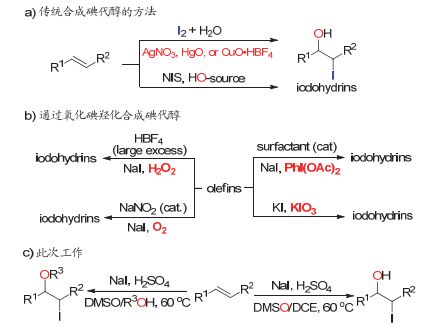
 图1
烯烃的碘羟化反应合成碘代醇
Figure1.
Approaches to iodohydrins from olefins
图1
烯烃的碘羟化反应合成碘代醇
Figure1.
Approaches to iodohydrins from olefins
引用本文:
李昕伟, 宋颂, 焦宁. 二甲基亚砜参与的烯烃的氧化碘羟化反应[J]. 化学学报,
2017, 75(12): 1202-1206.
doi:
10.6023/A17100448

Citation: Li Xinwei, Song Song, Jiao Ning. Oxidative Iodohydroxylation of Olefins with DMSO[J]. Acta Chimica Sinica, 2017, 75(12): 1202-1206. doi: 10.6023/A17100448
Citation: Li Xinwei, Song Song, Jiao Ning. Oxidative Iodohydroxylation of Olefins with DMSO[J]. Acta Chimica Sinica, 2017, 75(12): 1202-1206. doi: 10.6023/A17100448
二甲基亚砜参与的烯烃的氧化碘羟化反应
English
Oxidative Iodohydroxylation of Olefins with DMSO
Abstract:
Halohydrins bearing a hydroxyl and halide functional group, are privileged building blocks in organic synthesis and could be conveniently converted to other significant organic intermediates such as azidoalcohols, aminoalcohols, and epoxides, all of which are widely used in the synthesis of highly value-added chemicals. Among the approaches to halohydrins, the halohydroxylation of olefins provides a direct and efficient approach. The synthesis of bromohydrins has achieved great progress in recent years. However, the approaches to iodohydrins are still very limited. Our previous studies revealed that DMSO could oxidize halo anions to halo cations under acidic conditions. As our continuous development DMSO-based reactions, we report the iodohydroxylation of olefins by using DMSO and HI generated in situ. In this transformation, DMSO performed versatile roles as an oxidant, a solvent and an oxygen source. This reaction featured with simple operation, mild reaction condition, and wild substrate scope, and provided an efficient method to synthesize iodohydrins. Furthermore, the iodoetheration of olefins was also realized by using DMSO and alcohol as the solvent. A representative procedure for this reaction is as following:The mixture of alkene (0.5 mmol), NaI (0.6 mmol), conc. H2SO4 (1.0 mmol), DMSO (1 mL) and DCE (1 mL) were stirred at 60℃ under air. TCL monitor the reaction, and the product had a clear spot in phosphomolybdic acid chromogenic agent. After the reaction was completed, saturated solution of Na2S2O3 (0.5 mL) was added into the system to consume the extra I2. After cooling down to room temperature, the mixture was diluted with water (10 mL) and extracted with ethyl acetate (10 mL×3). The combined organic extract was washed with saturated solution of NaCl (15 mL), dried over MgSO4, and evaporated in vacuo. The residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate) to afford the desired product.
-
Key words:
- DMSO
- / olefin
- / oxidation
- / iodohydroxylation
- / iodohydrin
-
1 引言
有机卤化物是常见且重要的有机化学物质, 广泛存在于自然界尤其是海洋中, 有超过4500种天然产物中含有卤素官能团[1].同时卤化物在现代工业和生活例如医药、农药、阻燃剂和新材料等领域具有很高的工业价值[2].除此之外, 有机卤化物因其较为活泼的反应性使化学合成更简单、更便捷.卤代醇同时具有羟基和卤素两种官能团, 是常见的有机卤化物和重要的有机合成砌块, 可以方便地转化为叠氮醇、氨基醇和环氧化物等高附加值化学品[3].
近年来, 许多高效合成卤代醇的反应陆续报道[4].在这些方法中, 烯烃的卤羟化反应是合成卤代醇最直接的方法.溴代醇和氯代醇的合成已经取得了可喜的进展[5].然而, 碘代醇的合成方法仍然有很大的局限性.传统烯烃的碘羟化方法合成碘代醇需要使用当量的金属试剂如AgNO3[6a], HgO[6b], ZnCl2[6c]和CuO[6d]等来活化具有腐蚀性、易升华的碘单质(图 1a), 这些活化试剂毒性高, 价格相对昂贵, 不利于大规模合成碘代醇.因此化学家发展了活性相对较高的稳定的碘正试剂例如I(py)BF4和NIS, 成功应用于碘代醇的合成中.这些试剂会产生大量的有机副产物从而降低反应的原子经济性[7].大自然尤其是海洋中存在丰富的含碘天然产物, 这些天然产物得以合成的关键是海水中的酶利用氧气将无机碘化物氧化为亲电性的碘正化合物.受此启发[8], 化学家利用氧化策略成功实现了烯烃的氧化碘羟化反应来合成碘代醇(图 1b).这类反应中的氧化剂KIO3[9a]和NaIO4[9b]具有潜在的爆炸性, 并且底物适用范围相对较窄, 极大地限制了这种方法的广泛应用. 2011年, Maiti小组实现了以NaI为碘源、表面活性剂催化、PhI(OAc)2为氧化剂的烯烃的氧化碘羟化反应[5b].值得一提的是, Barluenga[10a]和Stavber[10b]小组分别实现了H2O2或者O2作为绿色廉价氧化剂的烯烃的碘羟化反应.
图1
烯烃的碘羟化反应合成碘代醇
Figure1.
Approaches to iodohydrins from olefins
二甲基亚砜(DMSO)是一种低毒、廉价、功能多样的常用非质子极性溶剂[11], 广泛应用于多种人名反应如Swern氧化反应[12a], Pfitzner-Moffatt氧化反应[12b]和Corey-Chaykovsky反应[12c]中. DMSO因其独特的结构, 近年来在有机合成中被广泛用作氧源[13]、碳源[14]和硫源[15].我们组之前的研究表明DMSO在酸性条件下具备氧化卤负离子的能力[13a], 利用DMSO的这种性质, 我们实现了烯(炔)烃、芳烃、酮的卤(官能团)化反应.在此基础上[16], 在本文中, 我们将报道利用DMSO和原位产生的HI来实现烯烃的碘羟化反应(图 1c).在此转化过程中, DMSO起到了包括氧化剂、溶剂和氧源的多重作用.此反应在高效合成碘代醇的过程中具备操作简便、反应条件温和以及底物适用性广的优点.此外, 添加亲核性的醇类溶剂, 该策略还可以实现烯烃的碘醚化反应.
2 结果与讨论
2.1 反应条件优化
以2-乙烯基萘1a为模型底物, 我们研究了烯烃的碘羟化反应, 实验结果见表 1.参考我们课题组之前的报道[13a], 氢碘酸(HI)的水溶液被选为碘源, 在DMSO作为溶剂的情况下, 只得到了痕量的碘代醇2a(Entry 1).根据芳烃碘化反应的经验, 我们尝试了无机碘化物与硫酸现场生成碘化氢的策略(Entries 2~4), 发现:利用NH4I和H2SO4原位生成HI, 以66%的收率得到了产物2a(Entry 2);碘盐的阳离子对反应收率影响不大, 使用NaI时, 收率可达69% (Entry 3).我们认为碘化物与浓硫酸现场生成氢碘酸比直接使用氢碘酸溶液有利于反应, 是因为现场产生的氢碘酸局部浓度较大.用其他有机酸如甲磺酸(MsOH)或者三氟甲磺酸(TfOH)替代H2SO4时, 反应的收率明显下降(Entries 5~6).硫酸的用量对反应影响较大, 降低H2SO4用量为1.2当量时, 烯烃几乎不转化(Entry 7);提高H2SO4用量为3当量时, 反应的收率同2倍量一样(Entry 9).当用DCE同DMSO作为共溶剂(1:1)时, 2a的收率略有提高(Entry 10).
 表 1
反应条件的优化a
Table 1.
Optimization of the reaction conditionsa
表 1
反应条件的优化a
Table 1.
Optimization of the reaction conditionsa

Entry [I] Acid Solvent t/h Yieldb/% 1 HI — DMSO 24 Trace 2 NH4I H2SO4 DMSO 17 66 3 NaI H2SO4 DMSO 17 69 4 KI H2SO4 DMSO 17 66 5 NaI MsOH DMSO 24 20 6 NaI TfOH DMSO 24 42 7c NaI H2SO4 DMSO 48 0 8d NaI H2SO4 DMSO 20 64 9e NaI H2SO4 DMSO 11 69 10f NaI H2SO4 DMSO/DCE 11 72 11f NaI H2SO4 DMSO/MeCN 18 66 aReaction conditions: the solution of 1a (0.5 mmol), [I]=iodine source (0.6 mmol) and acid (1.0 mmol) in solvent (2 mL) was stirred at 60 ℃ under air.bIsolated yields.cConc. H2SO4 (0.6 mmol) was added.dConc. H2SO4 (0.8 mmol) was added.eConc. H2SO4 (1.5 mmol) was added. fDMSO/cosolvent=1/1. 2.2 底物拓展
在最优反应条件下(表 1, Entry 10), 我们考察了反应的底物适用范围(表 2).苯乙烯的碘羟化反应以73%的收率得到2b.苯环对位的取代基对反应影响较小, 给电子取代基如甲基(2c), 甲氧基(2d), 叔丁基(2e)能给出较高的收率, 最高可达85%.间位是给电子取代基甲基(2g)时, 收率可达73%;间位是强拉电子硝基(2i)时, 反应仍以58%的收率得到目标产物.通常邻位取代基具有一定的位阻效应, 在该碘羟化反应中, 苯环邻位取代基对反应几乎没有影响, 分别以82%和61%收率得到目标产物(2j, 2k).值得一提的是, 1, 1-二取代的烯烃1l在最优条件下可以顺利以67%的收率得到带有季碳中心的碘代醇2l.环状烯烃1m能以52%的收率高选择性地得到反式碘代醇2m.
表 2
苯乙烯类底物范围a
Table 2.
Substrate scope of styrenes

aReaction conditions: the solution of 1 (0.5 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in DMSO/DCE (1 mL/1 mL) was stirred at 60 ℃ under air. Isolated yields. 除了苯乙烯衍生物, 脂肪烯烃也能以较好的收率和选择性转化为碘代醇(表 3).带有苯氧基的烯烃1n以70%的收率得到碘羟化目标产物2n, 同时没有检测到苯环上碘代产物的生成.对于单取代的烯烃(1o~1s), 能以中等到优秀的收率和较高的区域选择性(>5:1)得到碘代醇.对于1, 1-双烷基取代烯烃的碘羟化反应能以53%收率得到目标产物2t.
表 3
脂肪烯烃的底物范围a
Table 3.
Substrate scope of aliphatic olefins

aReaction conditions: the solution of 1 (0.5 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in DMSO/DCE (1 mL/1 mL) was stirred at 60 ℃ under air. Isolated yields. 实现了烯烃的碘羟化反应之后, 根据机理我们推测, 如果不使用DMSO为溶剂而将其用量降为当量, 换为亲核性的醇为溶剂时, 应该能够实现烯烃的氧化碘醚化反应.于是, 我们使用1.2当量DMSO为氧化剂, 乙醇为溶剂时, 能够以55%~92%的收率得到烯烃的碘醚化反应产物(表 4).其他醇如乙二醇为溶剂, 也可顺利得到目标产物.
表 4
烯烃的碘醚化反应a
Table 4.
The iodoetheration of olefins

aReaction conditions: the solution of 1 (0.5 mmol), DMSO (0.6 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in EtOH (2 mL) was stirred at 60 ℃ for 24 h under air. bEthanediol (2 mL) was used as the solvent. 2.3 反应机理
关于DMSO氧化碘负离子的机理目前还不是很清楚, 基于之前的报道, 我们提出了可能的反应机理(图 2).最初, NaI和H2SO4现场生成的HI被DMSO氧化为DMS•I2[5h, 17], 紧接着与DMSO结合生成[(DMSO)nI+]I− (n=1 or 2).在I+和烯烃1之间发生亲电加成反应生成碘嗡中间体4.进一步, DMSO作为亲核试剂进攻4生成烷氧硫醚正离子6, 然后在氢碘酸的作用下生成产物碘代醇2同时再生碘亲电试剂[13g, 16, 17].
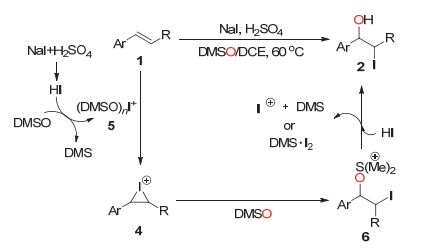 图2
可能的反应机理
Figure2.
Proposed mechanism
图2
可能的反应机理
Figure2.
Proposed mechanism
3 结论
我们揭示了一种烯烃的氧化碘羟化反应, 该反应使用廉价的碘化物和常用溶剂DMSO为氧化剂, 为碘代醇的合成提供了新的方法. DMSO在此反应中起到了关键性的作用, 扮演着氧化剂、溶剂和氧源等多种角色.这种方法可以通过换为更具亲核性的溶剂实现烯烃的碘醚化反应.
4 实验部分
烯烃碘羟化反应的一般操作步骤如下: 0.5 mmol底物烯烃, 0.6 mmol NaI, 1.0 mmol浓H2SO4, 1.0 mL DMSO和1.0 mL DCE在60℃下, 空气氛围中搅拌.薄层色谱检测反应过程.反应完成后, 加入0.5 mL饱和Na2S2O3水溶液来消耗过量的碘单质.将反应体系冷却至室温后, 加入10 mL水稀释, 用10 mL×3的乙酸乙酯萃取, 合并有机相, 有机相用饱和氯化钠水溶液洗涤, 无水MgSO4干燥, 旋转蒸发脱除溶剂后, 粗产品用石油醚和乙酸乙酯作为洗脱剂进行柱层析分离, 即可得到目标碘代醇产物2.
-
-
[1]
Gribble, G. W. J. Chem. Educ. 2004, 81, 1441. doi: 10.1021/ed081p1441
-
[2]
Dagani, M. J.; Barda, H. J.; Benya, T. J.; Sanders, D. C. Bromine Compounds in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002.
-
[3]
Neilson, A. H. Organic Bromine and Iodine Compounds in The Handbook of Environmental Chemistry, Springer, Berlin, Heidelberg, 2003.
-
[4]
(a) Smith, J. G.; Fieser, M. In Fieser and Fieser's Reagent for Organic Synthesis, Vol. 1~12, John Wiley and Sons, New York, 1990.(b) Grogan, G. Annu. Rep. Prog. Chem., Sect. B 2009, 105, 206.(c) Mahdi, T.; Stephan, D. W. J. Am. Chem. Soc. 2014, 136, 15809.
-
[5]
(a) Sels, B. F.; De Vos, D. E.; Jacobs, P. A. J. Am. Chem. Soc. 2001, 123, 8350.(b) Pandit, P.; Gayen, K. S.; Khamarui, S.; Chatterjee, N.; Maiti, D. K. Chem. Commun. 2011, 47, 6933.(c) Dewkar, G. K.; Narina, S. V.; Sudalai, A. Org. Lett. 2003, 5, 4501.(d) Yang, X.; Wu, J.; Mao, X.; Jamison, T. F.; Hatton, T. A. Chem. Commun. 2014, 50, 3245.(e) Darensbourg, D. J.; Wilson, S. J. J. Am. Chem. Soc. 2011, 133, 18610.(f) Kozhushkov, S. I.; Yufit, D. S.; de Meijere, A. Adv. Synth. Catal. 2005, 347, 255.(g) Sels, B.; De Vos, D.; Buntinx, M.; Pierard, F.; Kirsch-De Mesmaeker, A.; Jacobs, P. Nature 1999, 400, 855.(h) Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070.
-
[6]
(a) Hajra, S.; Karmakar, A.; Bhowmick, M. Tetrahedron 2005, 61, 2279.(b) Schmid, G. H.; Gordon, J. W. J. Org. Chem. 1983, 48, 4010.(c) Mahajan, V. A.; Shinde, P. D.; Gajare, A. S.; Karthikeyan, M.; Wakharkar, R. D. Green Chem. 2002, 4, 325.(d) Barluenga, J.; Rodriguez, M. A.; Campos, P. J.; Asensio, G. J. Chem. Soc., Chem. Commun. 1987, 1491.
-
[7]
(a) Barluenga, J.; Gonzalez, J. M.; Campos, P. J.; Asensio, G. Angew. Chem. 1985, 97, 341.(b) Rao, D. S.; Reddy, T. R.; Babachary, K.; Kashyap, S. Org. Biomol. Chem. 2016, 14, 7529.
-
[8]
For selected reviews on enzyme-catalyzed oxidative halogenations, see:(a) Vaillancourt, F. H.; Yeh, E.; Vosburg, D. A.; Garneau-Tsodikova, S.; Walsh, C. T. Chem. Rev. 2006, 106, 3364.(b) Wischang, D.; Brücher O.; Hartung, J. Coord. Chem. Rev. 2011, 255, 2204.(c) Lewis, J. C.; Coelho, P. S.; Arnold, F. H. Chem. Soc. Rev. 2011, 40, 2003.
-
[9]
(a) Agrawal, M. K.; Adimurthy, S.; Ganguly, B.; Ghosh, P. K. Tetrahedron 2009, 65, 2791.(b) Chakraborty, N.; Santra, S.; Kundu, S. K.; Hajra, A.; Zyryanov, G. V.; Majee, A. RSC Adv. 2015, 5, 56780.
-
[10]
(a) Barluenga, J.; Marco-Arias, M.; Gonzalez-Bobes, F.; Ballesteros, A.; Gonzalez, J. M. Chem.-Eur. J. 2004, 10, 1677.(b) Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Adv. Synth. Catal. 2008, 350, 2921.
-
[11]
(a) Martin, D. ; Weise, A. ; Niclas, H. J. Angew. Chem., Int. Ed. 1967, 6, 318. (b) Sun, P. ; Yang, D. ; Wei, W. ; Jiang, L. ; Wang, Y. ; Dai, T. ; Wang, H. Org. Chem. Front. 2017, 4, 1367. (c) Yang, X. -Q. ; Huang, K. -M. ; Jia, G. -Z. Acta Chim. Sinica 2008, 66, 1107(in Chinese). (杨晓庆, 黄卡玛, 贾国柱, 化学学报, 2008, 66, 1107. )(d) Xie, K. ; Qiao, S. ; Cheng, C. Acta Chim. Sinica 2009, 67, 231(in Chinese). (谢昆, 乔澍, 程聪, 化学学报, 2009, 67, 231. )(e) Liu, J. ; Wang, W. ; Wang, R. ; Gu, L. Chin. J. Chem. 2015, 33, 559. (f) Yang, J. ; Yin, W. ; Liu, R. ; Chu, C. Chin. J. Chem. 2012, 30, 2786.
-
[12]
(a) Omura, K.; Sharma, A. K.; Swern, D. J. Org. Chem. 1976, 41, 957.(b) Pfitzner, K. E.; Moffatt, J. G. J. Am. Chem. Soc. 1963, 85, 3027.(c) Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1962, 84, 867.
-
[13]
(a) Song, S.; Huang, X.; Liang, Y.-F.; Yuan, Y.; Li, X.; Jiao, N. Green Chem. 2015, 17, 2727.(b) Tomita, R.; Yasu, Y.; Koike, T.; Akita, M. Angew. Chem., Int. Ed. 2014, 53, 7144.(c) Mupparapu, N.; Khan, S.; Battula, S.; Kushwaha, M.; Gupta, A. P.; Ahmed, Q. N.; Vishwakarma, R. A. Org. Lett. 2014, 16, 1152.(d) Wu, X.; Gao, Q.; Liu, S.; Wu, A. Org. Lett. 2014, 16, 2888.(e) Gao, Q.; Wu, X.; Liu, S.; Wu, A. Org. Lett. 2014, 16, 1732.(f) Ashikari, Y.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2011, 133, 11840.(g) Ashikari, Y.; Nokami, T.; Yoshida, J. Org. Lett. 2012, 14, 938.(h) Xu, R.; Wan, J.-P.; Mao, H.; Pan, Y. J. Am. Chem. Soc. 2010, 132, 15531.(i) Liang, Y.-F.; Wu, K.; Song, S.; Li, X.; Huang, X.; Jiao, N. Org. Lett. 2015, 17, 876.(j) Liang, Y.-F.; Li, X.; Wang, X.; Zou, M.; Tang, C.; Liang, Y.; Song, S.; Jiao, N. J. Am. Chem. Soc. 2016, 138, 12271.
-
[14]
(a) Jiang, X.; Wang, C.; Wei, Y.; Xue, D.; Liu, Z.; Xiao, J. Chem.-Eur. J. 2014, 20, 58.(b) Qian, J.; Zhang, Z.; Liu, Q.; Liu, T.; Zhang, G. Adv. Synth. Catal. 2014, 356, 3119.(c) Ren, X.; Chen, J.; Chen, F.; Cheng, J. Chem. Commun. 2011, 47, 6725.(d) Lv, Y.; Li, Y.; Xiong, T.; Pu, W.; Zhang, H.; Sun, K.; Liu, Q.; Zhang, Q. Chem. Commun. 2013, 49, 6439.(e) Wu, X.-F.; Natte, K. Adv. Synth. Catal. 2016, 358, 336.(f) Jones-Mensah, E.; Karki, M.; Magolan, J. Synthesis 2016, 48, 1421.(g) Shen, T.; Huang, X.; Liang, Y.-F.; Jiao, N. Org Lett. 2015, 17, 6186.
-
[15]
(a) Liu, F.-L.; Chen, J.-R.; Zou, Y.-Q.; Wei, Q.; Xiao, W.-J. Org. Lett. 2014, 16, 3768.(b) Mal, K.; Sharma, A.; Maulik, P. R.; Das, I. Chem.-Eur. J. 2014, 20, 662.(c) Gao, Q.; Wu, X.; Li, Y.; Liu, S.; Meng, X.; Wu, A. Adv. Synth. Catal. 2014, 356, 2924.(d) Chu, L.; Yue, X.; Qing, F.-L. Org. Lett. 2010, 12, 1644.(e) Yin, G.; Zhou, B.; Meng, X.; Wu, A.; Pan, Y. Org. Lett. 2006, 8, 2245.(f) Hu, G.; Xu, J.; Li, P. Org. Lett. 2014, 16, 6036.(g) Gao, X.; Pan, X.; Gao, J.; Huang, H.; Yuan, G.; Li, Y. Chem. Commun. 2015, 51, 210.(h) Jiang, Y.; Loh, T.-P. Chem. Sci. 2014, 5, 4939.(i) Gao, X.; Pan, X.; Gao, J.; Jiang, H.; Yuan, G.; Li, Y. Org. Lett. 2015, 17, 1038.
-
[16]
(a) Song, S.; Li, X.; Sun, X.; Yuan, Y.; Jiao, N. Green Chem. 2015, 17, 3285.(b) Song, S.; Sun, X.; Li, X.; Yuan, Y.; Jiao, N. Org. Lett. 2015, 17, 2886.
-
[17]
(a) Gao, Q.; Liu, S.; Wu, X.; Wu, A. Org. Lett. 2014, 16, 4582.(b) Liu, S.; Xi, H.-L.; Zhang, J.-J.; Wu, X.; Gao, Q.-H.; Wu, A.-X. Org. Biomol. Chem. 2015, 13, 8807.(c) Gao, Q.; Zhang, J.; Wu, X.; Liu, S.; Wu, A. Org. Lett. 2015, 17, 134.(d) Gao, Q.; Liu, S.; Wu, X.; Zhang, J.; Wu, A. Org. Lett. 2015, 17, 2960.(e) Zhang, J.; Gao, Q.; Wu, X.; Geng, X.; Wu, Y.-D.; Wu, A. Org. Lett. 2016, 18, 1686.(f) Fan, W.; Yang, Z.; Jiang, B.; Li, G. Org. Chem. Front. 2017, 4, 1091.(g) Zhang, Z.; Xie, C.; Tan, X.; Song, G.; Wen, L.; Gao, H.; Ma, C. Org. Chem. Front. 2015, 2, 942.(h) Wu, W.; An, Y.; Li, J.; Yang, S.; Zhu, Z.; Jiang, H. Org. Chem. Front. 2017, 4, 1751.(i) Wu, Y.-D.; Geng, X.; Gao, Q.; Zhang, J.; Wu, X.; Wu, A.-X. Org. Chem. Front. 2016, 3, 1430.
-
[1]
-
表 1 反应条件的优化a
Table 1. Optimization of the reaction conditionsa
Entry [I] Acid Solvent t/h Yieldb/% 1 HI — DMSO 24 Trace 2 NH4I H2SO4 DMSO 17 66 3 NaI H2SO4 DMSO 17 69 4 KI H2SO4 DMSO 17 66 5 NaI MsOH DMSO 24 20 6 NaI TfOH DMSO 24 42 7c NaI H2SO4 DMSO 48 0 8d NaI H2SO4 DMSO 20 64 9e NaI H2SO4 DMSO 11 69 10f NaI H2SO4 DMSO/DCE 11 72 11f NaI H2SO4 DMSO/MeCN 18 66 aReaction conditions: the solution of 1a (0.5 mmol), [I]=iodine source (0.6 mmol) and acid (1.0 mmol) in solvent (2 mL) was stirred at 60 ℃ under air.bIsolated yields.cConc. H2SO4 (0.6 mmol) was added.dConc. H2SO4 (0.8 mmol) was added.eConc. H2SO4 (1.5 mmol) was added. fDMSO/cosolvent=1/1.  下载: 导出CSV
下载: 导出CSV
表 2 苯乙烯类底物范围a
Table 2. Substrate scope of styrenes
aReaction conditions: the solution of 1 (0.5 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in DMSO/DCE (1 mL/1 mL) was stirred at 60 ℃ under air. Isolated yields.
下载: 导出CSV
表 3 脂肪烯烃的底物范围a
Table 3. Substrate scope of aliphatic olefins
aReaction conditions: the solution of 1 (0.5 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in DMSO/DCE (1 mL/1 mL) was stirred at 60 ℃ under air. Isolated yields.
下载: 导出CSV
表 4 烯烃的碘醚化反应a
Table 4. The iodoetheration of olefins
aReaction conditions: the solution of 1 (0.5 mmol), DMSO (0.6 mmol), NaI (0.6 mmol) and conc. H2SO4 (1.0 mmol) in EtOH (2 mL) was stirred at 60 ℃ for 24 h under air. bEthanediol (2 mL) was used as the solvent.
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 24
- 文章访问数: 4554
- HTML全文浏览量: 1260
 下载:
下载:
