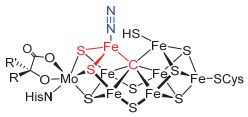
图 1.
开轴型铁钼辅基结构
Figure 1.
Open axial N2-binding mode of FeMo-cofactor

固氮酶可在温和条件下催化氮气还原为氨.该惰性小分子活化反应可在普通生理条件下进行, 这引发了人们对其催化机理的极大研究兴趣[1].到目前为止, 钼铁固氮酶的静息态结构已得到阐析[2], 但催化过程中固氮酶如何与氮气分子作用、如何促进氮气的还原转化仍然是知之甚少的问题[3].一般认为, 铁钼蛋白中的铁钼辅基(FeMo-cofactor)是钼铁固氮酶的催化活性中心, 是氮气配位和还原转化反应发生的位点.铁钼辅基是含有Fe7MoS9C组成的铁-钼-硫-碳簇合物[2].其中心碳原子的发现使得含碳配体配位的铁分子氮配合物受到研究者的极大关注[3b, 4].结合静息态下铁钼辅基的结构, 人们提出了诸多铁钼辅基配位活化氮气分子的可能模式[3, 5].在所谓的开轴型(open axial)模式中, 人们提出对铁钼辅基的质子化和还原会导致其腰部的一个铁—硫键断裂, 从而产生三角锥形FeS2C结构[5].该配位不饱和的铁位点配位氮气分子形成四配位的(N2)FeS2C结构[5](图 1).考虑到硫原子的弱场特征以及铁原子本身的较小的晶体场分裂能的特点, 人们推测(N2)FeS2C中铁位点可能为高自旋态.含碳配体配位的高自旋型铁分子氮配合物因此引起了生物金属有机模拟研究的兴趣[3, 4].
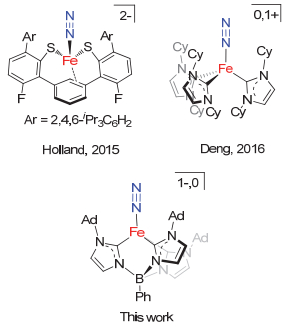
从配体场强弱考虑, 碳基配体多为强场配体.因此, 常见的含碳基配体配位的铁分子氮配合物多为低自旋型配合物[6].而结构得到表征的含碳配体配位高自旋铁分子氮配合物仅有Holland报道的芳烃-双硫酚配位的零价铁配合物[(2-S-1, 3-(6-F-3-(2, 4, 6-iPr3C6H2)C6H2)2-C6H4)Fe(N2)][4c]和我们报道的三(氮杂环卡宾)配位的零价铁和一价铁配合物[(ICy)3Fe(N2)]0, 1+ (ICy=1, 3-dicyclohexylimidazol-2-ylidene)][4e](图 2).氮杂环卡宾(N-heterocyclic carbene, NHC)是具有强给电子能力的碳基配体[7].从这一点来看一价和零价铁分子氮配合物[(ICy)3Fe(N2)]0, 1+展现的高自旋型电子结构特征十分特别[4e].我们推测可能原因之一是ICy的立体特性导致:不同卡宾配体上环己基间的空间排斥使得配位碳原子难以充分靠近铁中心, 致使配体场相对较弱, 形成高自旋配合物.受此启发, 我们推测通过立体位阻控制, 三齿氮杂环卡宾配体可能也可以支撑高自旋型铁分子氮配合物.让人意外的是Meyer和Smith等[8]在氮桥联和硼桥联三齿氮杂环卡宾铁配合物方面虽做了系统深入的研究, 却从未有相应三齿氮杂环卡宾稳定的铁分子氮配合物的报道.在本工作中, 我们利用N-金刚烷基取代的苯基硼桥联的三氮杂环卡宾[phenyltris(3-(1-adamant- ylimidazol-2-ylidene))borate, [PhB(AdIm)3]1-)为配体开展的高自旋态一价和零价铁分子氮配合物[PhB(AdIm)3Fe(N2)]和[K(18-C-6)(THF)][PhB(AdIm)3-Fe(N2)]的合成、表征和反应性质研究.
与[(ICy)3Fe]相比, Meyer和Smith等常用的氮桥联和硼桥联三齿氮杂环卡宾配体与铁配位后所形成的配位空腔可能较为宽敞, 缺乏对配位氮气分子的立体保护, 可能不利于分子氮配合物的稳定.同时考虑到Harman等[9]实现的高自旋型一价铁分子氮配合物[(TpAd, Me)Fe(N2)] (TpAd, Me=tris(3-adamantyl-5-methyl- pyrazol)borate)的合成, 我们决定采用N-上取代基为位阻较大的金刚烷基的苯基硼桥联三氮杂环卡宾([PhB-(AdIm)3]1-)为配体来开展相应的高自旋型铁分子氮配合物的合成研究.
采用与苯基硼桥三(N-叔丁基咪唑)盐类似的合成方法[10], 我们通过PhBCl2与3 equiv. N-金刚烷基咪唑、2 equiv. Me3SiOTf的反应以40%的收率合成得到了苯基硼桥三(N-金刚烷基咪唑)的三氟甲磺酸盐[PhB(HAdIm)3][OTf]2, 并对其进行了1H NMR, 13C NMR和元素分析的表征.常温下, 该三咪唑盐在四氢呋喃中和3 equiv.的二异丙基氨基锂(lithium diisopropylamide, LDA)反应可生成苯基硼桥三(N-金刚烷基氮杂环卡宾)配体.原位生成的氮杂环卡宾配体随后与0.5 equiv.的亚铁氯化物[Fe(tmeda)Cl2]2作用则可生成亚铁配合[PhB(AdIm)3FeCl] (1) (Scheme 1).配合物1为白色固体, 分离产率为70%.其在C6D6中的核磁氢谱在δ -37~75范围内显示有九组顺磁性信号峰.这显示了该配合物分子在溶液中的C3对称性, 与相应的叔丁基取代卡宾的亚铁氯化物[PhB(tBuIm)3FeCl]相一致[11]. Evans方法测得配合物1的溶液相磁矩为μeff=5.2(1) μB(室温, C6D6溶液), 显示其高自旋型电子结构特征(S=2).
在实现了苯基硼桥联三氮杂环卡宾亚铁配合物的合成的基础之上, 我们进一步探索了亚铁配合物1与石墨钾在氮气氛下的反应, 由此实现了一价和零价铁分子氮配合物的合成.
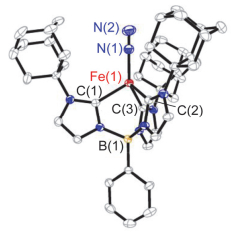
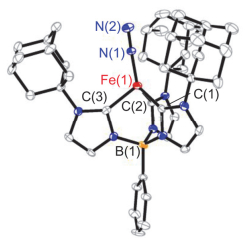
以四氢呋喃作溶剂, 配合物1在氮气氛下与1 equiv. KC8反应可以77%的产率生成一价铁分子氮配合物[PhB(AdIm)3Fe(N2)] (2) (Scheme 1).配合物2为绿色晶体.单晶X射线衍射显示铁中心除与[PhB(AdIm)3]1-上的三个卡宾配位外, 还与一分子氮气配位, 形成四面体构型(图 3). [PhB(AdIm)3]1-与铁中心配位形成的三角锥的三个C—Fe—C键角较为接近[92.3(2)°, 94.6(1)°和91.9(2)°], 三个Fe—C(carbene)键键长为0.2076(4), 0.2071(4)和0.2037(4) nm, 平均值为0.2061(4) nm, 与四面体型一价铁氮杂环卡宾配合物[(IEt2Me2)4Fe][BPh4][0.2058(4) nm][12]和[(IMe2Me2)4Fe][BPh4] [0.2047(2) nm][12]中Fe—C键长相当, 而略短于一价铁配合物[(ICy)3Fe(N2)][BPh4] [0.2087(3) nm][4e]和亚铁氯化物[PhB(tBuIm)3FeCl] (0.209 nm)[11]中Fe—C键长.配合物2中Fe—N键长为0.1854(4) nm, N—N键长为0.1046(5) nm, 与[(ICy)3Fe(N2)][BPh4]中相应键长一致[4e].配合物2的零场57Fe-穆斯堡尔谱展示出非对称双峰, 其同质异能位移值δ=0.59 mms-1, 四极距裂分值ΔEQ=1.31 mms-1(辅助材料中图S1).这两个数值与[(ICy)3Fe(N2)]-[BPh4]的57Fe-穆斯堡尔谱数值相当(δ=0.54 mms-1, ΔEQ=1.60 mms-1)[4e].配合物2的溶液相磁矩μeff=4.3(1) μB.这些数据显示配合物2为高自旋型一价铁分子氮配合物.值得一提的是尽管配合物2的N—N键长与自由氮气中键长相当, 配合物2的N—N伸缩振动峰出现在1928 cm-1处, 远低于低自旋一价铁分子氮配合物中数值, 如[((2-Ph2PC6H4)3Si)Fe(N2)] (2041 cm-1)[13], [((Cy2PCH2CH2)3P)Fe(N2)][BPh4] (2059 cm-1)[14]和[(THF)2MgCl(μ-H)(μ-(N)-PNP)Fe(N2)] (2084 cm-1)[15], 甚至比已报道的高自旋一价铁分子氮配合物[(ICy)3Fe(N2)][BPh4] (1967 cm-1)[4e]和[(TPAd, Me)Fe(N2)] (1959 cm-1)[9]更低.配合物2较小的N—N伸缩振动波数意味着在该配合物体系中, 配位的N2分子已被显著活化, 这可能与苯基硼桥联三氮杂环卡宾配体的强给电子能力相关[16].
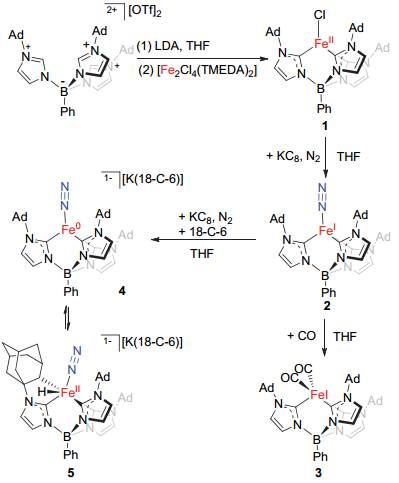
晶体结构显示配合物2中氮气分子位于三个金刚烷基环绕形成的空腔中.立体位阻保护对该高自旋型一价铁分子氮配合物的稳定可能至关重要.考察配合物2中氮气分子的配体取代反应发现2与膦配体PPh3和PMe3在加热条件下(80 ℃)也不反应.这可能是由于三个金刚烷基环绕形成的空腔空间较小, 膦配体难以与铁中心配位导致.配合物2与CO的反应则可迅速发生, 生成橙色的一价铁二羰基配合物[PhB(AdIm)3Fe(CO)2] (3) (Scheme 1).配合物3经过核磁共振氢谱、溶液相磁化率测试、紫外-可见吸收光谱、红外光谱以及元素分析表征.其红外光谱在1935和1855 cm-1处有两个特征振动峰, 与Smith等[17]报道的双羰基一价铁配合物[PhB(MesIm)3Fe(CO)2] (vCO=1957, 1886 cm-1)相近.配合物3较小的溶液相磁矩[μeff=1.8(1) μB]说明其低自旋型电子结构特征.
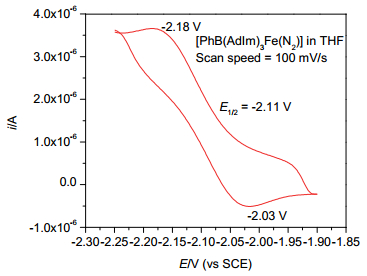
为考察配合物2的氧化还原性质, 我们测试了其循环伏安曲线.电化学测试显示配合物2的四氢呋喃溶液在较低电势下展示出近似可逆的氧化还原峰(图 4).该电化学过程可归属为[PhB(AdIm)3Fe(N2)]1-/0的氧化还原过程, 其半波电势E1/2=-2.11 V (vs. SCE)与配合物[(ICy)3Fe(N2)]0/1+的半波电势相当[4e].利用KC8为还原剂, 在四氢呋喃中将配合物2进一步还原, 再加入18-C-6后重结晶, 可以31%的分离产率得到深红色晶状配合物[K(18-C-6)THF][PhB(AdIm)3Fe(N2)] (4) (Scheme 1).单晶X射线衍射确认配合物4为配合物2经历单电子还原后生成的零价铁分子氮配合物.
如图 5所示, 配合物4的阴离子中铁中心呈四面体构型, 氮气分子以末端形式配位.需要指出的是与2相比, 4中铁中心的配位构型呈现出明显畸变.这反映在三个C-Fe-N角度展示出较大差异, B—Fe—N角度也更偏离180°(表 1).这可能与四面体构型下d8体系存在较大的姜-泰勒畸变有关.配合物4中Fe—N键键长为0.1735(7) nm, Fe—C键键长为0.1974(9) nm, 与配合物2相比分别缩短0.011和0.008 nm.配合物4中N—N键长为0.1149(11) nm, 比配合物2长近0.01 nm, 与已有报道的零价铁分子氮配合物[(2-S-1, 3-(6-F-3-(2, 4, 6-iPr3-C6H2)C6H2)2C6H4)Fe(N2)] [0.1132(7) nm][4c], [(ICy)3-Fe(N2)] [0.1138(10) nm][4e], [((2-Ph2PCH2SiMe2)3C)Fe-(N2)Na(THF)3]] [0.1147(4) nm][18]和[(depe)2Fe(N2)]- [K(benzo[15]crown-5)2] [0.1139(13) nm][19]中的N—N键长相当.与其相对较长的N—N键一致, 配合物4的N—N键伸缩振动峰出现在1807 cm-1处, 显著低于一价铁配合物2的N—N键伸缩振动频率, 也明显低于低自旋型零价铁末端分子氮配合物中相应伸缩振动频率, 如[((Cy2PCH2CH2)3P)Fe(N2)] (1996 cm-1), [20] [(depe)2Fe-(N2)][K(benzo[15]crown-5)2] (1956 cm-1)[19]和[((2-Ph2P-CH2SiMe2)3C)Fe(N2)][21] (1927 cm-1).该N—N伸缩振动频率也比已知的高自旋零价铁末端分子氮配合物[(2-S-1, 3-(6-F-3-(2, 4, 6-iPr3C6H2)C6H2)2C6H4)Fe(N2)] (1928 cm-1)[4c]和[(ICy)3Fe(N2)] (1853和1841 cm-1)[4e]中N—N伸缩振动频率低.
 下载:
导出CSV
下载:
导出CSV
| 2 | 4 | |
| N—N/nm | 0.1046(5) | 0.1149(11) |
| Fe—N/nm | 0.1845(4) | 0.1735(7) |
| Fe—C/nm | 0.2076(4) | 0.1982(9) |
| 0.2071(4) | 0.1994(9) | |
| 0.2037(4) | 0.1947(9) | |
| C-Fe-N | 122.1(2) | 121.6(4) |
| 125.3(2) | 130.6(4) | |
| 122.2(2) | 116.2(4) | |
| B-Fe-N/(°) | 178.2 | 172.5 |
值得指出的是配合物4在KBr片中测得的红外光谱除在1807 cm-1处有特征峰外, 在1958和1926 cm-1处也出现了较强的红外吸收峰(辅助材料中图S11). 1926 cm-1处的吸收峰与一价铁配合物2中的N—N伸缩振动峰位置一致, 推测为配合物4与外在氧化性物种作用生成配合物2所致.而1958 cm-1处的吸收峰的位置与低自旋型二价铁末端分子氮配合物[(ICy)2(ICy')Fe(N2)(H)]的N—N伸缩振动相近(1966 cm-1)[4e].由此, 我们推测配合物4可能通过分子内碳氢键氧化加成反应生成二价铁分子氮配合物5 (Scheme 1).与该推测一致, 配合物4在氘代四氢呋喃中的核磁共振氢谱(辅助材料中图S7)在δ -12.4处存在特征的Fe-H信号[4e], 同时合成得到的配合物4的深红色固体的57Fe-穆斯堡尔谱信号峰的同质异能位移(δ=0.13 mms-1)与[(ICy)2(ICy')Fe(N2)(H)] (δ=0.05 mms-1)[4e]较接近(辅助材料中图S2).这些数据显示零价铁配合物4可能在溶液中甚至在固相下可通过分子内碳-氢键活化反应与配合物5发生可逆转化(Scheme 1).该转化与我们前期发现的单齿氮杂环卡宾配位的零价铁分子氮配合物[(ICy)3Fe(N2)]与环金属化产物[(ICy)2(ICy')Fe(N2)(H)]转化类似[4e].可惜的是多次重结晶尝试均未能获得配合物5的单晶.
如前所述, 配合物2, 4, [(ICy)3Fe(N2)][BPh4]和[(ICy)3Fe(N2)]是一类新颖的以碳基配体为支撑配体的高自旋型铁分子氮配合物.这类配合物拥有的较低的N—N伸缩振动频率表明其中配位氮分子已被活化.为进一步研究这类分子氮配合物中配位氮分子的转化反应, 我们考察它们与质子酸的反应以及作为催化剂在氮气还原硅基化反应中的表现.
比色法检测显示, 低温下(-78 ℃)在乙醚溶液中一价铁分子氮配合物2与三氟甲磺酸或无水HCl乙醚溶液反应仅生成微量的肼(≤4(2)%)和氨(<1%).零价铁分子氮配合物4与这些强酸的反应则可以相对较高的产率生成肼(≤9(3)%)和氨(≤5(3)%) (表 2).这一差异显示零价铁分子氮配合物中配位氮分子的确更容易被转化为还原产物, 与前述表征数据所显示的零价铁分子氮配合物中配位氮分子明显被活化的结果相一致.这些硼桥联三卡宾配位的铁分子氮配合物的质子化反应与已报道的膦配体配位的铁分子氮配合物的反应相比显示出一定的差别. Peters等[13]曾发现一价铁配合物[((2-Ph2P-C6H4)3Si)Fe(N2)]与10 equiv. HBF4或HCl的反应以17%和7%的产率(per Fe atom)生成肼; Leigh等[22]发现零价铁分子氮配合物[(dmpe)2Fe(N2)]与过量HCl反应以12%的产率生成氨(per Fe atom); Tyler等[23]发现[(DMeOPrPE)2Fe(N2)]与6 equiv. HOTf反应生成氨(15%)和肼(2%); George等[24]则指出[(N(CH2CH2PPh2)3)-Fe(N2)]与过量HBr反应则主要生成肼(11%), 氨的产率仅为(3%).高自旋型铁分子氮配合物的质子化反应中相对较低的氨和肼的产率可能是与这些高自旋型铁分子氮配合物较强的还原性更容易导致质子还原反应的发生有关.与2和4的反应不同, 单齿卡宾配位的铁分子氮配合物[(ICy)3Fe(N2)][BPh4]和[(ICy)3Fe(N2)]的质子化反应中肼是主要的氮气还原产物, 其中以三氟甲磺酸为质子化试剂时肼的产率分别为9(2)%和18(2)% (表 2).单齿卡宾配位的铁分子氮配合物较高的肼的产率显示了氮杂环卡宾配体对铁分子氮配合物反应性的影响.配体的齿数和电荷数是可能的影响因素, 但导致差别的具体原因还需进一步研究.
下载:
导出CSV
 |
|||
| Fe—N2 | HX | Yield/% of N2H4 | Yield/% of NH3 |
| 2 | HClb | 2(1) | <1 |
| 2 | HOTf | 4(2) | <1 |
| 4 | HClb | 9(3) | 5(3) |
| 4 | HOTf | 5(3) | 3(2) |
| [(ICy)3Fe(N2)][BPh4] | HCl | 2(1) | <1 |
| [(ICy)3Fe(N2)][BPh4] | HOTf | 9(2) | <1 |
| [(ICy)3Fe(N2)] | HCl | 2(1) | <1 |
| [(ICy)3Fe(N2)] | HOTf | 18(2) | 2(1) |
| a Yields are relative to per mol iron, and are the averaged data of 2 runs. b 7.6 mol•L-1 HCl in Et2O. | |||
将氮气还原硅基化生成硅基胺是少有几种氮气催化转化方式之一[25]. Shiina[26]于1972年报道简单的金属盐FeCl3可以催化Li与N2和ClSiMe3反应生成N(SiMe3)3, 但TON值仅为2.3.近年来随着新的催化剂的发展, 氮气还原硅基化反应体系的TON有明显的提升.具有代表性的体系有Nishibayashi等[27]报道的二茂铁桥联的双膦配体配位的金属钼配合物trans-[(depf)2Mo(N2)2] (226 equiv. N(SiMe3)3/摩尔钼或75 equiv. N(SiMe3)3/摩尔金属)、Peters等[4d]报道的零价铁配合物[(Et2-cAAC)2] (24 equiv. N(SiMe3)3/摩尔铁)、Mezailles等[28]报道的金属钼催化剂[(P(CH2CH2-PCy2)3)Mo(NN(SiMe3)2)] (15 equiv. N(SiMe3)3/摩尔钼), Ohki等[29]报道的[Fe4(μ-H)4(μ3-H)2(N(SiMe3)2)2(PEt3)4] (40 equiv. N(SiMe3)3/摩尔铁), Lu等[30]报道的三胺三膦双钴配合物体系(98 equiv. N(SiMe3)3/摩尔钴), 和我们[31]报道的氮杂环卡宾配位的钴配合物M[(ICy)2Co(N2)2] (M=K, Rb, Cs; 120 equiv. N(SiMe3)3/摩尔钴).与已有铁催化剂相比, 氮杂环卡宾配位的铁配合物1, 2, 4, [(ICy)3Fe(N2)][BPh4]和[(ICy)3Fe(N2)]催化的反应均展示出相对较高的TON值.在2000 equiv. KC8存在下, 以乙醚作溶剂, 苯基硼桥联三卡宾铁配合物催化氮气与2000 equiv. Me3SiCl反应生成(Me3Si)3N的TON可达87;应用单齿卡宾配位的铁配合物的催化体系TON值约为35(表 3).这些催化体系所展现出的较高的TON值可能与氮杂环卡宾配体与铁中心结合能力较强, 能较好稳定低价铁物种相关[8].相对于单齿卡宾配合物体系, 苯基硼桥联的三卡宾铁配合物表现出的较高的TON值说明强给电性螯合配体在发展可催化氮气还原硅化反应的铁配合物催化剂方面具有潜能.
下载:
导出CSV
 |
|||
| Entry | Cat. | TON | Yield/% of N(SiMe3)3 |
| 1 | 1 | 83(11) | 12(2) |
| 2 | 2 | 84(21) | 12(4) |
| 3 | 4 | 87(11) | 13(3) |
| 4 | [(ICy)3Fe(N2)] | 35(2) | 5(1) |
| 5 | [(ICy)3Fe(N2)][BPh4] | 34(2) | 5(1) |
| a The reactions were run using 0.005 mmol of the catalyst in 10 mL of Et2O with the addition of the reducing reagent prior to catalyst. TONs and yields were the averaged data of three runs. b Turnover numbers were calculated as the molar ratio of N(SiMe3)3 to iron. c Yields of N(SiMe3)3 were based on Me3SiCl and determined by GC with cyclododecane as the internal standard. | |||
在发现苯基硼桥联三卡宾铁配合物的高效催化表现的基础上, 我们对配合物2和4与等物质的量的Me3SiCl的反应进行了初步研究, 以期揭示相关催化反应机理.结果发现配合物2与等物质的量的Me3SiCl反应生成二价铁配合物1, 而配合物4与等物质的量Me3SiCl反应则生成二价铁配合物1与一价铁配合物2的混合物.在这两个反应中均未实现硅基取代含氮配合物中间体的分离.这一结果说明催化反应中的氮分子的硅基化可能是由原位产生的三甲基硅自由基对配位氮分子的进攻而实现[30, 31].相关反应机理研究还在进行中.
报道了苯基硼桥联三卡宾配体支撑的铁分子氮配合物合成、表征及其催化的氮气还原硅烷化反应.利用N-金刚烷基取代的苯基硼桥联三氮杂环卡宾为配体实现了高自旋一价铁分子氮配合物2和零价铁分子氮配合物4的合成, 其N—N红外伸缩振动频率分别为1928和1807 cm-1, 均为同等价态铁末端氮气配合中最低值.这些苯基硼桥联三卡宾铁配合物可高效催化石墨钾和氮气, Me3SiCl反应生成(Me3Si)3N, 催化体系的TON值达87, 为铁配合物催化剂中较高值.这些结果说明具有强给电子能力的多齿氮杂环卡宾配体在金属催化氮气活化转化方面具有的潜在价值.
根据类似物[PhB(HtBuIm)3][OTf]2的合成方法稍作修改[10].将N-金刚烷基咪唑(8.9 g, 44.0 mmol)溶解在40 mL甲苯中, 另取200 mL Schlenk瓶加入100 mL甲苯和PhBCl2 (2.3 g, 14.7 mmol).真空线通氮气氛下将N-金刚烷基咪唑的甲苯溶液缓慢滴加到PhBCl2的甲苯溶液中.混合液缓慢搅拌15 min后, 再逐滴加入TMSOTf (6.5 g, 29.2 mmol).滴加完成后将混合物升温至100 ℃, 继续搅拌12 h.反应结束后, 冷却至室温, 过滤, 得黄色固体.固体经过乙醚(100 mL)洗涤, 真空抽干, 即可得产物[PhB(HAdIm)3][OTf]2, 为白色固体产物, 重5.9 g, 产率41%. 1H NMR (CD3CN, 400 MHz) δ: 8.08 (s, 3H), 7.69 (s, 3H), 7.46~7.44 (m, 3H), 7.24 (s, 3H), 7.15~7.13 (m, 2H), 2.24 (s, 9H), 2.15 (s, 18H), 1.81~1.73 (m, 18H); 13C NMR (CDCl3, 101 MHz) δ: 176.9, 173.9, 170.1, 165.8, 160.8, 160.1, 101.4, 83.2, 76.4, 70.6. Anal. calcd for C47H59BF6N6O6S2: C 56.85, H 5.99, N 8.46; found C 56.62, H 6.00, N 8.31.
在100 mL Schlenk瓶中加入[PhB(HAdIm)3][OTf]2 (1.02 g, 1.0 mmol)和60 mL四氢呋喃.混合物在-28 ℃下冷冻2 h后再在剧烈搅拌下加入LDA (365 mg, 3.4 mmol).继续搅拌4 h后, 加入[Fe(tmeda)Cl2]2 (250 mg, 0.5 mmol).反应体系在室温下搅拌12 h后, 抽干溶剂.黄色残余物中加入50 mL甲苯搅拌萃取.砂芯硅藻土过滤后得到的滤液, 减压下抽去溶剂, 得到灰白色固体.该固体经过乙醚(20 mL)漂洗后再溶于四氢呋喃形成饱和溶液.四氢呋喃溶液于-28 ℃下冷冻可得白色固体, 即为配合物1, 重650 mg, 产率70%. UV-vis (THF) λmax: 302, 335, 1340 nm; 1H NMR (400 MHz, C6D6, 294 K) δ: 74.89 (s, Δv1/2=48.47 Hz), 61.01 (s, Δv1/2=88.59 Hz), 43.15 (s, Δv1/2=66.48 Hz), 21.71 (s, Δv1/2=18.35 Hz), 18.76 (s, Δv1/2=17.20 Hz), 4.56 (s, Δv1/2=20.67 Hz), 3.42 (br), 1.08 (br, overlap with residual solvent peaks), -36.42 (s, Δv1/2=364.91 Hz). Anal. calcd for C45H56BCl- FeN6•THF: C 68.82, H 7.54, N 9.83; found C 68.50, H 7.53, N 9.82. Magnetic susceptibility (C6D6, 294 K): μeff=5.2(1) μB.
将[PhB(AdIm)3FeCl] (500 mg, 0.60 mmol)称量置于100 mL Schlenk瓶内, 加入50 mL四氢呋喃溶解后得到无色溶液.将溶液于-25 ℃下冷冻2 h后在剧烈搅拌下缓慢加入KC8 (97 mg, 0.72 mmol).反应体系逐渐恢复至室温后继续搅拌12 h.停止搅拌后将反应液通过硅藻土过滤, 得到绿色溶液.真空泵下减压除去溶剂得到浅绿色固体.绿色固体用10 mL四氢呋喃溶解后室温下乙醚扩散重结晶可得配合物2的绿色晶状固体, 重320 mg, 产率77%. UV-vis (THF) λmax: 270, 750, 1180, 1517 nm. 1H NMR (400 MHz, C6D6, 294 K) δ: 32.52 (very br), 28.25 (very br), 8.65 (s, Δv1/2=128.14 Hz), 3.86 (s, Δv1/2=64.74 Hz), 1.56 (br, overlap with residual solvent peaks), 1.41 (br, overlap with residual solvent peaks), 1.24 (br, overlap with residual solvent peaks); IR (KBr) vNN: 1928 cm-1; IR (THF) vNN: 1933 cm-1. Anal. calcd for C45H56B- FeN8•THF: C 69.42, H 7.61, N 13.22; found C 69.83, H 7.38, N 13.79. Magnetic susceptibility (C6D6, 294 K): μeff=4.3(1) μB.
手套箱内称量[PhB(AdIm)3Fe(N2)] (400 mg, 0.5 mmol)于100 mL Schlenk瓶内, 加入50 mL四氢呋喃溶解后得到绿色溶液.液氮冷冻下, 真空除去体系内N2, 再通入干燥过的CO (101 kPa).反应体系逐渐恢复至室温后继续搅拌8 h, 得棕黄色溶液.减压除去溶剂得到的棕黄色固体用10 mL四氢呋喃溶解后通过乙醚室温下扩散重结晶可得配合物3的深棕色晶状固体, 重125 mg, 产率32%. UV-vis (THF) λmax: 295, 330, 405, 775 nm. 1H NMR (400 MHz, C6D6) δ: 30.02 (s, Δv1/2=339.12 Hz), 14.78 (s, Δv1/2=144.13 Hz), 9.50 (s, Δv1/2=91.31 Hz), 7.02 (br, overlap with residual solvent peaks), 6.15 (s, Δv1/2=30.17 Hz), 5.87 (s, Δv1/2=40.08 Hz), 3.72 (br, overlap with residual solvent peaks), 2.11 (s, Δv1/2=17.46 Hz), 1.12 (br, overlap with residual solvent peaks); IR (KBr) vCO: 1935, 1855 cm-1. Anal. calcd for C47H56BFeN6O2: C 70.24, H 7.02, N 10.46; found C 70.32, H 7.40, N 10.30. Magnetic susceptibility (C6D6, 294 K): μeff=1.8(1) μB.
手套箱内称量[PhB(AdIm)3Fe(N2)] (650 mg, 0.8 mmol)于50 mL Schlenk瓶内, 加入20 mL四氢呋喃搅拌溶解得到绿色溶液.另取100 mL Schlenk瓶, 称量加入KC8 (135 mg, 1.0 mmol)于其中, 加入50 mL四氢呋喃搅拌得到悬浊液.将两Schlenk瓶绑好, 在真空线下抽换气, 液氮乙醇浴预冷至-78 ℃后将绿色溶液导入KC8悬浊液内, 维持低温搅拌2 h后将配制好的18-C-6 (422 mg, 1.6 mmol)的四氢呋喃(5 mL)溶液导入反应体系内.反应体系恢复至室温后继续搅拌4 h, 砂芯硅藻土过滤, 得到棕红色溶液.减压除去溶剂后得到的棕色固体用20 mL四氢呋喃溶解后经乙醚扩散重结晶可得配合物4的深棕色晶状固体, 重280 mg, 产率31%. 1H NMR (400 MHz, THF-d8) δ: 35.65 (very br), 33.40 (very br), 7.98, 7.30, 7.21, 7.06, 6.82, 6.63, 6.60, 6.47, 5.92, 4.46, 3.45, 2.37, 2.29, 2.17, -5.42 (very br), -12.43 (s, Fe-H); IR (KBr) vNN: 1808 cm-1, 1958 cm-1. Anal. calcd for C61H87BFeKN8O7: C 63.70, H 7.62, N 9.74; found C 64.02, H 8.24, N 9.43.配合物4易发生分子内C—H键活化反应转化为5.因此上述表征数据应为4和5的混合物.
将铁分子氮配合物(0.08 mmol)溶解于Et2O (2 mL)中, -78 ℃下加入质子酸(HCl的无水Et2O溶液或无水HOTf, 1.28 mmol酸).低温反应2 h后, 用水(3 mL×3)萃取, 通过比色法分析NH3和N2H4含量[32, 33].
25 mL Schlenk瓶内, 称量加入KC8 (1.35 g, 10 mmol), 铁配合物(0.005 mmol)和Me3SiCl (1.08 g, 10 mmol).加入Et2O (10 mL)后反应体系在氮气氛下室温搅拌24 h.经砂芯硅藻土过滤后得淡黄色滤液.滤液中再加入正十二烷(40 mg, 0.24 mmol)作为内标.溶液用GC检测确定(Me3Si)3N的含量.
辅助材料(Supporting Information) 所合成的配体和配合物的核磁共振谱图、穆斯堡尔谱图、紫外可见光谱、红外谱图及晶体数据等材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.配合物2和4的晶体结构数据已上传至英国剑桥晶体数据库(Cambridge Crystallographic Data Centre), CCDC号码为1827402和1827403.
(a) Winter, H. C. ; Burris, R. H. Annu. Rev. Biochem. 1976, 45, 409. (b) Bulen, W. A. ; LeComte, J. R. Proc. Natl. Acad. Sci. U. S. A. 1966, 56, 979. (c) Mortenson, L. E. Fed. Proc. 1965, 24, 233. (d) Mortenson, L. E. Biochim. Biophys. Acta 1966, 127, 18. (e) Hageman, R. V. ; Burris, R. H. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 2699. (f) Dean, D. R. ; Bolin, J. T. ; Zheng, L. J. Bacteriol. 1993, 175, 6737. (g) Howard, J. B. ; Rees, D. C. Annu. Rev. Biochem. 1994, 63, 235. (h) Kim, J. ; Rees, D. C. Biochemistry 1994, 33, 389. (i) Wang, Y. -S. ; Li, J. -L. Prog. Nat. Sci. 2000, 10, 481. (王友绍, 李季伦, 自然科学进展, 2000, 10, 481.
(a) Spatzal, T. ; Aksoyoglu, M. ; Zhang, L. ; Andrade, S. L. A. ; Schleicher, E. ; Weber, S. ; Rees, D. C. ; Einsle, O. Science 2011, 334, 940. (b) Lancaster, K. M. ; Roemelt, M. ; Ettenhuber, P. ; Hu, Y. ; Ribbe, M. W. ; Neese, F. ; Bergmann, U. ; DeBeer, S. Science 2011, 334, 974. (c) Lancaster, K. M. ; Hu, Y. ; Bergmann, U. ; Ribbe, M. W. ; DeBeer, S. J. Am. Chem. Soc. 2013, 135, 610. (d) Wiig, J. A. ; Hu, Y. ; Lee, C. C. ; Ribbe, M. W. Science 2012, 337, 1672. (e) Wiig, J. A. ; Lee, C. C. ; Hu, Y. ; Ribbe, M. W. J. Am. Chem. Soc. 2013, 135, 4982. (f) Zhang, C. -X. ; Fan, H. -J. ; Liu, Q. -T. Prog. Chem. 1997, 9, 266. (张纯喜, 樊红军, 刘秋田, 化学进展, 1997, 9, 266. ) (g) Chen, Q. -L. ; Chen, H. -B. ; Cao, Z. -X. ; Zhou, Z. -H. ; Wan, H. -L. ; Li, Y. ; Li, J. -L. Sci. China, Chem. 2014, 44, 1849. (陈全亮, 陈洪斌, 曹泽星, 周朝晖, 万惠霖, 李颖, 李季伦, 中国科学: 化学, 2014, 44, 1849.
(a) Hoffman, B. M. ; Lukoyanov, D. ; Yang, Z. -Y. ; Dean, D. R. ; Seefeldt, L. C. Chem. Rev. 2014, 114, 4041. (b) Čorić, I. ; Holland, P. L. J. Am. Chem. Soc. 2016, 138, 7200.
(a) Rittle, J. ; Peters, J. C. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15898. (b) Creutz, S. E. ; Peters, J. C. J. Am. Chem. Soc. 2014, 136, 1105. (c) Čorić, I. ; Mercado, B. Q. ; Bill, E. ; Vinyard, D. J. ; Holland, P. L. Nature 2015, 526, 96. (d) Ung, G. ; Peters, J. C. Angew. Chem., Int. Ed. 2015, 54, 532. (e) Ouyang, Z. -W. ; Cheng, J. ; Li, L. -L. ; Bao, X. -L. ; Deng, L. Chem. -Eur. J. 2016, 22, 14162.
(a) Kästner, J. ; Blöchl, P. E. J. Am. Chem. Soc. 2007, 129, 2998. (b) Spatzal, T. ; Perez, K. A. ; Einsle, O. ; Howard, J. B. ; Rees, D. C. Science 2014, 345, 1620.
(a) Hazari, N. Chem. Soc. Rev. 2010, 39, 4044. (b) Crossland, J. L. ; Tyler, D. R. Coord. Chem. Rev. 2010, 254, 1883. (c) Ohki, Y. ; Seino, H. Dalton Trans. 2016, 45, 874. (d) Danopoulos, A. A. ; Wright, J. A. ; Motherwell, W. B. Chem. Commun. 2005, 784. (e) Pugh, D. ; Wells, N. J. ; Evans, D. J. ; Danopoulos, A. A. Dalton Trans. 2009, 7189. (f) Yu, R. P. ; Darmon, J. M. ; Hoyt, J. M. ; Margulieux, G. W. ; Turner, Z. R. ; Chirik, P. J. ACS Catal. 2012, 2, 1760. (g) Bartholomew, E. R. ; Volpe, E. C. ; Wolczanski, P. T. ; Lobkovsky, E. B. ; Cundari, T. R. J. Am. Chem. Soc. 2013, 135, 3511.
(a) Bourissou, D. ; Guerret, O. ; Gabbaï, F. P. ; Bertrand, G. Chem. Rev. 2000, 100, 39. (b) Glorius, F., N-Heterocyclic Carbenes in Transition Metal Catalysis, Topics in Organometallic Chemistry, Vol. 21, Springer, Berlin, 2007. (c) Hahn, F. E. ; Jahnke, M. C. Angew. Chem., Int. Ed. 2008, 47, 3122.
(a) Ingleson, M. J. ; Layfield, R. A. Chem. Commun. 2012, 48, 3579. (b) Riener, K. ; Haslinger, S. ; Raba, A. ; Högerl, M. P. ; Cokoja, M. ; Herrmann, W. A. ; Kühn, F. E. Chem. Rev. 2014, 114, 5215.
McSkimming, A.; Harman, W. H. J. Am. Chem. Soc. 2015, 137, 8940. doi: 10.1021/jacs.5b06337
Cowley, R. E.; Bontchev, R. P.; Duesler, E. N.; Smith, J. M. Inorg. Chem. 2006, 45, 9771. doi: 10.1021/ic061299a
Scepaniak, J. J.; Fulton, M. D.; Bontchev, R. P.; Duesler, E. N.; Kirk, M. L.; Smith, J. M. J. Am. Chem. Soc. 2008, 130, 10515. doi: 10.1021/ja8027372
(a) Ouyang, Z. -W. ; Meng, Y. ; Cheng, J. ; Xiao, J. ; Gao, S. ; Deng, L. Organometallics 2016, 35, 1361. (b) Ohki, Y. ; Hoshino, R. ; Tatsumi, K. Organometallics 2016, 35, 1368.
Mankad, N. P.; Whited, M. T.; Peters, J. C. Angew. Chem., Int. Ed. 2007, 46, 5768. doi: 10.1002/(ISSN)1521-3773
Gilbert-Wilson, R.; Field, L. D.; Colbran, S. B.; Bhadbhade, M. M. Inorg. Chem. 2013, 52, 3043. doi: 10.1021/ic3024953
Hounjet, L. J.; Adhikari, D.; Pink, M.; Carroll, P. J.; Mindiola, D. J. Z. Anorg. Allg. Chem. 2015, 641, 45. doi: 10.1002/zaac.201400173
Smith, J. M. Comments Inorg. Chem. 2008, 29, 189. doi: 10.1080/02603590802590080
Hickey, A. K.; Chen, C.; Pink, M.; Smith, J. M. Organometallics 2015, 34, 4560. doi: 10.1021/acs.organomet.5b00646
Lee, Y.; Mankad, N. P.; Peters, J. C. Nat. Chem. 2010, 2, 558. doi: 10.1038/nchem.660
Komiya, S.; Akita, M.; Yoza, A.; Kasuga, N.; Fukuoka, A.; Kai, Y. J. Chem. Soc., Chem. Commun. 1993, 787. doi: 10.1039/c39930000787
Gilbert-Wilson, R.; Field, L. D.; Colbran, S. B.; Bhadbhade, M. M. Inorg. Chem. 2013, 52, 3043. doi: 10.1021/ic3024953
Creutz, S. E.; Peters, J. C. J. Am. Chem. Soc. 2014, 136, 1105. doi: 10.1021/ja4114962
(a) Hills, A. ; Hughes, D. A. ; Jimenez-Tenorio, M. ; Leigh, G. J. ; Rowley, A. T. J. Chem. Soc., Dalton Trans. 1993, 3041. (b) Hall, D. A. ; Leigh, G. J. J. Chem. Soc., Dalton Trans. 1996, 3539.
Gilbertson, J. D.; Szymczak, N. K.; Tyler, D. R. J. Am. Chem. Soc. 2005, 127, 10184. doi: 10.1021/ja053030g
George, T. A.; Rose, D. J.; Chang, Y.; Chen, Q.; Zubieta, J. Inorg. Chem. 1995, 34, 1295. doi: 10.1021/ic00109a046
李嘉鹏, 殷剑昊, 俞超, 张文雄, 席振峰, 化学学报, 2017, 75, 733. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346110.shtmlLi, J.-P.; Yin, J.-H.; Yu, C.; Zhang, W.-X.; Xi, Z.-F. Acta Chim. Sinica 2017, 75, 733. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346110.shtml
Shiina, K. J. Am. Chem. Soc. 1972, 94, 9266. doi: 10.1021/ja00781a068
Tanaka, H.; Sasada, A.; Kouno, T.; Yuki, M.; Miyake, Y.; Nakanishi, H.; Nishibayashi, Y.; Yoshizawa, K. J. Am. Chem. Soc. 2011, 133, 3498. doi: 10.1021/ja109181n
Liao, Q.; Saffon-Merceron, N.; Mezailles, N. Angew. Chem., Int. Ed. 2014, 53, 14206. doi: 10.1002/anie.v53.51
Araake, R.; Sakadani, K.; Tada, M.; Sakai, Y.; Ohki, Y. J. Am. Chem. Soc. 2017, 139, 5596. doi: 10.1021/jacs.7b01965
Siedschlag, R. B.; Bernales, V.; Vogiatzis, K. D.; Planas, N.; Clouston, L. J.; Bill, E.; Gagliardi, L.; Lu, C. C. J. Am. Chem. Soc. 2015, 137, 4638. doi: 10.1021/jacs.5b01445
Gao, Y.-F.; Li, G.-Y.; Deng, L. J. Am. Chem. Soc. 2018, 140, 2239. doi: 10.1021/jacs.7b11660
Weatherburu, M. W. Anal. Chem. 1967, 39, 971. doi: 10.1021/ac60252a045
Watt, G. W.; Chrisp, J. D. Anal. Chem. 1952, 24, 2006. doi: 10.1021/ac60072a044

图式 1 铁分子氮配合物2和4的合成路线与反应性质
Scheme 1 Synthetic route of iron dinitrogen complexes 2 and 4 and their reactivities

图 3 配合物[PhB(AdIm)3Fe(N2)] (2)的分子结构
Figure 3 Molecular structure of [PhB(AdIm)3Fe(N2)] (2)

图 4 配合物[PhB(AdIm)3Fe(N2)] (2)在0.05 mol•L-1 [(nBu)4N][BPh4]的四氢呋喃溶液中的循环伏安图
Figure 4 Cyclic voltammogram of [PhB(AdIm)3Fe(N2)] (2) measured at room temperature in THF with 0.05 mol•L-1 [(nBu)4N][PF6] as electrolyte

图 5 配合物4的阴离子[PhB(AdIm)3Fe(N2)]-结构
Figure 5 Structure of the anion [PhB(AdIm)3Fe(N2)]- in 4
表 1 Fe-N2配合物2和4的主要结构数据
Table 1. Key structure data of 2 and 4
| 2 | 4 | |
| N—N/nm | 0.1046(5) | 0.1149(11) |
| Fe—N/nm | 0.1845(4) | 0.1735(7) |
| Fe—C/nm | 0.2076(4) | 0.1982(9) |
| 0.2071(4) | 0.1994(9) | |
| 0.2037(4) | 0.1947(9) | |
| C-Fe-N | 122.1(2) | 121.6(4) |
| 125.3(2) | 130.6(4) | |
| 122.2(2) | 116.2(4) | |
| B-Fe-N/(°) | 178.2 | 172.5 |
 下载: 导出CSV
下载: 导出CSV
表 2 Fe-N2配合物的质子化反应生成肼和氨的产率a
Table 2. Yields of hydrazine and ammonia formed in the protonation reactions of iron-N2 complexes
|
|||
| Fe—N2 | HX | Yield/% of N2H4 | Yield/% of NH3 |
| 2 | HClb | 2(1) | <1 |
| 2 | HOTf | 4(2) | <1 |
| 4 | HClb | 9(3) | 5(3) |
| 4 | HOTf | 5(3) | 3(2) |
| [(ICy)3Fe(N2)][BPh4] | HCl | 2(1) | <1 |
| [(ICy)3Fe(N2)][BPh4] | HOTf | 9(2) | <1 |
| [(ICy)3Fe(N2)] | HCl | 2(1) | <1 |
| [(ICy)3Fe(N2)] | HOTf | 18(2) | 2(1) |
| a Yields are relative to per mol iron, and are the averaged data of 2 runs. b 7.6 mol•L-1 HCl in Et2O. | |||
下载: 导出CSV
表 3 铁-氮杂环卡宾配合物催化氮气硅基化a
Table 3. Catalytic performance of iron-NHC complexes in catalytic silylation of N2
|
|||
| Entry | Cat. | TON | Yield/% of N(SiMe3)3 |
| 1 | 1 | 83(11) | 12(2) |
| 2 | 2 | 84(21) | 12(4) |
| 3 | 4 | 87(11) | 13(3) |
| 4 | [(ICy)3Fe(N2)] | 35(2) | 5(1) |
| 5 | [(ICy)3Fe(N2)][BPh4] | 34(2) | 5(1) |
| a The reactions were run using 0.005 mmol of the catalyst in 10 mL of Et2O with the addition of the reducing reagent prior to catalyst. TONs and yields were the averaged data of three runs. b Turnover numbers were calculated as the molar ratio of N(SiMe3)3 to iron. c Yields of N(SiMe3)3 were based on Me3SiCl and determined by GC with cyclododecane as the internal standard. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: