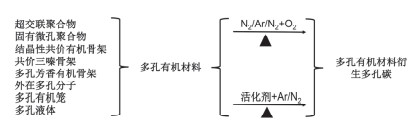
图式 1.
典型的制备多孔有机材料衍生的多孔碳的流程示意图
Scheme 1.
Typic synthesis procedure for porous organic materials derived porous carbon

化石燃料的大量消耗, 引起的环境污染问题以及能源短缺问题日益突出, 寻找并开发清洁、可持续利用的新能源, 是行之有效的解决方法[1].多孔碳材料是一类具有不同尺寸孔结构的碳材料, 具有较高的比表面积, 较大的孔体积, 杰出的物理化学稳定性, 优异的机械性能等优点, 可以广泛应用于气体的存储[2]、分离[3]、锂离子电池[4]、燃料电池[5]等领域.
制备多孔碳的原料来源广泛, 常见的有生物质[6, 7], 高分子聚合物[8], 金属有机骨架[9~12], 多孔有机材料[13]等.多孔有机材料作为制备多孔碳的原料之一, 主要由C、H、O、N等轻质元素组成.较高的比表面积, 良好的稳定性, 以及自身较高的碳含量, 使得多孔有机材料很适合作为制备多孔碳的前驱体.而且, 与其他常见的多孔碳来源相比(例如生物质), 多孔有机材料自身结构的可调控性, 精确的化学计量比, 使其衍生的多孔碳的结构更容易控制[14], 且重复性更好.同时, 多孔有机材料种类丰富, 包括超交联聚合物(HCPs)[15, 16], 固有微孔聚合物(PIMs)[17, 18], 结晶性共价有机骨架(COFs)[19, 20], 共轭微孔聚合物(CMPs)[21~23], 共价三嗪骨架(CTFs)[24, 25], 多孔芳香有机骨架(PAFs)[26~28], 外在多孔分子[29~31], 多孔有机笼[32, 33], 多孔液体[34]等, 可以根据不同的需求制备不同功能的多孔碳材料.此外, 多孔有机材料不仅可以作为制备多孔碳的前驱体, 还可以作为模板, 额外引入碳源, 进而制备出性能更加优异的多孔碳.
多孔碳材料自身的结构性质可以影响其应用, 不同的制备方法得到的多孔碳形貌, 孔结构各不相同.常见的由多孔有机材料制备多孔碳的方法主要可以分为两种:一种是非活化法碳化多孔有机材料, 即直接在惰性氛围下碳化多孔有机材料, 不添加任何活化剂, 通常非活化法碳化多孔有机前驱体制备多孔碳的方法, 被认为是一种简单、高效、环保的方法, 适用于大规模制备多孔碳材料; 另一种是将多孔有机材料与活化剂混合, 然后在惰性氛围内, 适当的温度下进行碳化制备多孔材料(图式 1是由多孔有机材料碳化制备多孔碳的方法).与非活化碳化多孔有机材料制备多孔碳的方法相比, 向多孔有机材料中添加活化剂制备的多孔碳材料一般具有相对较高的比表面, 孔尺寸分布更均一, 使其在能源存储, 能量转化领域有很好的应用.
本文综述了最近由多孔有机材料制备多孔碳的方法, 并且介绍了多孔有机材料衍生的多孔碳材料在气体吸附、存储、分离、锂离子电池、锂硫电池、燃料电池等方面的应用.文章的最后, 结合当前的研究状况, 提出了多孔有机材料衍生的多孔碳材料所面临的机遇与挑战, 并展望了多孔有机材料衍生的多孔碳材料的未来, 希望对多孔有机材料制备多孔碳材料的发展起到一定的推动作用.
非活化法是指在适当的温度条件下, 在惰性氛围下热解多孔有机材料制备多孔碳材料.非活化法碳化相对更加绿色环保, 经济; 缺点是非活化法碳化需要较高的温度, 并且产率较低.
非活化碳化法主要分为以下几部分: (1)制备相应的多孔碳前驱体(多孔有机材料); (2)程序升温在惰性氛围下进行热解.有时根据实验需要适当引入少量空气(或氧气)可以降低碳化温度[35].
利用非活化法碳化多孔有机材料, 科研人员制备了很多性能优异的多孔碳材料.
Ben课题组[35]报道的在N2/O2体积比98.5/1.5的氛围下, 通过调节不同的碳化温度(350, 380, 400, 450 ℃), 直接碳化具有较高比表面积的PAF-1, 成功地制备出几种产率较高的多孔碳材料. PAF-1-350, PAF-1-380, PAF-1-400, PAF-1-450的产率分别是95.4%, 90.0%, 80.0%, 62.8%.随着碳化温度的增加, 孔径发生收缩从PAF-1最初的1.4 nm收缩至PAF-1-450的1.0 nm.通过77 K下N2吸附测试计算比表面, 作者发现随着碳化温度升高, 比表面呈下降趋势. PAF-1, PAF-1-350, PAF-1-380, PAF-1-400, PAF-1-450的比表面分别是5500, 4033, 2881, 2292, 1191 m2•g-1, 前四种材料的吸附曲线均有滞后环, 说明这四种材料骨架部分存在柔性特征, 然而PAF-1-450的吸附曲线是明显的I型吸附曲线, 说明随着碳化温度的增加, 材料中的柔性部分消失.有意思的是, 这种孔尺寸的收缩更利于对CO2的吸附.作者发现PAF-1-450在273 K, 1 bar下CO2吸附量可达4.5 mmol•g-1, 而PAF-1只有2.1 mmol•g-1, 作者认为这是由于碳化后, PAF-1-450的CO2吸附热增加, 换句话说, 就是碳化后增强了主体材料与客体分子间的相互作用力.全碳骨架的表面会形成电场, 与四极矩作用强的CO2有较强的相互作用.同时作者对孔尺寸, 孔体积与CO2的吸附之间的关系进行分析, 作者发现虽然PAF-1-450的微孔体积(0.35 cm3•g-1)低于PAF-1 (1.44 cm3•g-1), 但PAF-1-450的CO2的吸附表现却更佳.这是因为PAF-1-450的微孔体积主要是0.8 nm至1.2 nm的微孔组成, 而PAF-1, 因为合成中的缺陷使其具有较宽的孔分布, PAF-1的微孔体积主要是1.2 nm至2.0 nm的微孔组成.在室温条件下与PAF-1相比, 由较小的微孔组成的PAF-1-450可以更高效地存储超过两层的CO2.通过直接碳化PAF-1, 作者发现制备出具有适当的孔尺寸和较高的吸附热的多孔碳材料, 可以显著地提高CO2吸附能力.随后作者利用理想吸附溶液理论(IAST)计算CO2/N2选择性高达209 (CO2/N2体积比15/85条件下).良好的气体吸附剂不仅应具有较高的吸附能力, 良好的物化稳定性对于实际应用同样很重要.作者通过热重分析发现PAF-1-450热失重5%在471 ℃, 此外PAF-1-450在常见的有机溶剂中不会发生溶解, 例如DMF, 丙酮, 乙醇等溶剂.即使在1 mol/L HCl中煮沸7天, PAF-1-450的多孔性依然可以保持.以上的优点充分说明PAF-1-450可以作为良好的、高效的气体吸附剂.
随后, Ben课题组[36]通过调节不同的碳化温度(700, 800, 900 ℃), 直接在氩气氛围下碳化含氮的多孔有机骨架JUC-Z2, 成功地制备了一系列的氮掺杂的多孔碳材料, 其中JUC-Z2-900表现出优异的气体吸附能力, 尤其是在273 K, 1 bar下CH4的吸附量可达2.7 mmol•g-1, 同时, 273 K下, JUC-Z2-900的CO2/H2选择性高达66.作者认为JUC-Z2-900具有较好的气体吸附能力, 主要是源于以下几个原因:经过碳化含氮的JUC-Z2制备的多孔碳中含有呈碱性的N, 可以与呈酸性的CO2之间有相对较强的吸引力; 碳化后, 材料的孔尺寸会从最初的1.18 nm收缩至0.60 nm左右, 通过之前的一些报道可知, 稍大于探针分子的动态分子直径, 会使孔壁与探针分子之间相互作用增强.对于低压下有较好的吸附H2能力, 主要是因为碳化得到的材料H2吸附热增加, H2吸附热的增加可能是由于碳化后材料较窄的孔尺寸分布和N掺杂的缘故.对于吸附CH4来说, 材料的孔尺寸大小与一个或者两个CH4动力学直径(0.38 nm)相匹配是最佳的.综上可知碳化含N的JUC-Z2制备的JUC-Z2-900, 具有较高的气体吸附能力及气体选择性, 这使其有较好的应用前景.
非活化法碳化不只是直接将多孔有机材料作为碳源(前驱体), 还可以将多孔有机材料作为模板, 额外引入碳源, 在惰性氛围下碳化制备多孔碳材料.通过引入额外的碳源, 碳化后得到的多孔碳的孔径更加狭小, 更利于气体分子与孔壁相互作用[37, 38], 可以显著地提高气体的吸附能力.
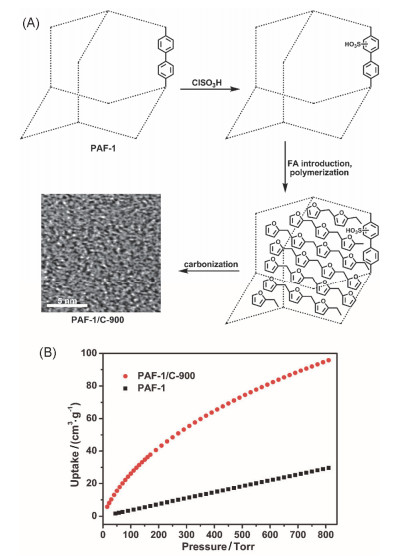
2013年, Ma课题组[37]将PAF-1进行磺酸化修饰, 然后将其浸泡在呋喃甲醇中, 利用磺酸的催化作用, 使呋呋喃甲醇在PAF-1的孔道中聚合.最后经过高温惰性氛围下直接碳化得到PAF-1/C-900(图 1). 295 K, 1 bar下的PAF-1/C-900可以吸附CO2高达4.1 mmol•g-1, 优于相同条件下的PAF-1-450.这是因为引入额外碳源, 经过高温碳化后制备得到超微孔(约0.54 nm)的碳材料, 较小的孔可以增强与CO2的相互作用, 使其CO2吸附能力显著增强.
活化法是指碳化的过程中加入相应的活化剂, 经过碳化制备多孔碳材料.根据活化剂的不同可以分为物理活化和化学活化, 有时也可将物理与化学活化相结合.
物理活化制备多孔碳材料主要是在热解过程中, 在惰性氛围下, 同时加入所需要的物理活化试剂.其中常见的物理活化剂包括水蒸汽, 二氧化碳等.与化学活化剂相比, 物理活化剂污染小, 后处理方法相对简单, 但物理活化的产率不高, 而且物理活化过程常常需要较高的温度进行碳化.
物理活化的过程是利用气体分子与碳发生反应.例如, 水蒸气活化的机理: C+H2O=H2+CO, C+2H2O=2H2+CO2; CO2活化机理: C+CO2=2CO, 将小孔中的“堵塞物”除去, 然后进一步活化, 打开小孔, 同时使原来的未堵塞的通孔变得更开放, 伴随着碳骨架的重排, 选择性的形成一些微孔组织.
化学活化主要是通过向主体材料中引入一定量的活化剂, 常见的是将主体材料与活化剂进行搅拌或超声, 使其分散均匀, 然后经过热解得到多孔碳材料.与物理活化相比, 化学活化制备多孔碳的产率高, 碳化温度相对较低, 得到的多孔碳比表面积较高.但是碳化后的材料通常需要用适当酸性的酸进行后处理, 以便除去残留在材料中的金属氧化物, 防止较大的或者团聚的金属氧化物堵塞孔道.同时, 化学活化剂对设备腐蚀性较大, 从某种程度上来说这增加了应用成本.
常见的化学活化剂主要有氢氧化钾、氯化锌、硝酸镁、氢氧化钠、柠檬酸钾等, 其中氢氧化钾是最常用的化学活化剂, 以氢氧化钾作为活化剂碳化制备的多孔碳材料, 一般具有相对较高的比表面积, 产率较高, 并且氢氧化钾作为硬模板, 可以适度的造孔[39~42].氢氧化钾在活化过程中会与碳发生反应(6KOH+2C=2K2CO3+3H2+2K), 可以降低反应的活化能.
常见的制备多孔碳的方法有硬模板法, 软模板法.硬模板法制备多孔碳是一种制备有序多孔碳的方法, 通过选用不同的模板可以制备出不同孔尺寸的有序的多孔碳.常用的硬模板有分子筛, 介孔硅, 胶体粒子等.制备出所需结构的多孔碳后, 需要将模板移除, 因为硬模板多是无机材料, 需要用到酸碱蚀刻模板, 这无疑会对环境造成一定程度的污染.另一种用于制备有序多孔碳的方法是软模板法, 该法是将结构导向剂与有机碳前驱体组装, 然后碳化得到所需多孔碳.该方法的优点是不需要移除模板, 但是对模板的热稳定要求很高, 否则高温下模板剂会发生分解.与这两种模板法相比, 碳化稳定性较好的, 含碳量较高多孔有机材料制备多孔碳, 不仅产率高, 而且孔尺寸大小可控, 实验重复性更高, 对环境更友好.碳化自身含有杂原子的多孔有机材料还可以制备掺杂的多孔碳, 无需额外引入掺杂剂, 这种掺杂的多孔碳在多个领域都有很好的应用.值得注意的是, 杂原子的掺杂, 例如氮, 随着碳化温度的增加, 氮会挥发, 所以制备氮杂的多孔碳需要合理地选择碳化条件.多孔有机材料因为自身的苯环较多, 骨架刚性较好, 得到的多孔碳物化稳定性都较好, 并且不溶于常见的有机溶剂.
多孔有机材料衍生的多孔碳材料具有较高的比表面积、优越的导电性和导热性等优点, 在气体吸附[43~53]、分离[35]、存储[39, 54]、超级电容器[55~60]、锂离子电池[41]等领域有着广泛的应用.
近年来, 由于化石燃料的大量燃烧造成的环境污染以及能源短缺等问题日益加剧, 已经严重地影响了人们的正常生活, 因此, 寻找高效的吸附剂用于温室气体的捕获与封存, 以及开发利用H2、CH4等新型清洁能源, 缓解能源危机已经迫在眉睫.碳化多孔有机材料制备的多孔碳材料, 骨架表面会形成电场[35, 36, 39], 可以增强与气体分子间的相互作用, 提高材料的气体吸附能力.结合其具有较高的比表面积等优点, 使多孔有机材料衍生的多孔碳被广泛地应用于气体吸附、分离及存储.
2012年, Ben课题组[35]报道了在N2与O2混合气氛下直接碳化PAF-1, 得到四种相对产率较高的多孔碳材料.随着碳化温度的增加, 多孔碳材料的比表面积会下降.对于含有纯碳骨架的PAF-1-450来说, 碳化后可以在其骨架表面形成一个电场, 增加了与CO2之间的亲和力.经过碳化后孔径由PAF-1之前的1.4 nm收缩到1.0 nm, 收缩后的孔增加了CO2与多重孔壁之间的碰撞. 273 K, 1 bar的条件下PAF-1-450的CO2吸附可达4.5 mmol•g-1, 明显高于相同条件下的PAF-1.利用克劳修斯-克拉柏龙方程计算得到CO2的吸附热为27.8 kJ• mol-1, PAF-1只有15.6 kJ•mol-1, 充分说明碳化后增强了吸附剂与吸附质之间的相互作用.利用理想吸附溶液理论(IAST)计算得到在体积比为15/85的条件下CO2/N2的选择性为209;在体积比为15/85和压力范围0<p<40的条件下, CO2/CH4的选择性为7.8~8.9, 这种高的选择性, 使得PAF-1-450完全可以应用于垃圾填埋时的气体分离; 在273 K, 1 bar的条件下, 体积比为20/80的CO2/H2的选择性高达392, 以上实验证明PAF-1-450在CO2的捕获与封存以及分离方面有应用前景(图 2).

另一种多孔有机材料JUC-Z2, 因为其骨架中本身含有氮原子, 氮原子呈碱性, 碳化后相当于原位掺杂氮[35], 利于对酸性CO2气体分子的吸附, 碳化后极大地提高了CO2的吸附量.其中JUC-Z2-900在273 K, 1 bar下CO2的吸附量高达5.0 mmol•g-1, 这一数值高于目前所报道的大多数多孔碳材料, 表明其在CO2的存储与封存方面有着潜在的应用价值.
2015年, Feng等[43]报道的在惰性氛围下直接碳化含B、N的交联聚合物, 制备得到产率很高的B/N共掺杂的多孔碳.通过X射线光电子能谱检测B的含量约为3.21%, N的含量约为5.72%, 结合其较高的比表面的优点, 使其在273 K, 1 bar表现出不错的CO2吸附能力(3.25 mmol•g-1).此外, 在298 K下CO2/CH4也有着较好的分离选择性.
2013年, Ben等[39]的报道, 经过高温热解KOH活化后的PAF-1, 可以得到一系列的具有双孔分布的多孔碳材料(0.6 nm和1.2 nm).其中K-PAF-1-750具有最高的BET比表面积(2926 m2•g-1, 孔体积是1.14 m3•g-1).因为K-PAF-1-750具有较高的比表面积和1 nm左右的微孔特点, 使其不论在低压吸附还是高压存储方面都有杰出的表现. 77 K, 1 bar下K-PAF-1-750可以吸附H2高达3.06 wt%; 高压气体存储实验表明其在298 K, 40 bar下可以存储CO2 30 mmol•g-1; 298 K, 35 bar下存储CH4可达12.9 mmol•g-1; 77 K, 48 bar下存储H2达到6.68 wt%. K-PAF-1-750高压下对于CO2和CH4的存储高于最初的PAF-1, 同时K-PAF-1-750是第一例在低压吸附和高压存储条件下都有很好表现的材料.作者认为氢氧化钾在热解过程中不仅可以作为活化剂, 它自身也可以作为硬模板阻止孔的坍陷.多孔碳结构中的0.6 nm的孔来源于氢氧化钾的造孔作用, 而1.2 nm的孔是PAF-1本身的孔(1.4 nm)的收缩形成的. 0.6 nm的孔利于低压下的气体吸附, 1.2 nm的孔利于高压下的气体存储.因为K-PAF-1-750较高的比表面积和独特的双孔分布特点, 使其在能源的存储方面有非常好的表现.
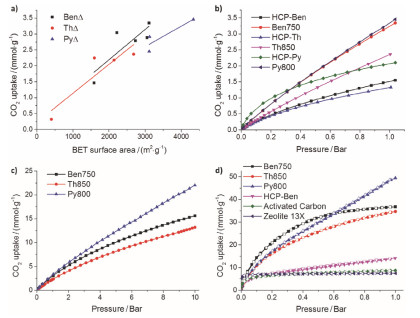
Cooper课题组[54]在2016年报道的利用氢氧化钾活化HCPs, 得到了Ben750, Th850, Py800三种多孔碳材料, 比表面积分别是3105, 2682, 4334 m2•g-1.与初始的多孔有机材料前驱体相比, 比表面积有着明显提高(HCP-Ben, 1382 m2•g-1; HCP-Th, 484 m2•g-1; HCP-Py, 322 m2•g-1), 其中Py800是目前报道的多孔有机材料衍生的多孔碳材料中比表面积最高的材料.对于三种材料在298 K下, 1到10 bar下的CO2的吸附测试, 发现CO2的吸附量基本与三种碳材料的比表面积成正比, 即CO2吸附能力, Py800>Ben750>Th850.并且在10 bar下, 三种材料对CO2的吸附并没有饱和, 换句话说, 增加压力还可以存储更多的CO2. 77 K, 1 bar下, Py800有最好的H2吸附能力, 可以吸附3.6 wt%.在77 K, 10 bar下Py800可以存储5.6 wt%的H2, 这个值超过了当前报道的大多数多孔碳材料(图 3).
Xu课题组[50]报道, 将COF-5与ZnCl2混合研磨, 在氩气氛围下分别在700, 900, 1000 ℃下碳化3 h, 得到粗产品, 用0.1 mol/L HCl和水洗掉残余的ZnO和B2O3, 干燥得到硼掺杂的具有微孔-介孔-大孔的多级孔碳材料.因为所得的材料中含有硼, 可以增加主体材料与H2之间的结合力, 所以该法制备的多孔碳有很好的H2存储能力.
碳化多孔有机材料制备的多孔碳, 具有良好的物化稳定性, 较高的比表面积, 丰富的孔结构等优点, 使其在气体的吸附、分离、存储领域有很好的应用前景.但是高效地吸附气体对多孔碳来说仍然是一种挑战.根据之前的报道, 可以发现作为有效的气体吸附剂, 多孔碳应具备以下特征: (1)含有较多的较窄的孔.根据Ben等[35]在研究碳化PAF-1制备多孔碳用于气体的吸附、分离应用时, 发现较小的微孔(小于1 nm)有利于低压下的CO2等气体吸附, 这是因为吸附剂的孔尺寸与CO2动力学直径可以更好的匹配, 增加CO2与孔壁的相互作用.例如, Ben等[27]报道的PAF-1在高压下表现出很好的气体存储能力, 但是低压下吸附能力较弱, 这是因为高压下气体的存储与材料的比表面和孔体积有关, 而低压下气体的吸附主要与孔尺寸小于1 nm的微孔的含量有关, 而PAF-1的微孔主要集中在1~2 nm.对于氢氧化钾活化碳化PAF-1制备的多孔碳材料的储存气体研究[39], 可知通过使用活化剂与骨架本身的碳发生反应, 得到一些超微孔(约0.6 nm), 使其在低压下可以有效地吸附CO2等气体, 同时材料中自身收缩的微孔(1.2 nm)可以有效地扩散气体.对于H2而言, 低温、低压下的H2吸附与CO2类似, 较小的孔可以接触较多的H2. (2)适当的气体吸附热.通常气体的吸附、分离、存储效果不仅与吸附剂自身的孔尺寸有关.吸附热是另一重要影响因素.较高的吸附热意味着主客体之间有较强的相互作用.通过之前的一些报道可知, 多孔碳材料以及杂原子掺杂的多孔碳相对于初始的前驱体来说, 气体的吸附热一般都会增加, 这意味着主体材料与气体分子之间的相互作用增强.通常高的吸附热有益于气体的吸附过程, 低的吸附热有益于脱附过程.
随着经济的快速发展, 化石燃料的大量消耗导致的能源短缺与环境污染问题日益严重.新能源的开发与能源的存储将是解决能源问题行之有效的方法.
电化学电容器又称超级电容器, 是一种高效且无污染的新型储能设备.超级电容器具有功率密度高, 充电速度快, 能量转换效率高, 使用寿命长, 安全无污染等优点, 被广泛地应用于电子通讯, 新能源汽车以及航天航空等领域.
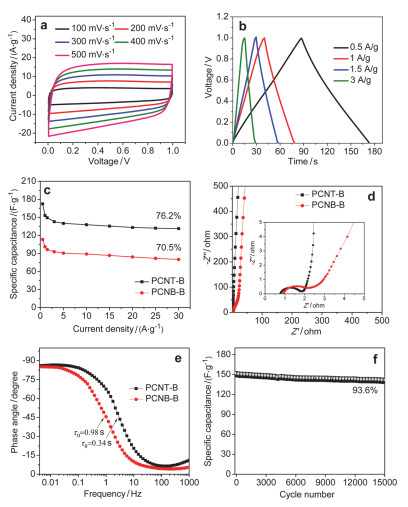
电极材料是超级电容器的关键组成部分, 电极材料性能的好坏也将直接影响超级电容器的性能.多孔碳材料作为超级电容器的电极材料已有半个世纪的历史了, 其中, 较高的比表面积, 合理的孔分布是影响超级电容器容量的重要因素.因此, 具有高的比表面和可调控的孔径分布的多孔有机材料衍生的多孔碳在超级电容器中具有广阔的应用前景. Jiang课题组[55]报道的在高温下碳化低成本管状的HCPs, 直接制备出高质量的多孔碳纳米管, PCNTs(如图 4所示). PCNTs具有一维孔道, 比表面积可以达到1000 m2•g-1. PCNTs的前驱体HCPTs可以通过搅拌加热、溶剂热、超声等方法合成, 前驱体的合成与其他的多孔有机材料的合成相比, 相对简单.而且可以通过控制芳香单体的尺寸, 从而控制碳管的直径.因为PCNTs自身的多孔性以及较高的比表面积, 作者探究了该材料作为超级电容器的潜质, 在6 mol/L KOH中, 0.5 A•g-1电流密度下, 比电容可达172 F•g-1, 并且经过15000次循环后, 比容量仍可保留93.6%, 表明该材料具有较好的稳定性, 适用于电化学电容器(图 5).

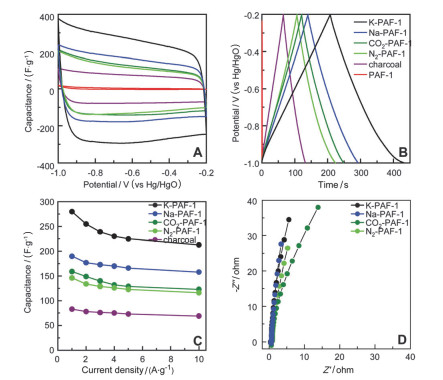
结合K-PAF-1-750高比表面积, 高的孔体积的优点, Ben课题组[40]探索了K-PAF-1-750在超级电容器领域的应用.如图 6所示, 通过活化法碳化后制备出N2-PAF-1, CO2-PAF-1, Na-PAF-1, K-PAF-1.在相同的碳化温度下, K-PAF-1-750比表面积最高, 这是因为在碳化过程KOH起到重要的作用.在6.0 mol/L KOH电解液中, 扫速为10 mV•s-1的循环伏安测试条件下, K-PAF-1-750的比电容量高达260 F•g-1, 与物理活化后得到的N2-PAF-1, CO2-PAF-1相比, 电容几乎提高一倍, 这是因为经过KOH活化后得到的K-PAF-1-750微孔孔体积很高(1.14 cm3•g-1), 几乎是其他材料(N2-PAF-1, CO2-PAF-1, Na-PAF-1)的二倍还多, 通过静电相互作用, 可以存储更多的电荷.作者通过选择阳离子尺寸不同有机电解液, 发现电解质堆积的密度会直接影响低电流密度下的电容值; 在高电流密度下, 电容存储能力主要受到扩散效应的影响.
2013年Banerjee课题组[38]报道的以含氮的PAF-6作为模板, 然后室温下, 氮气保护下, 将PAF-6浸泡在呋喃甲醇中24 h. 90 ℃保温24 h, 得到FA@PAF-6.然后将其在150 ℃下加热8 h使呋喃甲醇聚合.用无水乙醇洗去表面附着的呋喃甲醇.重复上述实验操作一次, 使PAF-6可以担载最大量的呋喃甲醇.然后在1000 ℃(或者800 ℃)下, 氩气氛围下碳化3 h, 得到POF-C-1000(或POF-C-800).石墨化程度较高的POF-C-1000的比表面积是785 m2•g-1, POF-C-1000具有较高的含氮量, 约为6 wt%.电化学测试发现POF-C-1000有很高的起始电位, 大约是40 mV, 高于商业利用的的Pt/C(20 wt%)电极(0.1 mol/L KOH溶液中, 扫速是50 mV•s-1).线性扫速伏安测试中, 0.1 mol/L KOH溶液中, 扫速是10 mV•s-1, POF-C-1000的电流密度是5.2 mA•cm-2, 略低于Pt/C电极.因为材料中含有杂原子, 作者探究了POF-C-1000在低温低压条件下的H2和CO2的吸附, 发现都有不错的表现.这说明通过原位碳化含氮的多孔有机材料可以制备多种功能化的多孔碳材料.
多孔有机材料衍生的多孔碳材料还可以应用于锂离子电池.作为锂离子电池正极材料, 多孔有机材料衍生的多孔碳材料应具备以下几个特点: (1)电极材料的电性能应该稳定, 即在锂离子的进入与脱出过程中, 自由能基本不变; (2)扩散效率高; (3)嵌入过程应可逆; (4)导电率高; (5)热稳定好, 并且不与锂离子发生化学反应.
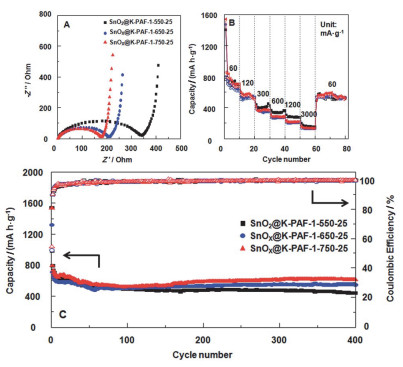
Ben课题组[41]将氢氧化钾活化的PAF-1作为主体材料, 与锡和氧化锡复合, 得到表现良好的锂离子电池正极材料.其中SnOx@K-PAF-1-750-25展现出较高的稳定性, 经过充放电循环400次, 电池容量仍可以保持在608 mAh•g-1.即使在很高的电流密度下, 电池容量也可以保持在400 mAh•g-1.实验证明通过碳化微孔PAF-1得到多孔碳材料具有独特的双微孔结构, 可以有效地容纳SnO2和Sn/SnO2纳米粒子, 有效地抑制在充放电过程的SnO2和Sn/SnO2纳米粒子体积的膨胀和收缩, 并且可以阻止纳米粒子在长效循环中粉碎和聚集.同时金属离子嵌入在K-PAF-1中, 可以保障即使在更高的碳化温度下, 仍然可以保持这个复合材料结构的完整性(图 7).
在过去的几十年, Li-S电池被认为是最具潜质的电化学能源设备, 因其具有很高的理论容量(1650 mAh• g-1), 高的能量密度(2600 Wh•kg-1), 低成本, 低污染的优点, 吸引了科研人员的研究兴趣.将多孔有机材料衍生的多孔碳材料与硫复合, 应用于Li-S电池, 可以极大地提高锂硫电池的循环稳定性能和倍率性能.
Li课题组[61]在氩气氛围下, 500 ℃碳化PAF-49得到了多孔碳材料CPAF.将CPAF与硫复合可以应用于Li-S电池的负极. CPAF可以担载75 wt%的硫, 高的硫含量使得CPAF初始容量可以达到1211 mAh•g-1.在0.5 C下经过1000次循环后, 容量衰减只有0.074%, 这些实验结果表明CPAF复合材料是很好的硫支持体, 可以改善Li-S电池的循环性能.
此外, 包含杂原子的多孔碳材料, 可以有效地替代重金属催化剂作为电化学氧化还原反应中(ORR)的电催化剂[38, 62, 63].
Dai课题组[62]通过选择不同的含N单体, 设计合成了一系列的共价有机聚合物, 并以此作为前驱体材料, 碳化后制备了一系列可以控制氮位置的中空石墨化的多孔碳材料, 实验结果表明这几种材料在ORR反应中有不错的表现.通过线性扫描伏安测试, C-COP-4展示出最好的ORR活性. C-COP-4的起始电位与商用的Pt/C电极的起始电位相近, 并且C-COP-4半波电位可以达到0.78 eV.这说明通过碳化含杂原子的多孔有机材料制备含有杂原子的石墨化的多孔碳材料可以作为ORR反应的电催化剂.作者认为这是因为N的电负性强于C, 并且通过DFT理论计算部分电荷强度, 表明四种多孔碳材料中主要是通过N提供电子, 并且C-COP中因为含有三嗪基团, 结合其平面π共轭特点, 与文中报道的其他三种材料相比, 其有着最好的电催化表现.并且与现在商用的Pt/C电极相比, 也毫不逊色.
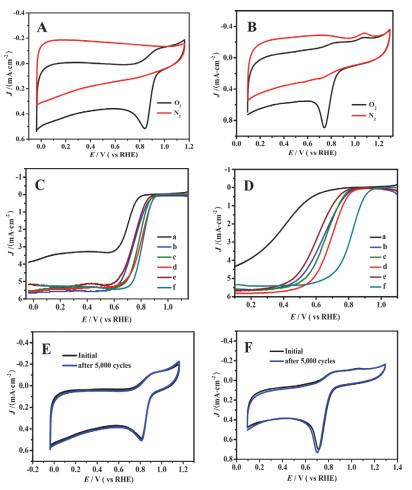
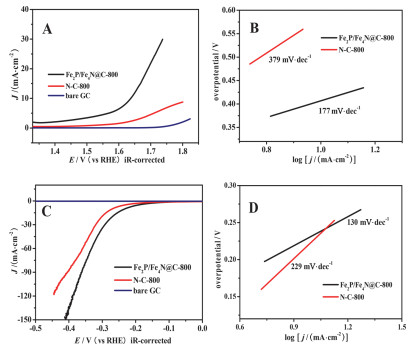
最近, Shan课题组[63]通过选用含有铁原子的四(对溴苯基)卟啉与1, 3, 5-苯三硼酸三乙酯通过Suzuki偶联反应制备出一种新的结构中含有卟啉基的COF.将其作为碳化的前驱体, 制备得到含有Fe2P和Fe4N纳米粒子嵌入的氮掺杂的多孔碳(Fe2P/Fe4N@N-doped carbons).因为前驱体中含有Fe, 直接在高温碳化下4 h (600~900 ℃), Fe的催化作用使Fe2P/Fe4N@N-doped carbons石墨化程度很高.在ORR的电化学测试中(图 8), 作者发现Fe2P/Fe4N@C-800(800 ℃下, 氮气氛围下碳化4 h制备的多孔碳)在碱性电解质中, 半波电位可以达到0.80 eV; 酸性碱性电解质中, 半波电位是0.68 eV, 与商用的Pt/C电极半波电位相近.同时该材料在水分解反应中[水分解反应包含两个半反应:析氢反应(HER)和析氧反应(OER)]也有不错的表现.在O2饱和的1 mol/L KOH溶液中, 进行OER反应, Fe2P/Fe4N@C-800表现出不错的电催化活性, 在1.74 V时, 有较高的电流密度是30 mA•cm-2; 在1.64 V时, 电流密度是10 mA•cm-2.经过计算Tafel斜率是177 mV•s-1, 这说明Fe2P/Fe4N@C-800是适用于OER反应的, 并且Fe2P/Fe4N@C-800可以与磷基的OER催化剂相媲美.此外, 在0.5 mol/L H2SO4电解液测试HER反应, Fe2P/Fe4N@C-800达到电流密度是10 mA•cm-2, 过电位只有-232 mV, Tafel斜率是130 mV•s-1 (见图 9).
CMP因其自身的共轭结构, 高的表面, 表面的可修饰性广泛地被用于气体吸附[21~23], 催化[64], 碘吸附[65]等领域.最近Liao等[66]设计利用含有咔唑基的有机单体聚合, 成功制备出新型的微孔材料, 引起广泛的关注.这三种新型的多孔有机材料自身含有丰富的氮, 比表面较高, 物化稳定性良好, 可以作为前驱体, 直接在惰性氛围下热解制备得到三种多孔碳, 分别是PTCT-C, PTCA-C, PBCP.因为前驱体自身骨架较刚性, 所以制备的碳产率非常高.作者对六种材料分别进行低压下的CO2吸附测试, 发现碳化后的材料的CO2吸附能力明显提高, 在相同测试条件下, 六种材料中PTCT-C有最好的CO2吸附能力.作者详细地解释了CO2吸附能力不同的原因. (1)PTCT-C含有较多的氮, 通过路易斯酸碱相互作用可以有助于吸收更多的CO2. (2)主体材料丰富的微孔组织有利于增加CO2与孔壁的相互作用. (3)适当的吸附热, 说明主客体之间的相互作用很强, 利于吸附更多的CO2.通过碳化聚咔唑制备的多孔碳材料在超级电容器应用方面也有很出色的表现.通过改变扫速, 循环伏安测试中均可以得到近似于正方形的曲线, 说明材料有很好的电容性能, 虽然曲线稍有起伏, 这是因为存在赝电容行为.结合恒电流充放电测试, 可以发现材料PTCT-C有最好的电容存储能力, 比电容高达558 F•g-1, 这是因为PTCT-C具有较高的比表面(1280 m2•g-1)和较高的氮含量(6.07 wt%), 此外, PTCT-C 0.7 nm的微孔可以有效地被填充, 从而形成双电层.良好的长效循环能力是实际应用的关键因素, PTCT-C在电流密度是2.0 A•g-1条加下, 循环1000次, 电容量仍可以保持在95%, 这使得PTCT-C完全可以作为电极材料应用于超级电容器等电化学能量转化领域.
多孔碳因其较高的比表面, 良好的稳定性, 可及的孔等优点, 已经被广泛地应用于超级电容器等能量转化领域.合理的设计并开发更多更好的多孔碳应用于能量转化领域是十分必要的.对于超级电容器来说, 主要可以分为两类, 而多孔碳主要用于电化学双层电容器(EDLCs)的电极材料.根据公式C=εrε0A/d (εr:电解液介电常数; ε0:真空介电常数; A:电极的面积; d:有效的双层厚度)可知, 理想的条件下EDLCs的比电容量主要与材料的比表面, 孔尺寸有关.合理的调节孔工程对于提高EDLCs的容量很有必要.如Ben等报道的一样, 当电极材料的孔尺寸大小与电解质相匹配时, 小孔可以有效地存储能量, 同时稍大的孔可以有效地传输, 这种协同作用可以有效地提高EDLCs的比电容量.碳化多孔有机材料制备的多孔碳材料应用于锂电池领域是当前的研究热点.这是因为多孔有机材料作为碳前驱体可以提高碳的产率, 同时利用多孔材料的“孔”的优势可以有效地限域活性成分, 并且可以在锂离子的嵌入和脱出的过程中起到缓冲作用.例如在高密度的锂硫电池研究中, 科研人员尝试过多种策略去提高锂硫电池的性质, 但是只有有限的几种策略是成功的, 因为科研人员发现限域效应是锂硫电池最重要的因素, 因此主体材料的孔特征对于锂硫电池的性能十分重要.通过使用具有可控的形貌的多孔碳作为基质, 将硫封装在其孔中, 可以有效的防止多硫化物流出并进入电解液中.同时纯碳的骨架表面形成的电场可以与极性的聚硫离子有很强的相互作用.丰富的微孔结构, 内部较大的空间以及内部交叉的微孔结构可以均匀地分散硫分子和聚硫离子, 从而对锂硫电池的性能有很好的提升.
近年来, 随着工业的发展, 环境污染问题和能源短缺问题引起全人类的关注, 急需找到有效的降低环境污染问题的方法.多孔有机材料衍生的多孔碳材料具有相对较高的比表面积, 可调控的孔结构, 可以作为气体吸附剂存储清洁能源; 高的热稳定性, 良好的导电性, 可以作为电极材料用于能量的转化.
目前, 碳化多孔有机材料衍生的多孔碳, 与气体分子间亲和力较强, 在室温下有很好的气体的吸附和分离能力.通过KOH活化后得到的多孔碳, 具有独特的双孔模型, 较小尺寸的孔可以有效地限域活性成分, 较大尺寸的孔可以快速地传质, 使其在气体吸附, 存储和超级电容器领域拥有广阔的应用前景.此外, 多孔有机材料衍生的多孔碳在锂离子电池, Li-S电池, 燃料电池等领域也有着不错的表现.
然而, 多孔有机材料衍生的多孔碳仍然面临着许多机遇与挑战.
(1) 多孔有机材料作为模板或前驱体, 是制备多孔碳的一个关键环节.如果可以大规模地制备多孔有机材料, 必然会推动多孔有机材料衍生的多孔碳的发展.
(2) 制备方法对于实际应用是至关重要的.开发高效、廉价的制备多孔碳的方法有利于快速实现其工业化.
(3) 如果可以制备具有自活化功能的多孔有机材料, 例如含有钾离子的骨架, 那么碳化的过程中就可以原位提供活化剂, 可以控制活化的位点, 从而得到性能优异的多孔碳材料.
(4) 探索如何利用多孔有机材料制备多级孔碳材料(包括微孔-介孔、微孔-大孔、介孔-大孔、微孔-介孔-大孔复合等), 对实际应用将会大有益处.
(5) 目前, 通过碳化多孔有机材料所制备的多孔碳材料大多是无序的, 限制了其应用.因此, 探索通过多孔有机材料制备有序的多孔碳材料是很有必要的.
Liu, T. Y.; Zhang, F.; Song, Y.; Li, Y. J. Mater. Chem. A 2017, 5, 17705. doi: 10.1039/C7TA05646J
Xia, Y. D.; Yang, Z. X.; Zhu, Y. Q. J. Mater. Chem. A 2013, 1, 9365. doi: 10.1039/c3ta10583k
Kyotani, T. Carbon 2000, 38, 269. doi: 10.1016/S0008-6223(99)00142-6
Yang, Y. F.; Jin, S.; Zhang, Z.; Du, Z. Z.; Liu, H. R.; Yang, J.; Xu, H. X.; Ji, H. X. ACS Appl. Mater. Interfaces 2017, 9, 14180. doi: 10.1021/acsami.6b14840
Yang, K.; Jiang, P.; Chen, J. T.; Chen, Q. W. ACS Appl. Mater. Interfaces 2017, 9, 32106. doi: 10.1021/acsami.7b09428
Dutta, S.; Bhaumik, A.; Wu, K. C. W. Energy Environ. Sci. 2014, 7, 3574. doi: 10.1039/C4EE01075B
Jiang, M.; Zhang, J. L.; Xing, L. B.; Zhou, J.; Cui, H. Y.; Si, W. J.; Zhuo, S. P. Chin. J. Chem. 2016, 34, 1093. doi: 10.1002/cjoc.v34.11
Hong, S. M.; Choi, S. W.; Kim, S. H.; Lee, K. B. Carbon 2016, 99, 354. doi: 10.1016/j.carbon.2015.12.012
Liu, B.; Shioyama, H.; Akita, T.; Xu, Q. J. Am. Chem. Soc. 2008, 130, 5390. doi: 10.1021/ja7106146
张贺, 李国良, 张可刚, 廖春阳, 化学学报, 2017, 75, 841. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346225.shtmlZhang, H.; Li, G. L.; Zhang, K. G.; Liao, C. Y. Acta Chim. Sinica 2017, 75, 841. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346225.shtml
黄刚, 陈玉贞, 江海龙, 化学学报, 2016, 74, 113. doi: 10.3969/j.issn.0253-2409.2016.01.016Huang, G.; Chen, Y. Z.; Jiang, H. L. Acta Chim. Sinica 2016, 74, 113. doi: 10.3969/j.issn.0253-2409.2016.01.016
Pei, X. K.; Chen, Y. F.; Li, S. Q.; Zhang, S. H.; Feng, X.; Zhou, J. W.; Wang, B. Chin. J. Chem. 2016, 34, 157. doi: 10.1002/cjoc.v34.2
Lu, S. L.; Jin, Y. H.; Gu, H. W.; Zhang, W. Sci. China. Chem. 2017, 60, 999. doi: 10.1007/s11426-017-9078-7
Sun, J. K.; Xu, Q. Energy Environ. Sci. 2014, 7, 2071. doi: 10.1039/c4ee00517a
Wood, C. D.; Tan, B. E.; Trewin, A.; Niu, H. J.; Bradshaw, D.; Rosseinsky, M. J.; Khimyak, Y. Z.; Campbell, N. L.; Kirk, R.; Stöckel, E.; Cooper, A. I. Chem. Mater. 2007, 19, 2034. doi: 10.1021/cm070356a
Wood, C. D.; Tan, B.; Trewin, A.; Su, F.; Rosseinsky, M. J.; Bradshaw, D.; Sun, Y.; Zhou, L.; Cooper, A. I. Adv. Mater. 2008, 20, 1916. doi: 10.1002/(ISSN)1521-4095
McKeown, N. B.; Budd, P. M. Chem. Soc. Rev. 2006, 35, 675. doi: 10.1039/b600349d
McKeown, N. B.; Budd, P. M.; Msayib, K. J.; Ghanem, B. S.; Kingston, H. J.; Tattershall, C. E.; Makhseed, S.; Reynolds, K. J.; Fritsch, D. Chem. Eur. J. 2005, 11, 2610. doi: 10.1002/(ISSN)1521-3765
Côté, A. P.; Benin, A. I.; Ockwing, N. W.; O'Keeffe, M.; Matzger, A. J.; Yaghi, O. M. Science 2005, 310, 1166. doi: 10.1126/science.1120411
Uribe-Romo, F. J.; Hunt, J. R.; Furukawa, H.; Klock, C.; O'Keeffe, M.; Yaghi, O. M. J. Am. Chem. Soc. 2009, 131, 4570. doi: 10.1021/ja8096256
Jiang, J. X.; Su, F.; Trewin, A.; Wood, C. D.; Campbell, N. L.; Niu, H.; Dickinson, C.; Ganin, A. Y.; Rosseinsky, M. J.; Khimyak, Y. Z.; Cooper, A. I. Angew. Chem. Int. Ed. 2007, 46, 8574. doi: 10.1002/anie.v46:45
Jiang, J. X.; Su, F.; Trewin, A.; Wood, C. D.; Niu, H.; Jones, J. T. A.; Khimyak, Y. Z.; Cooper, A. I. J. Am. Chem. Soc. 2008, 130, 8875. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
徐佳伟, 张崇, 王永昶, 蒋加兴, 汪锋, 化学学报, 2017, 75, 473. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346022.shtmlXu, J. W.; Zhang, C.; Wang, X. C.; Jiang, J. X.; Wang, F. Acta Chim. Sinica 2017, 75, 473. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346022.shtml
Kuhn, P.; Thomas, A.; Antonietti, M. Macromolecules 2009, 42, 319. doi: 10.1021/ma802322j
Kuhn, P.; Antonietti, M.; Thomas, A. Angew. Chem. Int. Ed. 2008, 47, 3450. doi: 10.1002/(ISSN)1521-3773
Ben, T.; Pei, C. Y.; Zhang, D. L.; Xu, J.; Deng, F.; Jing, X. F.; Qiu, S. L. Energy Environ. Sci. 2011, 4, 3991. doi: 10.1039/c1ee01222c
Ben, T.; Ren, H.; Ma, S.; Cao, D.; Lan, J.; Jing, X.; Wang, W.; Xu, J.; Deng, F. Angew. Chem. Int. Ed. 2009, 48, 9457. doi: 10.1002/anie.200904637
闫卓君, 元野, 刘佳, 李勤, 阮南中, 张大明, 田宇阳, 朱广山, 化学学报, 2016, 74, 67. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345319.shtmlYan, Z. J.; Yuan, Y.; Liu, J.; Li, Q.; Nguyen, N. T.; Zhang, D. M.; Tian, Y. Y.; Zhu, G. S. Acta Chim. Sinica 2016, 74, 67. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345319.shtml
Allcock, H. R.; Siegel, L. A. J. Am. Chem. Soc. 1964, 86, 5140. doi: 10.1021/ja01077a019
Sozzani, P.; Comotti, A.; Simonutti, R.; Meersmann, T.; Logan, J. W.; Pines, A. Angew. Chem. Int. Ed. 2000, 112, 2807. doi: 10.1002/(ISSN)1521-3757
Mastalerz, M.; Oppel, I. M. Angew. Chem. Int. Ed. 2012, 51, 5252. doi: 10.1002/anie.201201174
Tozawa1, T.; Jones, J. T. A.; Swamy, S. I.; Jiang, S.; Adams, D. J.; Shakespeare, S.; Clowes, R.; Bradshaw, D.; Hasell, T.; Chong, S. Y.; Tang, C.; Thompson, S.; Parker, J.; Trewin, A.; Bacsa, J.; Slawin, A. M. Z.; Steiner, A.; Cooper, A. I. Nature Mater. 2009, 8, 973. doi: 10.1038/nmat2545
Chen, L. J.; Reiss, P. S.; Chong, S. Y.; Holden, D.; Jelfs, K. E.; Hasell, T.; Little, M. A.; Kewley, A.; Briggs, M. E.; Stephenson, A.; Thomas, K. M.; Armstrong, J. A.; Bell, J.; Busto, J.; Noel, R.; Liu, J.; Strachan, D. M.; Thallapally, P. K.; Cooper, A. I. Nature Mater. 2014, 13, 954. doi: 10.1038/nmat4035
Giri, N.; Del Pópolo, M. G.; Melaugh, G.; Greenaway, R.; R tzke, K.; Koschine, T.; Pison, L.; Costa Gomes, M. F.; Cooper, A. I.; James, S. L. Nature 2015, 527, 216. doi: 10.1038/nature16072
Ben, T.; Li, Y. Q.; Zhu, L. K.; Zhang, D. L.; Cao, D. P.; Xiang, Z. H.; Yao, X. D.; Qiu, S. L. Energy Environ. Sci. 2012, 5, 8370. doi: 10.1039/c2ee21935b
李艳强, 贲腾, 裘式伦, 化学学报, 2015, 73, 605. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344998.shtmlLi, Y. Q.; Ben, T.; Qiu, S. L. Acta Chim. Sinica 2015, 73, 605. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344998.shtml
Zhang, Y. M.; Li, B. Y.; Williams, K.; Gao, W. Y.; Ma, S. Q. Chem. Commun. 2013, 49, 10269. doi: 10.1039/c3cc45252b
Pachfule, P.; Dhavale, V. M.; Kandambeth, S.; Kurungot, S.; Banerjee, R. Chem. Eur. J. 2013, 19, 974. doi: 10.1002/chem.201202940
Li, Y. Q.; Ben, T.; Zhang, B. Y.; Fu, Y.; Qiu, S. L. Sci. Rep. 2013, 3, 2420. doi: 10.1038/srep02420
Li, Y. Q.; Roy, S.; Ben, T.; Xu, S. X.; Qiu, S. L. Phys. Chem. Chem. Phys. 2014, 16, 12909. doi: 10.1039/c4cp00550c
Dong, Y.; Das, S.; Zhu, L. K.; Ben, T.; Qiu, S. L. J. Mater. Chem. A 2016, 4, 18822. doi: 10.1039/C6TA09384A
Li, Y. Q. Ph. D. Dissertation, Jilin University, Changchun, 2015 (in Chinese).(李艳强, 博士论文, 吉林大学, 长春, 2015.)
Zhao, W. X.; Han, S.; Zhuang, X. D.; Zhang, F.; Mai, Y. Y.; Feng, X. L. J. Mater. Chem. A 2015, 3, 23352. doi: 10.1039/C5TA06702B
Ashourirad, B.; Sekizkardes, A. K.; Altarawneh, S.; El-Kaderi, H. M. Chem. Mater. 2015, 27, 1349. doi: 10.1021/cm504435m
Yang, X.; Yu, M.; Zhao, Y.; Zhang, C.; Wang, X. Y.; Jiang, J. X. J. Mater. Chem. A 2014, 2, 15139. doi: 10.1039/C4TA02782E
Kou, J. H.; Sun, L. B. J. Mater. Chem. A 2016, 4, 17299. doi: 10.1039/C6TA07305K
Xu, Y. J.; Wu, S. P.; Ren, S. J.; Ji, J. Y.; Yu, Y.; Shen, J. J. RSC Adv. 2017, 7, 32496. doi: 10.1039/C7RA05551J
Gu, S.; He, J. Q.; Zhu, Y. L.; Wang, Z. Q.; Chen, D. Y.; Yu, G. P.; Pan, C. Y.; Guan, J. G.; Tao, K. ACS Appl. Mater. Interfaces 2016, 8, 18383. doi: 10.1021/acsami.6b05170
Alabadi, A.; Abbood, H. A.; Li, Q. Y.; Jing, N.; Tan, B. E. Sci. Rep. 2016, 6, 38614. doi: 10.1038/srep38614
Huang, Y. B.; Pachfule, P.; Sun, J. K.; Xu, Q. J. Mater. Chem. A 2016, 4, 4273. doi: 10.1039/C5TA10170K
Puthiaraj, P.; Ahn, W. S. J. Energ. Chem. 2017, 26, 965. doi: 10.1016/j.jechem.2017.07.012
Lee, Y. J.; Talapaneni, S. N.; Coskun, A. ACS Appl. Mater. Interfaces 2017, 9, 30679. doi: 10.1021/acsami.7b08930
Tian, Z. H.; Huang, J. J.; Zhang, X.; Shao, G. L.; He, Q. Y.; Cao, S. K.; Yuan, S. G. Microporous Mesoporous Mater. 2018, 257, 19. doi: 10.1016/j.micromeso.2017.08.012
Lee, J. S. M.; Briggs, M. E.; Hasell, T.; Cooper, A. I. Adv. Mater. 2016, 28, 9804. doi: 10.1002/adma.201603051
Wang, X. Y.; Mu, P.; Zhang, C.; Chen, Y.; Zeng, J. H.; Wang, F.; Jiang, J. X. ACS Appl. Mater. Interfaces 2017, 9, 20779. doi: 10.1021/acsami.7b05345
Feng, X. L.; Liang, Y. Y.; Zhi, L. J.; Thomas, A.; Wu, D. Q.; Lieberwirth, I.; Kolb, U.; Müllen, K. Adv. Funct. Mater. 2009, 19, 2125. doi: 10.1002/adfm.v19:13
Liang, Y. Y.; Feng, X. L.; Zhi, L. J.; Kolb, U.; Müllen, K. Chem. Commun. 2009, 809. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
Hao, L.; Luo, B.; Li, X. L.; Jin, M. H.; Fang, Y.; Tang, Z. H.; Jia, Y. Y.; Liang, M. H.; Thomas, A.; Yang, J. H.; Zhi, L. J. Energy Environ. Sci. 2012, 5, 9747. doi: 10.1039/c2ee22814a
Liu, X. H.; Zhou, L.; Zhao, Y. Q.; Bian, L.; Feng, X. T.; Pu, Q. S. ACS Appl. Mater. Interfaces 2013, 5, 10280. doi: 10.1021/am403175q
Kim, G. Y.; Yang, J.; Nakashima, N.; Shiraki, T. Chem. Eur. J. 2017, 23, 17504. doi: 10.1002/chem.v23.69
Zha, W. L.; Tu, W. L.; Li, Y.; Gao, H. Y.; Yu, J. G.; Zhao, Y. N.; Li, G. D. Electrochim. Acta 2016, 219, 143. doi: 10.1016/j.electacta.2016.09.133
Xiang, Z. H.; Cao, D. P.; Huang, L.; Shui, J. L.; Wang, M.; Dai, L. M. Adv. Mater. 2014, 26, 3315. doi: 10.1002/adma.v26.20
Fan, X. H.; Kong, F. T.; Kong, A. G.; Chen, A. L.; Zhou, Z. Q.; Shan, Y. K. ACS Appl. Mater. Interfaces 2017, 9, 32840. doi: 10.1021/acsami.7b11229
Liao, Y. Z.; Cheng, Z. H.; Zuo, W. W.; Thomas, A.; Faul, C. F. J. ACS Appl. Mater. Interfaces 2017, 9, 38390. doi: 10.1021/acsami.7b09553
Liao, Y. Z.; Weber, J.; Mills, B. M.; Ren, Z. H.; Faul, C. F. J. Macromolecules 2016, 49, 6322. doi: 10.1021/acs.macromol.6b00901
Wang, H. G.; Cheng, Z. H.; Liao, Y. Z.; Li, J. H.; Weber, J.; Thomas, A.; Faul, C. F. J. Chem. Mater. 2017, 29, 4885. doi: 10.1021/acs.chemmater.7b00857

图式 1 典型的制备多孔有机材料衍生的多孔碳的流程示意图
Scheme 1 Typic synthesis procedure for porous organic materials derived porous carbon

图 1 (A) PAF-1/C-900的合成原理示意图; (B) 295 K下PAF-1/C-900和PAF-1的CO2吸附曲线
Figure 1 (A) Schematic illustration of the procedures for the preparation of PAF-1/C-900. (B) CO2 adsorption isotherms of PAF-1/C-900 and PAF-1 at 295 K. Reproduced from Ref. 37 with permission from The Royal Society of Chemistry

图 2 (A) PAF-1及碳化后的多孔碳样品在273 K下的CO2吸附; (B) PAF-1及碳化后的多孔碳样品的CO2吸附热随着吸附CO2量变化关系图). (C) PAF-1及碳化后的多孔碳样品在273 K下的CH4吸附; (D) PAF-1及碳化后的多孔碳样品的CH4吸附热随着吸附CH4量变化关系图; (E) PAF-1及碳化后的多孔碳样品在77 K下的H2吸附; (F) PAF-1及碳化后的多孔碳样品的H2吸附热随着吸附H2量变化关系图
Figure 2 (A) CO2 adsorption (solid symbols) and desorption (open symbols) isotherms of PAF-1 and carbonized samples at 273 K; (B) QstCO2 of PAF-1 and carbonized samples as a function of the amount of CO2 adsorbed. (C) CH4 adsorption (solid symbols) and desorption (open symbols) isotherms of PAF-1 and carbonized samples at 273 K; (D) QstCH4 of PAF-1 and carbonized samples as a function of the amount of CH4 adsorbed. (E) H2 adsorption (solid symbols) and desorption (open symbols) isotherms of PAF-1 and carbonized samples at 77 K; (F) QstH2 of PAF-1 and carbonized samples as a function of the amount of H2 adsorbed. Reproduced from Ref. 35 with permission from The Royal Society of Chemistry

图 3 (a) 碳化的HCPs的CO2吸附与比表面之间的关系. (b) 298 K下0~1 bar的CO2吸附. (c) 298 K下0~10 bar的CO2吸附. (d) 195 K下0~1 bar的CO2吸附
Figure 3 (a) Correlation of CO2 uptake with BET surface area for carbonized HCPs at 1 bar. (b) CO2 sorption isotherms at 298 K over pressure range of 0~1 bar. (c) CO2 sorption isotherms at 298 K over pressure range of 0~10 bar. (d) CO2 adsorption-desorption isotherms at 195 K and 1 bar (the adsorption and desorption branches are labeled with filled and empty symbols, respectively). Adopted with permission from ref. 54 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

图 4 扫描电子显微镜(SEM)图片: (a) HCPT-B, (b) PCNT-B; 透射电子显微镜(TEM)图片: (c) HCPT-B, (b) PCNT-B; (e)和(f)分别是通过SEM和TEM图片分析得到PCNT-B的内径和外径的直方图
Figure 4 Scanning electron microscope (SEM) images: (a) HCPT-B, (b) PCNT-B; transmission electron microscope (TEM) images: (c) HCPT-B, (d) PCNT-B; (e) outer and (f) inner diameter distribution histograms of PCNT-B from analysis of SEM and TEM images. Reprinted with permission from Ref. 54 Copyright (2017) American Chemical Society

图 5 PCNT-B和PCNB-B的电化学表现: (a) PCNT-B在不同扫速下的CV曲线, (b) PCNT-B的充放电曲线; (c) PCNT-B和PCNB-B在不同电流密度下的比电容; (d) PCNT-B和PCNB-B的Nyquist图, 插图是高频区的放大图, (e) PCNT-B和PCNB-B的Bode图, (f) PCNT-B在电流密度是1.5 A•g-1的循环稳定性
Figure 5 Electrochemical performances of PCNT-B and PCNB-B: (a) CV curves of PCNT-B at various scan rates, (b) charge-discharge profiles of PCNT-B, (c) specific capacitances of PCNT-B and PCNB-B at various current densities, (d) Nyquist plots and the inset shows the close-up view of the high-frequency region, (e) Bode plots of phase angle vs frequency for PCNT-B and PCNB-B, and (f) cycling stability of PCNT-B at a current density of 1.5 A•g-1. Reprinted with permission from Ref. 55 Copyright (2017) American Chemical Society

图 6 (A) 碳化后的样品在扫速是0.05 V•s-1时的CV曲线; (B)碳化后的样品在扫速是1 A•g-1时的充放电; (C)碳化后的样品比电容与电流之间的关系图; (D)碳化后的样品在6 mol/L KOH中的Nyquist图(频率范围是105到10-2 Hz)
Figure 6 (A) CV curves of carbonized samples at a scan rate of 0.05 V•s-1; (B) galvanostatic charge-discharge curves of carbonized samples at 1 A•g-1; (C) capacitance versus current of carbonized samples; (D) Nyquist plots (frequency ranges from 105 to 10-2 Hz) of carbonized samples in 6 mol/L KOH. Reproduced from Ref. 40 with permission from the PCCP Owner Societies

图 7 (A) 电化学测试前, 样品的Nyquist图. (B)从60到3000 mA•g-1变化电流密度下的倍率性能的测试. (C)电流密度是400 mA•g-1条件下循环400圈的充放电曲线
Figure 7 (A) Nyquist plots before testing the electrochemical performance. (B) Rate capability with various current densities from 60 to 3000 mA•g-1. (C) Cycling performance showing the discharge capacity at a current density of 400 mA•g-1 for 400 cycles (solid symbols, discharge capacity; open symbols, coulombic efficiency). Reproduced from Ref. 41 with permission from The Royal Society of Chemistry

图 8 Fe2P/Fe4N@C-800在0.1 mol/L KOH电解质溶液(A)和0.1 mol/L HClO4电解质溶液(B)中的ORR曲线. (C)和(D)分别是0.1 mol/L KOH电解质溶液和0.1 mol/L HClO4电解质溶液中的RDE极化曲线, 转速为1600 r/min. (E)和(F)是Fe2P/Fe4N@C-800分别在O2饱和的0.1 mol/L KOH电解质溶液和0.1 mol/L HClO4电解质溶液中的经过5000次循环的CV曲线
Figure 8 (A and B) ORR CV curves on Fe2P/Fe4N@C-800 in 0.1 mol/L KOH (A) and 0.1 mol/L HClO4 (B) electrolytes. (C and D) RDE polarization curves on N-C-800 (a), Fe2P/Fe4N@C-600 (b), Fe2P/Fe4N@C-700 (c), Fe2P/Fe4N@C-800 (d), Fe2P/Fe4N@C-900 (e), and commercial Pt/C (f) (20 wt%) at 1600 r/min in 0.1 mol/L KOH (C) and 0.1 mol/L HClO4 (D) electrolytes at 1600 r/min. (E and F) CVs run for 5000 cycles recorded in O2-saturated in 0.1 mol/L KOH (E) and 0.1 mol/L HClO4 (F) electrolytes for Fe2P/Fe4N@C-800. Reprinted with permission from Ref. 57 Copyright (2017) American Chemical Society

图 9 (A) 在1 mol/L KOH电解质溶液中, 转速1600 r/min条件下的OER的LSV曲线. (B)不同的催化剂在OER中的Tafel斜率. (C)在0.5 mol/L H2SO4电解质溶液中, 转速1600 r/min条件下的HER的LSV曲线. (D)不同的催化剂在HER中的Tafel斜率
Figure 9 (A) The OER LSV curves over different catalysts at a rotation rate of 1600 r/min in 1 mol/L KOH solution. (B) The corresponding Tafel slopes of the OER for different catalysts. (C) The HER LSV curves over different catalysts at a rotation rate of 1600 r/min in 0.5 mol/L H2SO4 solution. (D) The corresponding Tafel slopes of the HER for different catalysts. Reprinted with permission from Ref. 63 Copyright (2017) American Chemical Society

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
 下载:
下载: