Received Date:
15 March 2018 Available Online:
15 July 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 51472037)
Abstract:
Synthesis of large scale ultrathin 2D structural TiO2 is challenging and meaningful in many fields of science and technology, because of their larger surface area and higher electron-hole pairs separation efficiency. In this work, a "bottomup" method is used to synthesize the large-size ultrathin TiO2 nanosheets at low temperature by liquid-phase. The mixture of tetrabutyl titanate and ethanol could hydrolyze in a dilute nitric acid solution with an ice-water bath and the obtained hydrolysates could peptize. After peptized, the hydrolysis products become to be very small TiO2 nanoclusters and they form a two-dimensional network structure via the orientation bonding formed by hydrogen bond. Continue to be aged at low temperature, the crystallization degree of samples will increase, and the networks eventually turn into TiO2 nanosheets. Effects of the concentration of nitric acid, ambient temperature and reactant concentration on the formation of two-dimensional structure TiO2 are studied in this work. The transmission electron microscope (TEM), ultraviolet-visible (UV-Vis) absorbance spectra, X-ray diffractometer (XRD), X-ray photoelectron spectroscopy (XPS) and Fourier Transform infrared spectroscopy (FTIR) are used to analyze the morphology, microstructure and properties of the samples, and the experiment of photocatalytic reduction of Cr(Ⅵ) is conducted to observe the photocatalytic activity of samples as well as to verify the effects of system parameters on the microstructure of TiO2 nanosheets. The results show that when the concentration of nitric acid is during 0.0217~0.0721 mol·L-1, the anatase TiO2 nanosheets that thinner than 1 nm could be obtained by peptizing and aging at 0~4℃. Excess nitric acid leads to crystalline and morphology transformation of TiO2, but lower concentration of nitric acid could prolong the peptizing time; increasing the ambient temperature will undermine the formation of two-dimensional structure; improving the amount of ethanol in the reactant will be helpful to disperse the hydrolysis products, and promote the process of the peptizing and formation of the two-dimensional structure.
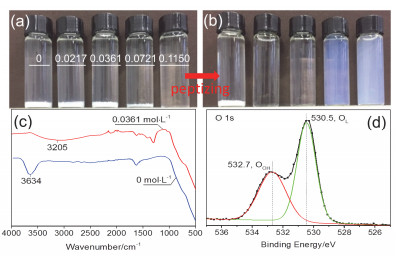
Figure 1.
(a) Hydrolysis and (b) peptizing status of TBT with different concentration of HNO3. The volume of HNO3 is 0, 0.0217, 0.0361, 0.0721 and 0.1150 mol·L-1 separately from left to right. (c) FT-IR spectrum of hydrolysates in the pure water and 0.0361 mol·L-1 HNO3 solution; (d) XPS spectrum of hydrolysate in the 0.0361 mol·L-1 HNO3 solution
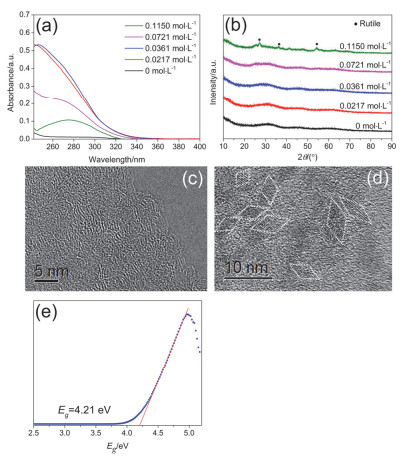
Figure 2.
(a) UV-vis absorption spectra and (b) XRD patterns of the systems peptized with different concentration of HNO3 for 7 days; TEM images of specimens with different concentration of HNO3: (c) 0.0217 and 0.0361 mol·L-1, (d) 0.1150 mol·L-1; (e) Tauc plot for the sol peptized with 0.0361 mol·L-1 HNO3 solution
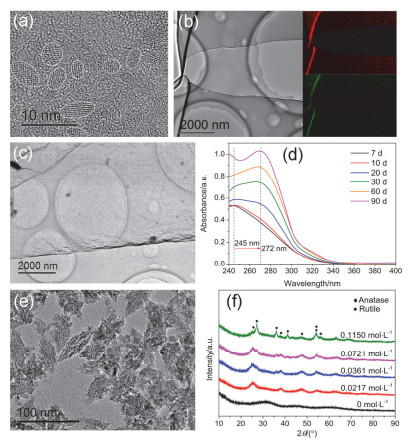
Figure 3.
TEM photos of TiO2 clusters aged for (a) 10 days, (b) 1 month, (c) 3 months and (d) the UV-vis absorption spectra of system during the aging time; (e) TEM image of the sample with 0.1150 mol·L-1 HNO3 solution aged for 1 month; (f) XRD patterns of samples aged for 1 month with different concentration of HNO3
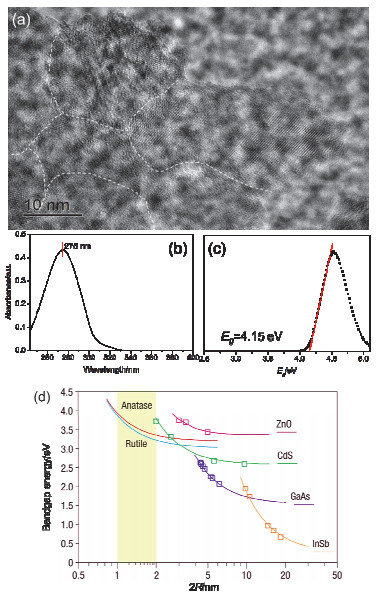
Figure 4.
(a) HRTEM image, (b) UV-vis absorption spectrum and (c) Tauc plots of the specimen aged for 4 months. (d) Calculated size dependence of the TiO2 bandgap energy[25]
Figure 5.
States of the solution after N719 adsorbed by TiO2with different forms: (a) P25, (b) TiO2 nanocrystal particles with a size about 10 nm, (c) TiO2 nanosheets
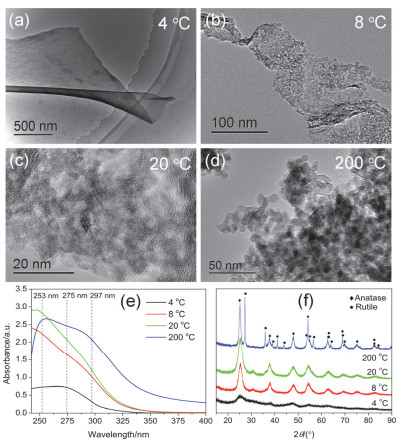
Figure 6.
TEM images of the specimens aged at different temperature: (a) 4 ℃, (b) 8 ℃, (c) 20 ℃, (d) 200 ℃; (e) UV-vis absorption spectra and (f) XRD patterns of the specimens aged at different temperature
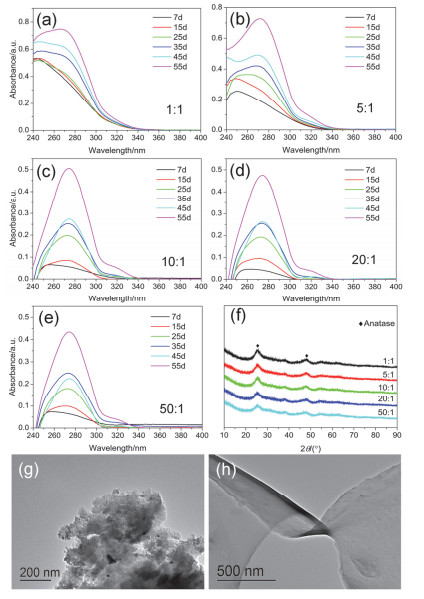
Figure 7.
UV-vis absorption spectra of the specimens with different volume ratio of ethanol to TBT: (a) 1:1, (b) 5:1, (c) 10:1, (d) 20:1, (e) 50:1; (f) XRD patterns of the specimens with different volume ratio of ethanol to TBT that aged for 1 month; TEM images of the specimens aged for 15 days with different volume ratio of ethanol to TBT: (g) 1:1, (h) 10:1
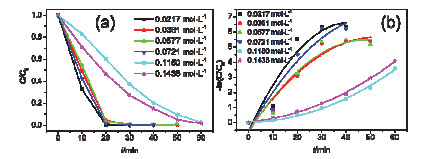
Figure 8.
(a) Photocatalytic reduction of Cr(Ⅵ) by the samples with different concentrations of HNO3 that aged for 35 days and (b) the corresponding reaction kinetics fitting
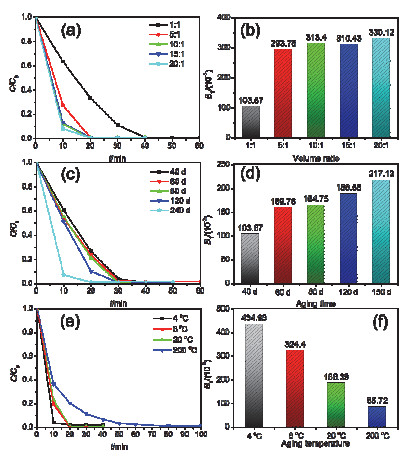
Figure 9.
Photocatalytic reduction of Cr(Ⅵ) by samples with different volume ratio of ethanol to TBT (a), different aging time (c), different aging temperature (e) and the corresponding reaction rate constants: (b), (d) and (f)
Figure 1
(a) Hydrolysis and (b) peptizing status of TBT with different concentration of HNO3. The volume of HNO3 is 0, 0.0217, 0.0361, 0.0721 and 0.1150 mol·L-1 separately from left to right. (c) FT-IR spectrum of hydrolysates in the pure water and 0.0361 mol·L-1 HNO3 solution; (d) XPS spectrum of hydrolysate in the 0.0361 mol·L-1 HNO3 solution
Figure 2
(a) UV-vis absorption spectra and (b) XRD patterns of the systems peptized with different concentration of HNO3 for 7 days; TEM images of specimens with different concentration of HNO3: (c) 0.0217 and 0.0361 mol·L-1, (d) 0.1150 mol·L-1; (e) Tauc plot for the sol peptized with 0.0361 mol·L-1 HNO3 solution
Figure 3
TEM photos of TiO2 clusters aged for (a) 10 days, (b) 1 month, (c) 3 months and (d) the UV-vis absorption spectra of system during the aging time; (e) TEM image of the sample with 0.1150 mol·L-1 HNO3 solution aged for 1 month; (f) XRD patterns of samples aged for 1 month with different concentration of HNO3
Figure 4
(a) HRTEM image, (b) UV-vis absorption spectrum and (c) Tauc plots of the specimen aged for 4 months. (d) Calculated size dependence of the TiO2 bandgap energy[25]
Figure 5
States of the solution after N719 adsorbed by TiO2with different forms: (a) P25, (b) TiO2 nanocrystal particles with a size about 10 nm, (c) TiO2 nanosheets
Figure 6
TEM images of the specimens aged at different temperature: (a) 4 ℃, (b) 8 ℃, (c) 20 ℃, (d) 200 ℃; (e) UV-vis absorption spectra and (f) XRD patterns of the specimens aged at different temperature
Figure 7
UV-vis absorption spectra of the specimens with different volume ratio of ethanol to TBT: (a) 1:1, (b) 5:1, (c) 10:1, (d) 20:1, (e) 50:1; (f) XRD patterns of the specimens with different volume ratio of ethanol to TBT that aged for 1 month; TEM images of the specimens aged for 15 days with different volume ratio of ethanol to TBT: (g) 1:1, (h) 10:1
Figure 8
(a) Photocatalytic reduction of Cr(Ⅵ) by the samples with different concentrations of HNO3 that aged for 35 days and (b) the corresponding reaction kinetics fitting
Figure 9
Photocatalytic reduction of Cr(Ⅵ) by samples with different volume ratio of ethanol to TBT (a), different aging time (c), different aging temperature (e) and the corresponding reaction rate constants: (b), (d) and (f)

 下载:
下载:





 下载:
下载:










 下载:
下载:
