程中有四个电压平台, 分别对应NaSn5, NaSn, Na9Sn4, Na15Sn4, 体积膨胀达到420%.与储锂的合金化过程类似, Na+嵌入金属材料晶格中会引起材料体积的巨大膨胀, 在这个过程中, 产生的应力会使材料颗粒发生裂纹并最终破碎, 导致容量不断衰减.同时, Sn自身较差的导电性也限制了其倍率性能的发挥.
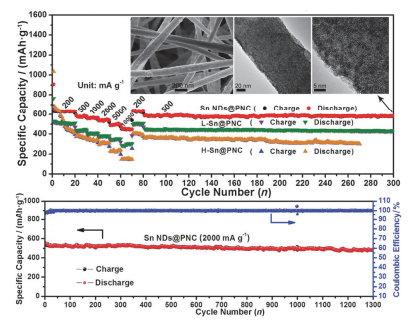
对于金属单质来说, 碳材料复合可以有效改善材料的储钠稳定性:碳材料的缓冲作用可以缓解金属材料在嵌钠过程中的体积膨胀, 提高循环稳定性; 同时碳材料的引入也可以提高电极的电导率, 改善材料的倍率性能. Dirican等[119]通过电纺PAN和聚甲基丙烯酸甲酯(PMMA)的前驱液制备出多孔碳纳米纤维, 然后在其表面电沉积一层SnO2颗粒, 最后再化学气相沉积一层碳层, 得到PCNF@SnO2@C复合纤维, 既提高了导电性, 又有效地保持了循环过程中的结构稳定, 因而得到了较好的钠电性能. Liang等[120]在电纺得到的氮掺杂碳纳米纤维表面通过水热生长了一层纳米花状SnO2.其中碳纤维既为SnO2的生长提供载体又可作为导电网络加速离子的传递, 因此在100 mA•g-1下循环100次后得到了270 mAh•g-1的储钠容量.陈军课题组[121]以PAN和SnCl2为前驱体, 电纺得到纤维后在N2保护下进行高温煅烧, 将SnCl2还原成Sn颗粒固定在碳纤维中.并且引入PMMA, 利用其在高温下挥发的特点使得到的碳纤维为多孔状态, 既增大了材料的比表面积, 又能有效地缓冲Na+脱嵌引起的体积膨胀.用作钠离子电池负极时, 在200 mA•g-1的电流下可逆容量为633 mAh•g-1, 在2 A•g-1的大电流条件下循环1300次后可逆容量仍然可以达到483 mAh•g-1(图 9), 在所报道的Sn做钠离子电池负极材料中倍率性能和循环稳定性最好. TiO2稳定性好, 因此引入TiO2也可以改善合金类材料的电化学性能. Mao等[122]先将Sn电纺引入碳纤维中, 然后用原子沉积法在外层包裹一层TiO2, 然后在空气中煅烧将碳部分分解得到电线状TiO2-Sn@CNFs的核壳结构, 在100 mA•g-1下循环400次得到413 mAh•g-1的可逆储钠容量.其中, Sn为活性物质主体, 碳既可增加导电性又能够作为一个体积膨胀的缓冲材料, TiO2外壳稳定的结构可以进一步限制体积变化, 并可防止Sn颗粒在循环过程中的团聚和极化, 因而可以提高复合材料的库伦效率.
图 9
同Sn类似, 合金材料Sb也具有较大的理论容量(Na3Sb, 660 mAh•g-1), 同时也面临体积膨胀的问题(390%).目前对Sb负极的一些研究已经取得了一定的成果, 而这些研究中Sb的循环稳定性的实现主要依赖于电解液中添加的FEC(氟代碳酸乙烯酯)对SEI膜的稳定作用, 不加FEC时Sb负极的循环性能不会超过20~50个循环[116, 123], 即使添加FEC, Sb负极的寿命也很差, 超过160个充放电循环的报道很少[124].碳复合是改善Sb负极电化学性能的有效方法. Zhu等[125]在电纺碳纤维外采用化学沉积的方法包裹一层Sb形成核壳结构, 其中Sb层的厚度为40~60 nm, Sb的质量约为复合纤维质量的34.6%.在0.2 A•g-1的电流密度下, 首次嵌钠容量为560 mAh•g-1且循环80次后仍能保持在538 mAh•g-1.当电流密度提高到1.5 A•g-1, 可逆容量仍可达到385 mAh•g-1, 展现出较好的储钠倍率性能. Zhu[124]和Wu[115]等分别将Sb嵌入碳纤维中合成了Sb/C纳米纤维, 利用碳纤维缓冲充放电过程中的体积膨胀并增加材料导电性, 均得到了较好的循环性能.前者在100 mA•g-1电流密度下首次嵌钠容量为422 mAh•g-1且经过300次的循环后容量仍保持在350 mAh•g-1; 后者更是在C/15倍率下可逆容量达到631 mAh•g-1, 并且经历400次循环后容量保持率高达90%.
由于Sn和Sb可以分别与3.75和3个Na+反应形成Na15Sn4和Na3Sb, 两者都具有较高的比容量同时都因体积膨胀较大而导致循环稳定差, 且两者存在电位差, 因此研究者们提出了制备SnSb合金的方法提高材料的储钠稳定性.根据Sn和Sb的理论储钠容量可以得到SnSb合金中, 当Sn与Sb的物质的量比为1:1时, 其理论容量为750 mAh•g-1.当两者形成合金时, 反应电位较高的Sb(0.55 V)优先与Na+反应形成Na3Sb, 此时Sn可以起到缓冲体积膨胀的作用, 之后Sn(0.2 V)与Na+反应时, 形成的Na3Sb可以缓冲体积膨胀, 因此SnSb合金可以提高储钠稳定性.但是由于Na+半径较大, 因此SnSb合金仍然无法完全避免体积膨胀的问题. Ji等[126]通过电纺PAN/PMMA/Sn(Ac)2/Sb(Ac)3辅以后续煅烧处理将SnSb合金颗粒(约30 nm)包入多孔碳纤维中, 并测试了这种CNF-SnSb纤维的钠电性能.在0.2C的倍率下循环200多次可逆容量达到345~350 mAh•g-1, 具有较高的容量保持率; 而在20C的超大倍率下容量仍能达到110 mAh•g-1, 展示了极其优异的倍率性能.同时研究了电解液中加入FEC对材料性能的影响. FEC的存在可以防止电解液的分解, 使形成的SEI膜更均匀, 从而可以降低Na+的传输阻力, 提高电池的性能. Chen等[127]用同样的方法得到了竹节状的SnSb/C纤维, 通过调节前驱体中氧化锡锑与PAN的比例得到不同的纤维并测试其在钠离子电池中的性能. Jia等[128]在电纺前驱体中加入不同量的氧化石墨烯, 得到SnSb@rGO@CNF纳米纤维, 利用氧化石墨烯的层状结构和柔性进一步提高材料导电性以及对体积膨胀的缓冲作用, 其中最优的材料在100 mA•g-1下循环200次容量保持率可达87.2%, 而相同条件下不含石墨烯的复合材料只有34.5%.
金属Bi的理论容量为385 mAh•g-1 (Na3Bi), 在水、空气中比较稳定, 易于制备, 是比较有潜力的负极材料. Jin等[129]从挂满水滴的蜘蛛网受到启发, 制备了Bi颗粒负载在碳纤维网上的结构, 此种制备方式极大地提高了活性物质Bi的负载量, 用作钠离子电池负极时, 在50 mA•g-1的电流密度下循环100次后容量为186 mAh•g-1.而Yin等[130]以BiCl3/Bi(NO3)3为Bi源和PAN进行电纺得到含Bi的碳纳米纤维, 其中Bi2O3颗粒的直径约为20 nm且在纤维中分布均匀.当用作钠离子电池负极时, 在100 mA•g-1下经过前十个循环容量迅速衰减后达到一个稳定的状态, 循环200次后容量可维持在430 mAh•g-1, 且在3200 mA•g-1的大电流下容量仍有230 mAh•g-1, 电池性能较Jin等有所提高.
4.6 单质磷及金属磷化物
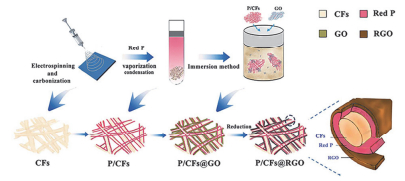
单质磷和碳一样也可以用作钠离子电池负极材料.磷是一种特殊的材料, 它本身导电性不好(红磷、白磷不导电, 黑磷可以导电), 但是储钠机理为合金反应, 理论容量高达2596 mAh•g-1 (Na3P), 工作电压低且安全(约0.4 V vs. Na+/Na)[131], 是非常有潜力的钠电负极材料, 但由于制备条件要求非常高, 研究相对较少.磷作为负极主要有两点限制因素:导电率低导致极化严重、活性物质难以有效利用以及嵌钠过程中巨大的体积膨胀(约300%).慈立杰等[132]在电纺碳纤维外雾化负载一层磷然后再包裹一层还原石墨烯得到P/CFs@RGO的多层复合结构(图 10), 既可以缓冲红磷在循环过程中的体积膨胀又可以有效防止红磷脱落.这种复合材料在50 mA•g-1下循环55次后可逆储钠容量保持在725 mAh•g-1, 并且在1000 mA•g-1的大电流密度下循环180次储钠容量仍有406.6 mAh•g-1.焦丽芳等[133]首次利用电纺方法将约97.7 nm的红磷颗粒包埋进氮掺杂碳纤维中, 其中红磷含量为51 wt%.在200 mA•g-1的电流密度下, 储钠容量可高达1308 mAh•g-1, 并且在1000次循环后容量保持率在81%.当以此种纤维为负极、Na3V2(PO4)2F3/C为正极组装全电池时, 工作电压高达约3.65 V并且能量密度可达161.8 Wh•kg-1.出色的钠电性能主要得益于红磷小颗粒均匀分布于氮掺杂碳纳米纤维中的结构便于离子电子的传输并可缓冲活性材料的体积膨胀.这种方法为高性能磷负极的制备提供了很好的借鉴.
图 10

除了碳复合的方法外, 还可以将磷与金属单质形成金属磷化物.金属单质与磷都是具有合金化反应机理的高比容量负极材料, 两者形成的金属磷化物依然具有非常高的理论容量(如SnP3为1616 mAh•g-1), 而且金属磷化物通过转换反应机理储存Na+, 通过将金属和磷重新结合成金属磷化物从而避免接下来由于合金化反应引起的金属和磷的团聚, 因此可以具有较好的循环稳定性[134], 所以金属磷化物也是非常有前景的负极材料.目前各种金属磷化物已经被用来作为钠离子电池负极材料, 比如SnP3[134], Sn4P3[135, 136], FeP[137, 138], NiP3[139], CoP[140], CuP2[141]等, 但据我们所知, 尚未见静电纺丝制备金属磷化物用于钠离子电池负极的报道.
表 3总结了用静电纺丝技术制备的钠离子电池负极材料的电化学性能.
表 3
表 3 静电纺丝钠离子电池负极材料Table 3. Electrospun anode materials for sodium-ion batteries下载:
导出CSV
Material Current density/(mA•g-1) Cycle Capacity/(mAh•g-1) Capacity retention Binder-free Reference Carbon nanofiber 200 200 169 97.7% [54] PAN-CNFs 100 100 ca. 200 — [55] Lignin based carbon nanofibers 100 200 247 90.2% √ [56] Fulvic acid based carbon nanofiber 100 100 248 91% [57] Humic acid-carbon nanofiber 100 100 249.6 92.8% [58] N-doped carbon nanofiber 1000 200 150 93.7% [60] Porous carbon nanofiber 500 1000 ca. 140 ca. 70% √ [29] Porous carbon nanofiber 500 1000 210 — √ [61] Porous carbon nanotubes 5000 1200 110 — [62] N-doped porous carbon nanofiber 5000 7000 210 99% √ [63] Cross-linked carbon nanofiber 5000 500 126 85% √ [64] Graphene-porous CFs 2000 1000 300.8 91% √ [66] Porous TiO2 hollow nanofiber 6700 4500 93 89.4% [76] N-doped mesoporous TiO2 NF 3350 500 110 94% [78] N-doped carbon/TiO2 nanofiber 100 100 313 ca. 93% √ [79] Titania and carbon fiber 125 100 134 — [80] TiO2/C nanofiber 200 1000 237.1 98.6% [81] TiO2@C nanofiber 30 100 238.1 100.3% [82] rGO A-TiO2 nanofiber 335 200 148.5 90% [83] TiO2-X/carbon nanofiber 16.6 — ca. 160 — [84] Na2Ti3O7/C nanofiber 178 100 99 — [85] Li4Ti5O12@C nanofiber 175 100 165 94% [86] Mesoporous NaTi2(PO4)3/C NF 26.6 — 126.7 — [92] Co3O4@C nanofiber 500 500 251.7 84.3% [94] Co3O4Carbon nanofiber 500 700 ca. 400 — [95] CuO@C nanofiber 500 500 401 — [96] Pead-like Fe2O3@N-doped porous CFs 2000 1500 396 — [97] Amorphous FeOx/CFs 500 500 277 100% [98] T-Nb2O5/C nanofiber 1000 5000 150 ca. 100% [99] MnFe2O4@C nanofiber 2000 4200 306 90% [59] Ni-doped MnCo2O4 porous nanotubes 1000 11000 109 81% [102] Vike-like MoS2/TiO2 nanofiber 100 30 ca. 473 64% [19] MoS2/carbon nanofiber 1000 500 198 77.95% [108] Single-layered MoS2 /CFs 1000 100 484 — [109] MoS2/C nanofiber 100 600 283.9 74.8% [110] NiSe2-rGO-C nanofiber 200 100 468 — [114] Hollow sphere FeSe2@GC-rGO 1000 150 412 82% [111] PCNF@SnO2@C nanofiber 50 100 374 82.7% [119] N-doped CFs@SnO2 100 100 270 — [120] Sn nanodots@porous CNF 2000 1300 483 90% [121] Pipe-wire TiO2-Sn@CNFs 100 400 413 84.3% [122] Sb@porous CNFs 200 80 538 96% [125] Sb nanoparticle@C fiber 100 300 350 — [124] Sb-C nanofiber 200 400 446 90% [115] Porous CNF-SnSb 100 205 345 99.4% [126] SnSb@C porous nanofiber 500 200 410 63.2% [127] SnSb@rGO@CNFs 100 200 422.1 87.2% [128] Spider-web-like Bi/CNF 50 100 186 53% √ [129] Bi2O3 NP/carbon nanofiber 100 200 271 63% √ [130] Red P/CF/graphene 50 55 725.9 75.7% √ [132] Red P@NCNFs 2000 1000 619 ca. 81% [133] 5. 静电纺丝材料用于钠离子全电池
众所周知, 无论是正极材料还是负极材料测试, 实验室组装的钠离子半电池一般是以金属钠为对电极.而在实际应用中, 金属钠做对电极会造成一定的安全隐患, 因此, 若要证明所制备的材料有实际应用的可能, 组装全电池是非常必要的一步.
一般全电池的电压平台是正负极材料电压平台之间的差值, 而全电池的电压平台会直接影响电池的能量密度.另外, 正负极材料的容量匹配也是组装全电池的关键.在实验室研究中, 研究者们很少同时合成正负极两种材料组装成全电池, 更多情况下是合成其中的一种, 然后选择另一种目前性能比较稳定的材料进行组装.即便如此, 这种全电池也为材料的实际应用向前推进了一步.近年来的研究中, 钠离子全电池也逐渐成为钠离子电极材料研究的一种标配. Zhu等[35]通过静电纺丝法制备了Na3V2(PO4)3/C复合纤维, 用作钠离子电池负极时在0.2C下得到了189.9 mAh•g-1的容量.由于Na3V2(PO4)3具有1.6 V和3.4 V两个电压平台, 因此Na3V2(PO4)3自身既可以作为正极又可以作为负极使用.将此Na3V2(PO4)3/C复合纤维同时作为正负极组装全电池时, 在0.01~4.00 V的电压范围内以及100 mA•g-1的电流密度下具有106.2 mAh•g-1的可逆容量.南开大学陈军课题组在钠离子电池电极材料方面做了许多工作, 几乎每个出色的电极材料都组装了全电池. 2015年, 他们课题组以包含Sn颗粒的电纺多孔碳纤维为负极, NaVPO4F/C为正极制备钠离子全电池[121], 并按照工业标准, 将正负极的活性物质比例控制在5.5:1, 然后在0.5~3.2 V的电压窗口下测试.在500 mA•g-1的电流密度下, 全电池的首次容量约为540 mAh•g-1, 首次库伦效率约达到99%, 并且在100次循环后, 容量仍有约460 mAh•g-1, 其容量保持率高达85.2%.随后, 他们又将MnFe2O4纳米颗粒均匀分布到多孔的氮掺杂碳纤维中作为负极, 并以Na3V2(PO4)2F3/C为正极制备软包钠离子全电池[59], 在500 mA•g-1下循环100次后容量稳定在约400 mAh•g-1, 并且全电池的能量密度达到77.8 Wh•kg-1, 为所制备的材料具有优异的储钠性能提供了更加有力的证明.焦丽芳课题组[133]也将电纺制备的柔性P@C复合纤维和NaVPO4F/C组装成软包钠离子全电池, 在1000 mA•g-1下其平均输出电压高达约3.65 V, 且得到了161.8 Wh•kg-1的能量密度, 在所报道的钠离子电池中工作电压和能量密度最高, 说明此材料具有很大的实际应用潜力.此外, Yin等[130]以静电纺丝得到的柔性Bi2O3纳米颗粒复合碳纤维为负极, 与Na3V2(PO4)2组装成钠离子全电池, 充满电的全电池在反复弯折后仍然可以点亮高色温红色LED灯, 且在400 mA•g-1下循环50次后容量约252 mAh•g-1, 为便携式设备和可穿戴产品开发提供了潜在可能.
虽然组装全电池可以说明材料在实际应用中的潜力, 但是静电纺丝技术制备的材料用于钠离子全电池的研究并不多, 在以后的工作中, 研究者们需要重视全电池的组装与测试, 以推进钠离子电池产业化的进程.
6. 结论与展望
综上所述, 静电纺丝作为一种简单高效的制备一维纳米纤维的技术, 应用在钠离子电池材料上具有许多优势.首先, 通过调整电纺以及后续煅烧过程的参数可以得到各种形貌的纤维; 其次, 由于电纺得到的纤维多为一维纳米结构, 通常具有比较好的机械强度和高的比表面积, 可以缩短扩散路径, 加快钠离子的嵌入过程, 从而可以提高电极性能; 再次, 通过电纺可以非常容易地获得大面积的柔性纤维薄膜, 不仅可以简化电池制备的工序, 提高电池的能量密度, 而且便于在可弯曲折叠的设备上使用.目前静电纺丝制备正极材料相对比较成熟, 但隔膜方面的研究非常缺乏, 相关工作亟需开展.在负极材料方面, 制备既容量高又稳定性好的材料一直是研究的热点和难点, 为此研究者们投入了大量的工作, 也取得了一些成果.未来可以通过静电纺丝制备一些新材料, 比如有机材料、二维碳化物、MOF等用于钠离子电池负极.
虽然静电纺丝在制备钠离子电池电极材料上已经取得了很大的进展, 但是仍然存在很大的发展空间.首先当前的研究主要集中在材料合成方面, 缺乏对储钠机理的深入研究, 而且几乎没有静电纺丝法制备金属磷化物用于钠离子电池负极的报道, 而金属磷化物具有高比容量和稳定性的优势, 因此此方向还有很大的研究空间.其次, 对于全电池的研究还很少, 需要研究者们在以后的研究中重视全电池性能的测试, 以促进钠离子电池的产业化进程.再者, 目前这些研究大都处在实验室阶段, 产率较低, 且电纺前驱体溶液通常包含有毒或腐蚀性有机溶剂, 因此如何将这些结构实现产业化且环境友好的制备仍然需要考虑.静电纺丝可以制备柔性纤维作为钠离子电池正极和负极材料, 因此也可以用来制备离子电导率高、电解质浸润性好的多孔性隔膜, 以便用来开发全柔性电池.我们相信, 随着制备经验的不断积累以及电纺产业的发展, 静电纺丝技术将会对未来钠离子电池的发展起到积极的促进作用.
-
-
[1]
Pan, H.; Hu, Y.; Chen, L. Energ. Environ. Sci. 2013, 6, 2338. doi: 10.1039/c3ee40847g
-
[2]
Hwang, J.; Myung, S.; Sun, Y. Chem. Soc. Rev. 2017, 46, 3529. doi: 10.1039/C6CS00776G
-
[3]
Kim, H.; Kim, H.; Ding, Z.; Lee, M.; Lim, K.; Yoon, G.; Kang, K. Adv. Energy Mater. 2016, 6, 1600943. doi: 10.1002/aenm.201600943
-
[4]
Samin, N.; Rusdi, R.; Kamarudin, N.; Kamarulzaman, N. Adv. Mater. Res. 2012, 545, 185. doi: 10.4028/www.scientific.net/AMR.545
-
[5]
Tanabe, D.; Shimono, T.; Kobayashi, W.; Moritomo, Y. Phys. Status Solidi-R. 2014, 8, 287. doi: 10.1002/pssr.v8.3
-
[6]
Park, K.; Yu, B.; Goodenough, J. Chem. Mater. 2015, 27, 6682. doi: 10.1021/acs.chemmater.5b02684
-
[7]
Billaud, J.; Clement, R.; Armstrong, A.; Canales-Vazquez, J.; Rozier, P.; Grey, C.; Bruce, P. J. Am. Chem. Soc. 2014, 136, 17243. doi: 10.1021/ja509704t
-
[8]
Kim, H.; Shakoor, R.; Park, C.; Lim, S.; Kim, J.; Jo, Y.; Cho, W.; Miyasaka, K.; Kahraman, R.; Jung, Y.; Choi, J. Adv. Funct. Mater. 2013, 23, 1147. doi: 10.1002/adfm.v23.9
-
[9]
Zheng, Q.; Liu, W.; Li, X.; Zhang, H.; Feng, K.; Zhang, H. J. Mater. Chem. A 2016, 4, 19170. doi: 10.1039/C6TA07109K
-
[10]
Zhang, Q.; Wang, W.; Wang, Y.; Feng, P.; Wang, K.; Cheng, S.; Jiang, K. Nano Energy 2016, 20, 11. doi: 10.1016/j.nanoen.2015.12.005
-
[11]
Zhao, J.; Gao, Y.; Liu, Q.; Meng, X.; Chen, N.; Wang, C.; Du, F.; Chen, G. Chemistry 2017.
-
[12]
Kubota, K.; Komaba, S. J. Electrochem. Soc. 2015, 162, A2538. doi: 10.1149/2.0151514jes
-
[13]
李慧, 吴川, 吴锋, 白莹, 化学学报, 2014, 72, 21 http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract343662.shtmlLi, H.; Wu, C.; Wu, F.; Bai, Y. Acta Chim. Sinica, 2014, 72, 21(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract343662.shtml
-
[14]
Zhang, Q.; Uchaker, E.; Candelaria, S.; Cao, G. Chem. Soc. Rev. 2013, 42, 3127. doi: 10.1039/c3cs00009e
-
[15]
Liu, D.; Cao, G. Energ. Environ. Sci. 2010, 3, 1218. doi: 10.1039/b922656g
-
[16]
Lee, J.; Lee, J.; Chung, K.; Jung, H.; Kim, H.; Mun, J.; Choi, W. Electrochim. Acta 2016, 200, 21. doi: 10.1016/j.electacta.2016.03.110
-
[17]
Cao, Y.; Xiao, L.; Sushko, M.; Wang, W.; Schwenzer, B.; Xiao, J.; Nie, Z.; Saraf, L.; Yang, Z.; Liu, J. Nano Lett. 2012, 12, 3783. doi: 10.1021/nl3016957
-
[18]
Luo, W.; Schardt, J.; Bommier, C.; Wang, B.; Razink, J.; Si-monsen, J.; Ji, X. J. Mater. Chem. A 2013, 1, 10662. doi: 10.1039/c3ta12389h
-
[19]
Ryu, W.; Jung, J.; Park, K.; Kim, S.; Kim, I. Nanoscale 2014, 6, 10975. doi: 10.1039/C4NR02044H
-
[20]
Liao, S.; Sun, Y.; Wang, J.; Cui, H.; Wang, C. Electrochim. Acta 2016, 211, 11. doi: 10.1016/j.electacta.2016.06.018
-
[21]
Fu, F.; Li, J.; Yao, Y.; Qin, X.; Dou, Y.; Wang, H.; Tsui, J.; Chan, K.; Shao, M. ACS Appl. Mater. Inter. 2017, 9, 16194. doi: 10.1021/acsami.7b02175
-
[22]
Zhang, Q.; Guo, Y.; Guo, K.; Zhai, T.; Li, H. Chem. Commun. 2016, 52, 6229. doi: 10.1039/C6CC01057A
-
[23]
Liu, Y.; Zhang, N.; Kang, H.; Shang, M.; Jiao, L.; Chen, J. Chemistry 2015, 21, 11878. doi: 10.1002/chem.v21.33
-
[24]
Wang, J.; Yang, G.; Wang, L.; Yan, W. J. Mater. Chem. A 2016, 4, 8620. doi: 10.1039/C6TA02655A
-
[25]
Mai, L.; Xu, L.; Han, C.; Xu, X.; Luo, Y.; Zhao, S.; Zhao, Y. Nano Lett. 2010, 10, 4750. doi: 10.1021/nl103343w
-
[26]
Ren, Y.; Yang, B.; Wei, H.; Ding, J. Solid State Ionics 2016, 292, 27. doi: 10.1016/j.ssi.2016.05.002
-
[27]
Greiner, A.; Wendorff, J. Angew. Chem. Int. Ed. 2007, 46, 5670.
-
[28]
Jung, J.; Lee, C.; Yu, S.; Kim, I. J. Mater. Chem. A 2016, 4, 703. doi: 10.1039/C5TA06844D
-
[29]
Li, W.; Zeng, L.; Yang, Z.; Gu, L.; Wang, J.; Liu, X.; Cheng, J.; Yu, Y. Nanoscale 2014, 6, 693. doi: 10.1039/C3NR05022J
-
[30]
向兴德, 卢艳莹, 陈军, 化学学报, 2017, 75, 154. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345659.shtmlXiang, X.; Lu, Y.; Chen, J. Acta Chim. Sinica 2017, 75, 154(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345659.shtml
-
[31]
Li, M.; Liu, L.; Wang, P.; Li, J.; Leng, Q.; Cao, G. Electrochim. Acta, 2017, 252, 523. doi: 10.1016/j.electacta.2017.09.020
-
[32]
Liu, J.; Tang, K.; Song, K.; Aken, P.; Yu, Y.; Maier, J. Nanoscale, 2014, 6, 5081. doi: 10.1039/c3nr05329f
-
[33]
Li, H.; Bai, Y.; Wu, F.; Li, Y.; Wu, C. J. Power Sources, 2015, 273, 784. doi: 10.1016/j.jpowsour.2014.09.153
-
[34]
Li, H.; Bai, Y.; Wu, F.; Ni, Q.; Wu, C. Solid State Ionics 2015, 278, 281. doi: 10.1016/j.ssi.2015.06.026
-
[35]
Zhu, Q.; Nan, B.; Shi, Y.; Zhu, Y.; Wu, S.; He, L.; Deng, Y.; Wang, L.; Chen, Q.; Lu, Z. J. Solid State Electrochem. 2017, 21, 2985. doi: 10.1007/s10008-017-3627-y
-
[36]
Kajiyama, S.; Kikkawa, J.; Hoshino, J.; Okubo, M.; Hosono, E. Chemistry, 2014, 20, 12636. doi: 10.1002/chem.v20.39
-
[37]
Barker, J.; Saidi, M.; Swoyer, J. Electrochem. Solid-State Lett. 2003, 6, A1. doi: 10.1149/1.1523691
-
[38]
Jin, T.; Liu, Y.; Li, Y.; Cao, K.; Wang, X.; Jiao, L. Adv. Energy Mater. 2017, 7, 1700087. doi: 10.1002/aenm.201700087
-
[39]
刘丽露, 戚兴国, 胡勇胜, 陈立泉, 黄学杰, 化学学报, 2017, 75, 218. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345805.shtmlLiu, L.; Qi, X.; Hu, Y.; Chen, L.; Huang, X. Acta Chim. Sinica, 2017, 75, 218(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345805.shtml
-
[40]
Fu, B.; Zhou, X.; Wang, Y. J. Power Sources 2016, 310, 102. doi: 10.1016/j.jpowsour.2016.01.101
-
[41]
Kalluri, S.; Seng, K.; Pang, W.; Guo, Z.; Chen, Z.; Liu, H.; Dou, S. ACS Appl. Mater. Inter. 2014, 6, 8953. doi: 10.1021/am502343s
-
[42]
Kalluri, S.; Pang, W.; Seng, K.; Chen, Z.; Guo, Z.; Liu, H.; Dou, S. J. Mater. Chem. A 2015, 3, 250. doi: 10.1039/C4TA04271A
-
[43]
Niu, C.; Meng, J.; Wang, X.; Han, C.; Yan, M.; Zhao, K.; Xu, X.; Ren, W.; Zhao, Y.; Xu, L.; Zhang, Q.; Zhao, D.; Mai, L. Nat. Commun. 2015, 6, 7402. doi: 10.1038/ncomms8402
-
[44]
Niu, Y.; Xu, M.; Dai, C.; Shen, B.; Li, C. Phys. Chem. Chem. Phys. 2017, 19, 17270. doi: 10.1039/C7CP02483E
-
[45]
Yu, T.; Lin, B.; Li, Q.; Wang, X.; Qu, W.; Zhang, S.; Deng, C. Phys. Chem. Chem. Phys. 2016, 18, 26933. doi: 10.1039/C6CP04958C
-
[46]
Gocheva, I.; Nishijima, M.; Doi, T.; Okada, S.; Yamaki, J.; Nishida, T. J. Power Sources 2009, 187, 247. doi: 10.1016/j.jpowsour.2008.10.110
-
[47]
Lu, Y.; Wang, L.; Cheng, J.; Goodenough, J. Chem. Commun. 2012, 48, 6544. doi: 10.1039/c2cc31777j
-
[48]
Zhao, R.; Zhu, L.; Cao, Y.; Ai, X.; Yang, H. Electrochem. Commun. 2012, 21, 36. doi: 10.1016/j.elecom.2012.05.015
-
[49]
Zhou, M.; Xiong, Y.; Cao, Y.; Ai, X.; Yang, H. J. Polym. Sci. Pol. Phys. 2013, 51, 114. doi: 10.1002/polb.23184
-
[50]
张思伟, 张俊, 吴思达, 吕伟, 康飞宇, 杨全红, 化学学报, 2017, 75, 163. doi: 10.11862/CJIC.2017.023Zhang, S.; Zhang, J.; Wu, S.; Lv, W.; Kang, F.; Yang, Q. Acta Chim. Sinica 2017, 75, 163(in Chinese). doi: 10.11862/CJIC.2017.023
-
[51]
Ge, P.; Fouletier, M. Solid State Ionics 1988, 28, 1172. http://www.sciencedirect.com/science/article/pii/0167273888903517
-
[52]
Doeff, M.; Ma, Y.; Visco, S.; Jonghe, L. C. D. J. Electrochem. Soc. 1993, 140, L169. doi: 10.1149/1.2221153
-
[53]
Zhang, B.; Kang, F.; Tarascon, J.; Kim, J. Prog. Mater Sci. 2016, 76, 319. doi: 10.1016/j.pmatsci.2015.08.002
-
[54]
Chen, T.; Liu, Y.; Pan, L.; Lu, T.; Yao, Y.; Sun, Z.; Chua, D.; Chen, Q. J. Mater. Chem. A 2014, 2, 4117. doi: 10.1039/c3ta14806h
-
[55]
Jin, J.; Shi, Z.; Wang, C. Electrochim. Acta 2014, 141, 302. doi: 10.1016/j.electacta.2014.07.079
-
[56]
Jin, J.; Yu, B.; Shi, Z.; Wang, C.; Chong, C. J. Power Sources 2014, 272, 800. doi: 10.1016/j.jpowsour.2014.08.119
-
[57]
Zhao, P.; Zhang, J.; Li, Q.; Wang, C. J. Power Sources 2016, 334, 170. doi: 10.1016/j.jpowsour.2016.10.029
-
[58]
Zhao, P.; Yu, B.; Sun, S.; Guo, Y.; Chang, Z.; Li, Q.; Wang, C. Electrochim. Acta 2017, 232, 348. doi: 10.1016/j.electacta.2017.02.159
-
[59]
Liu, Y.; Zhang, N.; Yu, C.; Jiao, L.; Chen, J. Nano Lett. 2016, 16, 3321. doi: 10.1021/acs.nanolett.6b00942
-
[60]
Zhu, J.; Chen, C.; Lu, Y.; Ge, Y.; Jiang, H.; Fu, K.; Zhang, X. Carbon 2015, 94, 189. doi: 10.1016/j.carbon.2015.06.076
-
[61]
Qi, Y.; Fan, W.; Nan, G. Mater. Lett. 2017, 189, 206. doi: 10.1016/j.matlet.2016.11.085
-
[62]
Chen, Z.; Wang, T.; Zhang, M.; Cao, G. Small 2017, 13, 1604045. doi: 10.1002/smll.201604045
-
[63]
Wang, S.; Xia, L.; Yu, L.; Zhang, L.; Wang, H.; Lou, X. Adv. Energy Mater. 2016, 6, 1502217. doi: 10.1002/aenm.201502217
-
[64]
Guo, X.; Zhang, X.; Song, H.; Zhou, J. J. Mater. Chem. A 2017, 5, 21343. doi: 10.1039/C7TA05621D
-
[65]
Xu, J.; Wang, M.; Wickramaratne, N.; Jaroniec, M.; Dou, S.; Dai, L. Adv. Mater. 2015, 27, 2042. doi: 10.1002/adma.v27.12
-
[66]
Liu, Y.; Fan, L.; Jiao, L. J. Mater. Chem. A 2017, 5, 1698. doi: 10.1039/C6TA09961K
-
[67]
Xiong, H.; Slater, M.; Balasubramanian, M.; Johnson, C.; Rajh, T. J. Phys. Chem. Lett. 2011, 2, 2560. doi: 10.1021/jz2012066
-
[68]
Bi, Z.; Paranthaman, M.; Menchhofer, P.; Dehoff, R.; Bridges, C.; Chi, M.; Guo, B.; Sun, X.; Dai, S. J. Power Sources 2013, 222, 461. doi: 10.1016/j.jpowsour.2012.09.019
-
[69]
Yang, X.; Wang, C.; Yang, Y.; Zhang, Y.; Jia, X.; Chen, J.; Ji, X. J. Mater. Chem. A 2015, 3, 8800. doi: 10.1039/C5TA00614G
-
[70]
Wu, L.; Buchholz, D.; Bresser, D.; Chagas, L.; Passerini, S. J. Power Sources 2014, 251, 379. doi: 10.1016/j.jpowsour.2013.11.083
-
[71]
Shi, X.; Zhang, Z.; Du, K.; Lai, Y.; Fang, J.; Li, J. J. Power Sources 2016, 330, 1. doi: 10.1016/j.jpowsour.2016.08.132
-
[72]
Zhang, Y.; Pu, X.; Yang, Y.; Zhu, Y.; Hou, H.; Jing, M.; Yang, X.; Chen, J.; Ji, X. Phys. Chem. Chem. Phys. 2015, 17, 15764. doi: 10.1039/C5CP01227A
-
[73]
Huang, J.; Yuan, D.; Zhang, H.; Cao, Y.; Li, G.; Yang, H.; Gao, X. RSC Adv. 2013, 3, 12593. doi: 10.1039/c3ra42413h
-
[74]
Pérez-Flores, J.; Baehtz, C.; Kuhn, A.; García-Alvarado, F. J. Mater. Chem. A 2014, 2, 1825. doi: 10.1039/C3TA13394J
-
[75]
Su, D.; Dou, S.; Wang, G. Chem. Mater. 2015, 27, 6022. doi: 10.1021/acs.chemmater.5b02348
-
[76]
Wu, Y.; Jiang, Y.; Shi, J.; Gu, L.; Yu, Y. Small 2017, 13, 1700129. doi: 10.1002/smll.v13.22
-
[77]
Hwang, J.; Myung, S.; Lee, J.; Abouimrane, A.; Belharouak, I.; Sun, Y. Nano Energy 2015, 16, 218. doi: 10.1016/j.nanoen.2015.06.017
-
[78]
Wu, Y.; Liu, X.; Yang, Z.; Gu, L.; Yu, Y. Small 2016, 12, 3522. doi: 10.1002/smll.201600606
-
[79]
Shen, J.; Hu, W.; Li, Y.; Li, L.; Lv, X.; Zhang, L. J. Alloys Compd. 2017, 701, 372. doi: 10.1016/j.jallcom.2017.01.100
-
[80]
Udomsanti, P.; Vongsetskul, T.; Limthongkul, P.; Tangboriboonrat, P.; Subannajui, K.; Tammawat, P. Electrochim. Acta 2017, 238, 349. doi: 10.1016/j.electacta.2017.03.156
-
[81]
Xiong, Y.; Qian, J.; Cao, Y.; Ai, X.; Yang, H. ACS Appl. Mater. Inter. 2016, 8, 16684. doi: 10.1021/acsami.6b03757
-
[82]
Ge, Y.; Zhu, J.; Lu, Y.; Chen, C.; Qiu, Y.; Zhang, X. Electrochim. Acta 2015, 176, 989. doi: 10.1016/j.electacta.2015.07.105
-
[83]
Yeo, Y.; Jung, J.; Park, K.; Kim, I. Sci. Rep. 2015, 5, 13862. doi: 10.1038/srep13862
-
[84]
Lee, N.; Jung, J.; Lee, J.; Jang, H.; Kim, I.; Ryu, W. Electrochim. Acta 2018, 263, 417. doi: 10.1016/j.electacta.2018.01.085
-
[85]
Zou, W.; Fan, C.; Li, J. Chin. J. Chem. 2017, 35, 79. doi: 10.1002/cjoc.v35.1
-
[86]
Liu, J.; Tang, K.; Song, K.; Aken, P.; Yu, Y.; Maier, J. Phys. Chem. Chem. Phys. 2013, 15, 20813. doi: 10.1039/c3cp53882f
-
[87]
Wu, C.; Kopold, P.; Ding, Y.; Aken, P.; Maier, J.; Yu, Y. ACS Nano 2015, 9, 6610. doi: 10.1021/acsnano.5b02787
-
[88]
Wang, D.; Liu, Q.; Chen, C.; Li, M.; Meng, X.; Bie, X.; Wei, Y.; Huang, Y.; Du, F.; Wang, C.; Chen, G. ACS Appl. Mater. Inter. 2016, 8, 2238. doi: 10.1021/acsami.5b11003
-
[89]
Hu, Q.; Yu, M.; Liao, J.; Wen, Z.; Chen, C. J. Mater. Chem. A 2018, 6, 2365. doi: 10.1039/C7TA10207K
-
[90]
Guo, D.; Qin, J.; Zhang, C.; Cao, M. Cryst. Growth Des. 2018, 18, 3291. doi: 10.1021/acs.cgd.7b01549
-
[91]
Fang, Y.; Xiao, L.; Qian, J.; Cao, Y.; Ai, X.; Huang, Y.; Yang, H. Adv. Energy Mater. 2016, 6, 1502197. doi: 10.1002/aenm.201502197
-
[92]
Liu, H.; Liu, Y. Ceram. Int. 2018, 44, 5813. doi: 10.1016/j.ceramint.2017.12.147
-
[93]
Li, W.; Zeng, L.; Wu, Y.; Yu, Y. Sci. China Mater. 2016, 59, 287. doi: 10.1007/s40843-016-5039-6
-
[94]
Mao, Z.; Zhou, M.; Wang, K.; Wang, W.; Tao, H.; Jiang, K. RSC Adv. 2017, 7, 23122. doi: 10.1039/C7RA02965A
-
[95]
Fu, B.; Zhou, X.; Wang, Y. Mater. Lett. 2016, 170, 21. doi: 10.1016/j.matlet.2016.01.132
-
[96]
Wang, X.; Liu, Y.; Wang, Y.; Jiao, L. Small 2016, 12, 4865. doi: 10.1002/smll.v12.35
-
[97]
Xia, G.; Gao, Q.; Sun, D.; Yu, X. Small 2017, 13. http://europepmc.org/abstract/MED/28722318
-
[98]
Xu, Z.; Yao, S.; Cui, J.; Zhou, L.; Kim, J. Energy Storage Mater. 2017, 8, 10. doi: 10.1016/j.ensm.2017.03.010
-
[99]
Yang, L.; Zhu, Y.; Sheng, J.; Li, F.; Tang, B.; Zhang, Y.; Zhou, Z. Small 2017, 1702588.
-
[100]
Lee, J.; Shin, H.; Lee, C.; Jung, K. Nanoscale Res. Lett. 2016, 11, 45. doi: 10.1186/s11671-016-1271-6
-
[101]
Guo, Y.; Zhu, Y.; Yuan, C.; Wang, C. Mater. Lett. 2017, 199, 101. doi: 10.1016/j.matlet.2017.04.069
-
[102]
Wu, L.; Lang, J.; Zhang, P.; Zhang, X.; Guo, R.; Yan, X. J. Mater. Chem. A 2016, 4, 18392. doi: 10.1039/C6TA08364A
-
[103]
Xiao, Y.; Lee, S.; Sun, Y. Adv. Energy Mater. 2017, 7, 1601329. doi: 10.1002/aenm.201601329
-
[104]
Kitajou, A.; Yamaguchi, J.; Hara, S.; Okada, S. J. Power Sources 2014, 247, 391. doi: 10.1016/j.jpowsour.2013.08.123
-
[105]
Qu, B.; Ma, C.; Ji, G.; Xu, C.; Xu, J.; Meng, Y. S.; Wang, T.; Lee, J. Y. Adv. Mater. 2014, 26, 3854. doi: 10.1002/adma.201306314
-
[106]
李攀, 刘建, 孙惟袆, 陶占良, 陈军, 化学学报, 2018, 76, 286. doi: 10.3866/PKU.WHXB201708172Li, P.; Liu, J.; Sun, W.; Tao, Z.; Chen, J. Acta Chim. Sinica 2018, 76, 286(in Chinese). doi: 10.3866/PKU.WHXB201708172
-
[107]
Ryu, W.; Jung, J.; Park, K.; Kim, S.; Kim, I. Nanoscale 2014, 6, 10975. doi: 10.1039/C4NR02044H
-
[108]
Chen, C.; Li, G.; Lu, Y.; Zhu, J.; Jiang, M.; Hu, Y.; Cao, L.; Zhang, X. Electrochim. Acta 2016, 222, 1751. doi: 10.1016/j.electacta.2016.11.170
-
[109]
Zhu, C.; Mu, X.; Aken, P.; Yu, Y.; Maier, J. Angew. Chem. Int. Ed. 2014, 53, 2152. doi: 10.1002/anie.201308354
-
[110]
Xiong, X.; Luo, W.; Hu, X.; Chen, C.; Qie, L.; Hou, D.; Huang, Y. Sci. Rep. 2015, 5, 9254. doi: 10.1038/srep09254
-
[111]
Cho, J.; Lee, J.; Kang, Y. Sci. Rep. 2016, 6, 23699. doi: 10.1038/srep23699
-
[112]
Ko, Y.; Choi, S.; Park, S.; Kang, Y. Nanoscale 2014, 6, 10511. doi: 10.1039/C4NR02538E
-
[113]
Zhang, K.; Hu, Z.; Liu, X.; Tao, Z.; Chen, J. Adv. Mater. 2015, 27, 3305. doi: 10.1002/adma.v27.21
-
[114]
Cho, J.; Lee, S.; Kang, Y. Sci. Rep. 2016, 6, 23338. doi: 10.1038/srep23338
-
[115]
Wu, L.; Hu, X.; Qian, J.; Pei, F.; Wu, F.; Mao, R.; Ai, X.; Yang, H.; Cao, Y. Energ. Environ. Sci. 2014, 7, 323. doi: 10.1039/C3EE42944J
-
[116]
Qian, J.; Chen, Y.; Wu, L.; Cao, Y.; Ai, X.; Yang, H. Chem. Commun. 2012, 48, 7070. doi: 10.1039/c2cc32730a
-
[117]
Komaba, S.; Matsuura, Y.; Ishikawa, T.; Yabuuchi, N.; Murata, W.; Kuze, S. Electrochem. Commun. 2012, 21, 65. doi: 10.1016/j.elecom.2012.05.017
-
[118]
Chevrier, V.; Ceder, G. J. Electrochem. Soc. 2011, 158, A1011. doi: 10.1149/1.3607983
-
[119]
Dirican, M.; Lu, Y.; Ge, Y.; Yildiz, O.; Zhang, X. ACS Appl. Mater. Inter. 2015, 7, 18387. doi: 10.1021/acsami.5b04338
-
[120]
Liang, J.; Yuan, C.; Li, H.; Fan, K.; Wei, Z.; Sun, H.; Ma, J. Nano-Micro Lett. 2017, 10.
-
[121]
Liu, Y.; Zhang, N.; Jiao, L.; Chen, J. Adv. Mater. 2015, 27, 6702. doi: 10.1002/adma.201503015
-
[122]
Mao, M.; Yan, F.; Cui, C.; Ma, J.; Zhang, M.; Wang, T.; Wang, C. Nano Lett. 2017, 17, 3830. doi: 10.1021/acs.nanolett.7b01152
-
[123]
Darwiche, A.; Marino, C.; Sougrati, M.; Fraisse, B.; Stievano, L.; Monconduit, L. J. Am. Chem. Soc. 2012, 134, 20805. doi: 10.1021/ja310347x
-
[124]
Zhu, Y.; Han, X.; Xu, Y.; Liu, Y.; Zheng, S.; Xu, K.; Hu, L.; Wang, C. ACS Nano 2013, 7, 6378. doi: 10.1021/nn4025674
-
[125]
Zhu, M.; Kong, X.; Yang, H.; Zhu, T.; Liang, S.; Pan, A. Appl. Surf. Sci. 2018, 428, 448. doi: 10.1016/j.apsusc.2017.09.154
-
[126]
Ji, L.; Gu, M.; Shao, Y.; Li, X.; Engelhard, M.; Arey, B.; Wang, W.; Nie, Z.; Xiao, J.; Wang, C.; Zhang, J.; Liu, J. Adv. Mater. 2014, 26, 2901. doi: 10.1002/adma.v26.18
-
[127]
Chen, C.; Fu, K.; Lu, Y.; Zhu, J.; Xue, L.; Hu, Y.; Zhang, X. RSC Adv. 2015, 5, 30793. doi: 10.1039/C5RA01729G
-
[128]
Jia, H.; Dirican, M.; Chen, C.; Zhu, J.; Zhu, P.; Yan, C.; Li, Y.; Dong, X.; Guo, J.; Zhang, X. ACS Appl. Mater. Inter. 2018.
-
[129]
Jin, Y.; Yuan, H.; Lan, J.; Yu, Y.; Lin, Y.; Yang, X. Nanoscale 2017, 9, 13298. doi: 10.1039/C7NR04912A
-
[130]
Yin, H.; Cao, M.; Yu, X.; Zhao, H.; Shen, Y.; Li, C.; Zhu, M. Mater. Chem. Front. 2017, 1, 1615. doi: 10.1039/C7QM00128B
-
[131]
Zhu, Y.; Wen, Y.; Fan, X.; Gao, T.; Han, F.; Luo, C.; Liou, S.; Wang, C. ACS Nano 2015, 9, 3254. doi: 10.1021/acsnano.5b00376
-
[132]
Ma, X.; Chen, L.; Ren, X.; Hou, G.; Chen, L.; Zhang, L.; Liu, B.; Ai, Q.; Zhang, L.; Si, P.; Lou, J.; Feng, J.; Ci, L. J. Mater. Chem. A 2018, 6, 1574. doi: 10.1039/C7TA07762A
-
[133]
Liu, Y.; Zhang, N.; Liu, X.; Chen, C.; Fan, L.; Jiao, L. Energy Storage Mater. 2017, 9, 170. doi: 10.1016/j.ensm.2017.07.012
-
[134]
Fan, X.; Mao, J.; Zhu, Y.; Luo, C.; Suo, L.; Gao, T.; Han, F.; Liou, S.; Wang, C. Adv. Energy Mater. 2015, 5, 1500174. doi: 10.1002/aenm.201500174
-
[135]
Jung, S.; Choi, J.; Han, Y. J. Mater. Chem. A 2018, 6, 1772. doi: 10.1039/C7TA07310K
-
[136]
Qian, J.; Xiong, Y.; Cao, Y.; Ai, X.; Yang, H. Nano Lett. 2014, 14, 1865. doi: 10.1021/nl404637q
-
[137]
Li, W.; Chou, S.; Wang, J.; Liu, H.; Dou, S. Chem. Commun. 2015, 51, 3682. doi: 10.1039/C4CC09604E
-
[138]
Wang, X.; Chen, K.; Wang, G.; Liu, X.; Wang, H. ACS Nano 2017, 11, 11602. doi: 10.1021/acsnano.7b06625
-
[139]
Fullenwarth, J.; Darwiche, A.; Soares, A.; Donnadieu, B.; Monconduit, L. J. Mater. Chem. A 2014, 2, 2050. doi: 10.1039/C3TA13976J
-
[140]
Ge, X.; Li, Z.; Yin, L. Nano Energy 2017, 32, 117. doi: 10.1016/j.nanoen.2016.11.055
-
[141]
Kim, S.; Manthiram, A. Chem. Commun. 2016, 52, 4337. doi: 10.1039/C5CC10585D
-
[1]
-
表 1 Na与Li的性质对比[1]
Na Li Cation radius 97 pm 68 pm Atomic weight 23.0 g•mol-1 6.9 g•mol-1 E0 vs. SHE -2.7 V -3.04 V A-O Coordination Octahedral Octahedral or tetrahedral Melting point 97.7 ℃ 180.5 ℃ Abundance 23.6×103 mg•kg-1 20 mg•kg-1 Distribution Everywhere 70% in South America Price, Carbonates ca. 2 RMB per kg ca. 40 RMB per kg  下载: 导出CSV
下载: 导出CSV
表 2 静电纺丝钠离子电池正极材料
Table 2. Electrospun cathode materials for sodium-ion batteries
Material Initial discharge/(mAh•g-1) Rate Cycle Capacity retention Cut-off voltage/V Reference NaTi2(PO4)3/C nanofiber 105 5C 500 93% 1.5~3.3 [31] Na3V2(PO4)3/C nanofiber 101 0.1C — — 2.7~3.8 [32] Na3V2(PO4)3/C nanofiber 106.8 0.2C 125 100% 2.5~4.0 [33] Na3V2(PO4)3/C nanorods 105.3 0.5C 50 92.6% 2.5~4.0 [34] Na3V2(PO4)3/C nanofiber 106.2 100 mA•g-1 300 34.8% 0.01~4 [35] Na3V2(PO4)3/C core-shell NWs 94 1C 50 74% 2.5~3.8 [36] NaVPO4F/C nanofiber — 2C 1000 96.5% 2.6~4.4 [38] Na0.44MnO2 NFs and NRs 120 0.42C 140 ca. 100% 1.5~4.0 [40] P2-type Na2/3(Fe1/2Mn1/2)O2 NFs ca. 195 0.1C 80 85.6% 1.5~4.2 [41] Li1+x(Mn1/3Ni1/3Fe1/3)O2 NFs ca. 88 0.1C 100 — 2~4.5 [42] Na0.7Fe0.7Mn0.3O2 nanotubes 82 500 mA•g-1 5000 70% 3.0~4.5 [43] Na6.24Fe4.88(P2O7)4@C@rGO ca. 99 40 mA•g-1 320 — 1.5~4.0 [44] Na2+2xFe2-x(SO4)3@porous CFs — 1C 300 95% — [45]
下载: 导出CSV
表 3 静电纺丝钠离子电池负极材料
Table 3. Electrospun anode materials for sodium-ion batteries
Material Current density/(mA•g-1) Cycle Capacity/(mAh•g-1) Capacity retention Binder-free Reference Carbon nanofiber 200 200 169 97.7% [54] PAN-CNFs 100 100 ca. 200 — [55] Lignin based carbon nanofibers 100 200 247 90.2% √ [56] Fulvic acid based carbon nanofiber 100 100 248 91% [57] Humic acid-carbon nanofiber 100 100 249.6 92.8% [58] N-doped carbon nanofiber 1000 200 150 93.7% [60] Porous carbon nanofiber 500 1000 ca. 140 ca. 70% √ [29] Porous carbon nanofiber 500 1000 210 — √ [61] Porous carbon nanotubes 5000 1200 110 — [62] N-doped porous carbon nanofiber 5000 7000 210 99% √ [63] Cross-linked carbon nanofiber 5000 500 126 85% √ [64] Graphene-porous CFs 2000 1000 300.8 91% √ [66] Porous TiO2 hollow nanofiber 6700 4500 93 89.4% [76] N-doped mesoporous TiO2 NF 3350 500 110 94% [78] N-doped carbon/TiO2 nanofiber 100 100 313 ca. 93% √ [79] Titania and carbon fiber 125 100 134 — [80] TiO2/C nanofiber 200 1000 237.1 98.6% [81] TiO2@C nanofiber 30 100 238.1 100.3% [82] rGO A-TiO2 nanofiber 335 200 148.5 90% [83] TiO2-X/carbon nanofiber 16.6 — ca. 160 — [84] Na2Ti3O7/C nanofiber 178 100 99 — [85] Li4Ti5O12@C nanofiber 175 100 165 94% [86] Mesoporous NaTi2(PO4)3/C NF 26.6 — 126.7 — [92] Co3O4@C nanofiber 500 500 251.7 84.3% [94] Co3O4Carbon nanofiber 500 700 ca. 400 — [95] CuO@C nanofiber 500 500 401 — [96] Pead-like Fe2O3@N-doped porous CFs 2000 1500 396 — [97] Amorphous FeOx/CFs 500 500 277 100% [98] T-Nb2O5/C nanofiber 1000 5000 150 ca. 100% [99] MnFe2O4@C nanofiber 2000 4200 306 90% [59] Ni-doped MnCo2O4 porous nanotubes 1000 11000 109 81% [102] Vike-like MoS2/TiO2 nanofiber 100 30 ca. 473 64% [19] MoS2/carbon nanofiber 1000 500 198 77.95% [108] Single-layered MoS2 /CFs 1000 100 484 — [109] MoS2/C nanofiber 100 600 283.9 74.8% [110] NiSe2-rGO-C nanofiber 200 100 468 — [114] Hollow sphere FeSe2@GC-rGO 1000 150 412 82% [111] PCNF@SnO2@C nanofiber 50 100 374 82.7% [119] N-doped CFs@SnO2 100 100 270 — [120] Sn nanodots@porous CNF 2000 1300 483 90% [121] Pipe-wire TiO2-Sn@CNFs 100 400 413 84.3% [122] Sb@porous CNFs 200 80 538 96% [125] Sb nanoparticle@C fiber 100 300 350 — [124] Sb-C nanofiber 200 400 446 90% [115] Porous CNF-SnSb 100 205 345 99.4% [126] SnSb@C porous nanofiber 500 200 410 63.2% [127] SnSb@rGO@CNFs 100 200 422.1 87.2% [128] Spider-web-like Bi/CNF 50 100 186 53% √ [129] Bi2O3 NP/carbon nanofiber 100 200 271 63% √ [130] Red P/CF/graphene 50 55 725.9 75.7% √ [132] Red P@NCNFs 2000 1000 619 ca. 81% [133]
下载: 导出CSV
-












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 67
- 文章访问数: 8277
- HTML全文浏览量: 1471
 下载:
下载:
