 图1
基于氮自由基的远端C—H键官能化反应
Figure1.
Remote C—H bond functionalization based on nitrogen-centered radical
图1
基于氮自由基的远端C—H键官能化反应
Figure1.
Remote C—H bond functionalization based on nitrogen-centered radical
引用本文:
宋颢, 刘小宇, 秦勇. 氮自由基化学新进展:光催化N-H键活化途径[J]. 化学学报,
2017, 75(12): 1137-1149.
doi:
10.6023/A17080384

Citation: Song Hao, Liu Xiaoyu, Qin Yong. Advances on Nitrogen-centered Radical Chemistry:A Photocatalytic N-H Bond Activation Approach[J]. Acta Chimica Sinica, 2017, 75(12): 1137-1149. doi: 10.6023/A17080384
Citation: Song Hao, Liu Xiaoyu, Qin Yong. Advances on Nitrogen-centered Radical Chemistry:A Photocatalytic N-H Bond Activation Approach[J]. Acta Chimica Sinica, 2017, 75(12): 1137-1149. doi: 10.6023/A17080384
氮自由基化学新进展:光催化N-H键活化途径
English
Advances on Nitrogen-centered Radical Chemistry:A Photocatalytic N-H Bond Activation Approach
Abstract:
Nitrogen-centered radicals are highly reactive intermediates, which provide new opportunities for designing new chemical reactions and preparing nitrogen-containing molecules. Direct generation of nitrogen-centered radicals via activation of N-H bonds under photocatalytic conditions has emerged as a green, efficient, and economical process, where significant progress has been made with methodology development in very recent years. In this paper, we highlight the important advances in this area that were reported since 2016.
-
Key words:
- nitrogen-centered radical
- / visible light
- / photocatalyst
- / single-electron transfer
- / cascade reaction
-
1 引言
氮自由基化学作为有机合成领域中的重要分支之一, 长期以来一直受到合成化学家们的关注[1].通过氮自由基反应方法学来实现多官能化含氮杂环骨架的构建是一种高效合成含氮化合物的重要途径.传统的氮自由基产生方法大多是基于N—X键(X为氧、氮或卤原子)的均裂来实现, 这些方法通常存在反应条件苛刻、操作复杂、缺乏原子经济性等缺陷[2].从其产生方式而言, N—H键无疑是产生氮自由基最为直接、经济的前体.然而, 由于大多数N—H键化学键裂解能较高, 因此将N—H键直接转化为氮自由基通常需要借助对环境危害较大的自由基引发剂或剧烈的反应条件[3].例如, 需要使用当量的强氧化剂、或在高温下发生热裂解反应、或使用高能量的UV灯照射光解等.如何通过对反应进行合理设计来实现温和条件下氮自由基的可控产生一直是氮自由基化学研究中的难题.
近年来, 由可见光促进的各类反应也因其反应条件温和、操作简单、绿色环保等优点在有机合成领域中得到广泛关注[4].基于光氧化还原催化策略, 高效产生碳自由基、杂原子自由基或离子等活泼中间体, 进而实现C—C键、C—N键、C—O键、C—P键、C—X键(X为卤原子)等化学键的构建, 成为近年来发展迅速的研究方向.在绿色化学的理念之下, 由可见光促进的光氧化还原催化策略不仅为氮自由基的温和产生提供了新思路, 也为基于氮自由基的新反应设计和含氮杂环化合物的合成提供了新工具.可见光促进的光氧化还原催化活化N—H键直接产生氮自由基是该领域面临的重要挑战, 其在近年来经历了快速发展, 部分研究进展在相关综述中进行了总结[5].本文主要针对2016年至今报道的几例最新的研究成果进行亮点介绍.
2 远端C—H键官能化反应
惰性C—H键的直接官能化反应是合成有机化学中一种便捷、经济、高效的成键方式[6].近年来, 通过光氧化还原催化条件进行惰性C—H键的直接官能化引起了研究者的关注[7]. 2016年, Knowles小组[8]和Rovis小组[9]几乎同时发现, 在蓝色LED光照射下, 酰胺的N—H键转化为酰胺氮自由基, 后者可经1, 5-氢原子转移(1, 5-HAT)过程攫取远端不活泼C(sp3)—H键上的氢原子, 所得碳自由基再进行Michael加成实现C—C键的构建.两者研究思路相同, 仅在底物选择方面略有差别.如图 1所示, Knowles小组以芳基取代的酰胺1为底物、以具有高度缺电子结构的[Ir(dF(CF3)ppy)2(5, 5'-d(CF3)-bpy)]PF6为催化剂, 以Bu4NOP(O)(OBu)2为碱来产生氮中心自由基; Rovis小组则是以三氟乙酰胺3为底物、以[Ir(dF-CF3ppy)2(dtbbpy)]PF6为光催化剂、K3PO4为碱来产生氮中心自由基.两个方法均表现出良好的底物普适性, 可与不同类型及不同取代形式的Michael受体发生反应, 生成相应的产物(如2a~2f, 4a~4f); 同时, 该反应对远程攫氢的位置具有良好的区域选择性, 主要只发生1, 5-氢迁移, 为官能基化的C—C键的高效构建提供了一种新方法.
图1
基于氮自由基的远端C—H键官能化反应
Figure1.
Remote C—H bond functionalization based on nitrogen-centered radical
上述两个反应可能的机理如图 2所示, 底物1或3在碱性条件下与蓝色LED光照射激发的催化剂*IrⅢ通过协同的质子耦合电子转移(PCET)过程[5c]或通过分步的去质子化/氧化过程, 将酰胺的N—H键转化为氮自由基5.后者经1, 5-氢迁移得到远端碳自由基6后, 再与Michael受体反应, 得到α羰基自由基7, 构建新的C—C键.随后, 7与还原态的IrⅡ作用, 经单电子转移(SET), 前者被还原为碳负离子8, 质子化后得到产物2或4; 同时IrⅡ被氧化为IrⅢ完成催化循环.
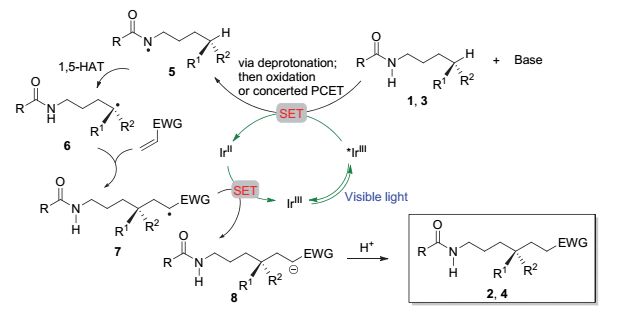 图2
远端C—H键官能化反应的可能机理
Figure2.
The proposed mechanism of remote C—H bond functionalization
图2
远端C—H键官能化反应的可能机理
Figure2.
The proposed mechanism of remote C—H bond functionalization
3 不对称共轭胺化和不对称远端C—H键官能化反应
Meggers等近年来发展了新型的“以金属为手性中心的”手性Lewis酸催化剂, 其在多种催化不对称反应中具有广泛应用[10].最近, 该小组受到Knowles等的研究工作[5c]启发, 将光照条件下通过PCET过程产生氮自由基与手性金属Lewis酸催化剂相结合, 分别实现了不对称共轭胺化[11]和不对称远端C—H键官能化反应[12].如图 3, 以氨基甲酸酯底物(9)为氮源, 在蓝色LED灯光照条件下产生氮自由基, 与α, β-不饱和-2-酰基咪唑衍生物(10)反应, 通过手性Lewis酸(Δ-RhO)的诱导生成不对称共轭胺化产物11[11].该方法具有良好的底物普适性, 其中烯酮10的β-位可为各种具有吸电子或给电子取代基团的芳环体系(如11a~11e).氨基甲酸酯底物(9)中氮原子的芳环上同样可耐受各种吸电子或给电子取代基(如11f~11j);其R1基团替换为多种烷基时亦给出良好的收率和优秀的对映选择性(如11k~11n).值得指出的是, N-烷基氨基甲酸酯或N-芳基酰胺在此体系下并不发生相应的不对称共轭胺化反应(11o和11p).
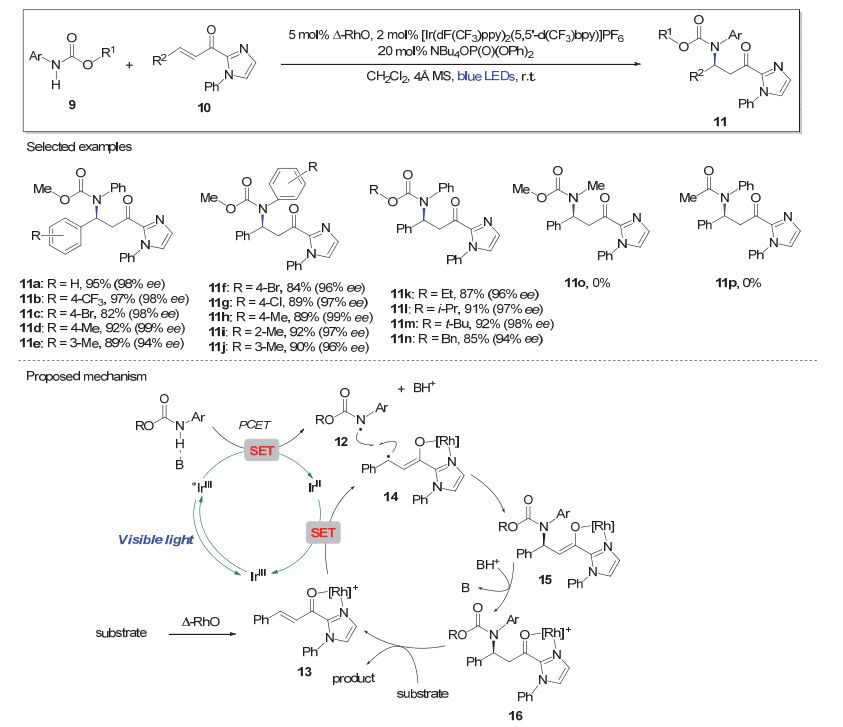 图3
不对称共轭胺化反应及其可能的机理
Figure3.
Asymmetric conjugate amination and its proposed mechanism
图3
不对称共轭胺化反应及其可能的机理
Figure3.
Asymmetric conjugate amination and its proposed mechanism
机理上来看, 氨基甲酸酯在光敏剂和有机碱的存在下, 受蓝色LED光照, 通过PCET过程将N—H键转化为氮自由基12.另一方面, 底物10与手性Lewis酸生成络合物13, 后者在还原态光敏剂(IrⅡ)的作用下转化为自由基中间体14. 12和14通过自由基偶联得到烯醇中间态15, 后者质子化后生成16. 16可释放出产物, 手性铑催化剂则进一步与底物10作用得到13, 完成催化循环.
Meggers小组发现, 当以酰胺17为底物时, 同样机理下产生的氮自由基可快速发生如图 2的1, 5-HAT过程, 在远端碳原子上产生碳自由基, 后者进一步参与到上述的手性Lewis酸催化循环(图 3), 最终得到不对称远端C—H键官能化反应的产物[12].如图 4, 底物17在相应光敏剂、碱及手性Lewis酸(△-RhO)的存在下与α, β-不饱和-2-酰基咪唑衍生物(18)反应, 通过蓝色LED光照射, 以中等到较好的收率及优异的对映选择性, 获得远端形成C—C键的产物19.该方法显示出较好的底物适用范围(如19a~19e), 其局限性主要体现在当烯酮18的β-位(R3)为烷基取代时, 反应不能发生(19f和19g).
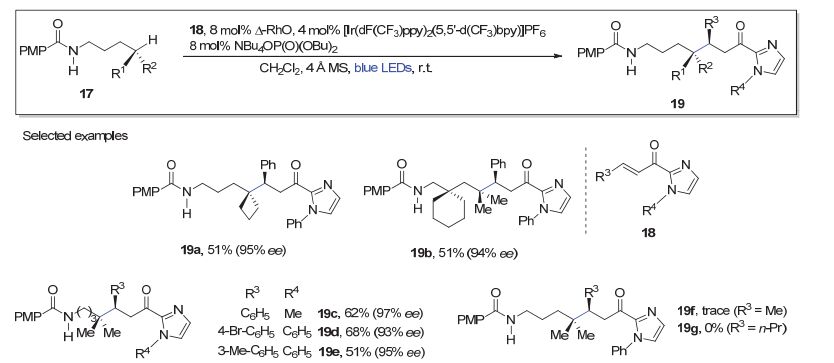 图4
不对称远端C—H键官能化反应
Figure4.
Asymmetric remote C—H bond functionalization reaction
图4
不对称远端C—H键官能化反应
Figure4.
Asymmetric remote C—H bond functionalization reaction
4 非活化烯烃的氢胺化反应
胺与烯烃的氢胺化反应是高效构筑C—N键的方法之一.虽然过渡金属催化的烯烃氢胺化反应在近年来的研究中得到了长足的发展[13], 然而普通烷基胺底物对非活化烯烃的直接氢胺化反应却未见报道.最近, Knowles小组[14]借助可见光介导的氮自由基阳离子反应历程实现了这一突破.如图 5, 该反应在催化量光敏剂[Ir(dF(Me)ppy)2dtbbpy]PF6和氢原子供体催化剂2, 4, 6-三异丙基苯硫酚(TRIP thiol)存在下, 通过蓝色LED灯照射, 烷基胺底物(20)对烯烃(21)进行氢胺化反应生成产物22.该反应在室温下进行, 条件温和, 底物适应性广, 具有优秀的反马氏区域选择性. 图 5给出了部分反应的例子, 其中普通烯烃(如22a和22b)和烯醇醚(如22c和22d)均可给出良好的产率; 更复杂的环状二级胺(如22e)和非环二级胺(如22f和22g)也表现出类似的反应性; 另外, 在相同的条件下, 分子内氢胺化反应可给出相应的含氮杂环产物(22h).
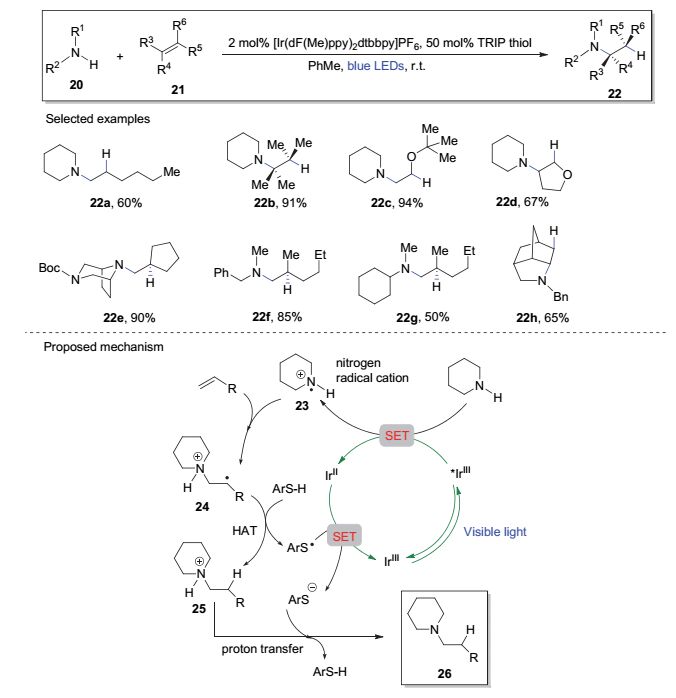 图5
非活化烯烃的氢胺化反应及其可能的机理
Figure5.
Hydroamination of unactivated alkenes and its proposed mechanism
图5
非活化烯烃的氢胺化反应及其可能的机理
Figure5.
Hydroamination of unactivated alkenes and its proposed mechanism
该反应可能的机理如图 5所示, 激发态的铱络合物与二级胺(如哌啶)作用, 通过SET生成氮自由基阳离子23, 后者与烯烃进行加成构建C—N键, 得到中间体24. 24在氢原子供体芳基硫酚的存在下发生氢原子转移(HAT), 所得的芳基硫自由基将还原态的IrⅡ氧化回到基态IrⅢ光敏剂.芳基硫负离子通过质子转移过程回到芳基硫酚, 同时给出产物26.
5 胺γ-位官能化反应
基于自由基的芳基迁移重排历程可被用于远端官能化反应, 传统上这通常需要利用化学计量且毒性较大的自由基引发剂或氧化剂, 在严苛的条件下来进行[15].最近Nevado等[16]借助可见光介导的芳基迁移过程, 在温和条件下实现了胺γ-位的官能化反应.如图 6, γ-位具有二芳基取代的三氟甲磺酰胺底物27在蓝色LED光照射, [Ir(dF-CF3ppy)2dtbpy]PF6为催化剂, K2HPO4作碱, 室温条件下, 可发生芳基迁移及进一步的γ-位官能化反应.具体来讲, 在反应体系中存在Michael受体的情况下, 该方法可在胺γ-位形成新的C—C键(如28a~28j); 相对应的是, 当反应中含有硫醇或烷基卤代物时, 可生成新的C—X键(X为氢或卤原子, 如28k和28l).该反应可能的机理如图 6所示, 底物中的N—H键在碱的作用下去质子化, 并进一步经过光催化剂介导的SET过程转化为氮自由基29.后者通过中间态30发生从碳到氮的芳基迁移重排过程, 得到碳自由基中间态31. 31进一步与反应体系中的Michael受体作用得到最终的产物, 磺酰基自由基则进一步参与到SET过程完成催化循环.作者在研究过程中观察到富电子的芳环更容易发生重排(如28h~28j), 推测这与29中氮自由基的缺电子特性密不可分.
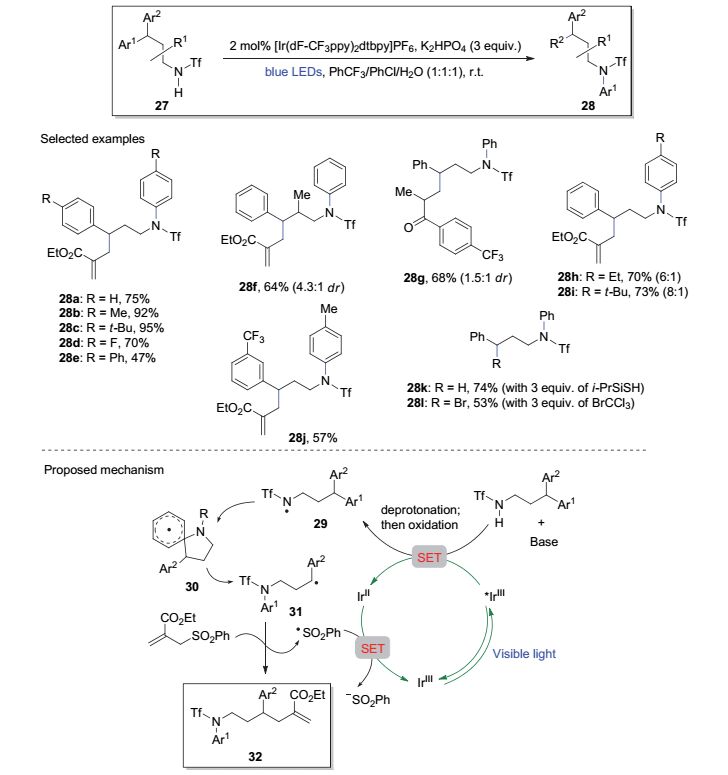 图6
胺γ-位官能化反应及其可能的机理
Figure6.
γ-Functionalization of amines and its proposed mechanism
图6
胺γ-位官能化反应及其可能的机理
Figure6.
γ-Functionalization of amines and its proposed mechanism
6 腙氮自由基加成反应
近年来, 华中师范大学肖文精和陈加荣课题组[17]对光催化条件下腙氮自由基的产生及其反应性进行了系统的研究. 2016年, 该小组[17b]通过对催化体系的合理设计及TEMPO添加剂的加入, 以[Ru(bpy)3]Cl2为光敏剂, K2CO3为碱, 将腙底物中N—H键转化为氮自由基, 进而以理想的区域选择性和中等以上的收率在温和条件下实现β, γ-不饱和腙33的6-endo氮自由基串联环化反应(图 7).在上述反应体系中, TEMPO并非自由基捕捉剂, 而是作为氢原子受体用于促进氢原子转移过程中氮杂烯丙基的去质子化.该光催化反应体系底物普适性良好, 在芳环(如34a~34e, 34i~34m)、芳杂环(如34f)或脂肪族(如34g~34h)取代体系中均可以中等以上的收率得到相应的1, 6-二氢哒嗪产物, 且不同电子效应的芳环取代基对反应没有明显影响.反应可能的机理如图 7所示, 腙33在碱性条件下去质子化得到氮负离子中间体后, 经SET过程, 被蓝色LED灯光照射激发的*RuⅡ催化剂氧化成氮自由基35. 35经6-endo环化反应得到苄基自由基36后, 通过TEMPO介导的氢原子转移过程, 完成1, 6-二氢哒嗪34的合成.同时, 还原态的RuⅠ被CHCl3或其他氧化剂氧化为基态的RuⅡ催化剂, 完成催化反应循环.
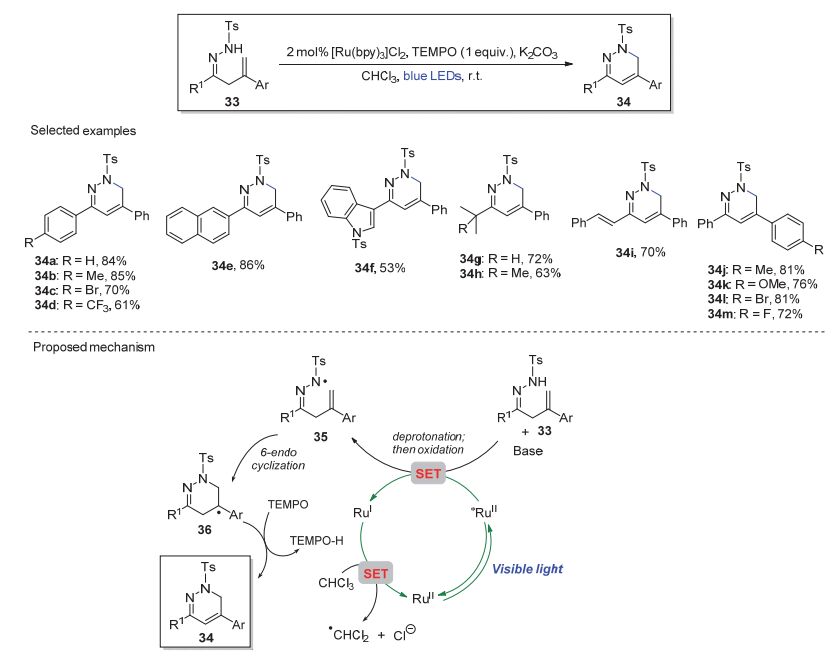 图7
基于氮自由基合成1, 6-二氢哒嗪及其可能的机理
Figure7.
Synthesis of 1, 6-dihydropyridazines based on nitrogen-centered radical and its proposed mechanism
图7
基于氮自由基合成1, 6-二氢哒嗪及其可能的机理
Figure7.
Synthesis of 1, 6-dihydropyridazines based on nitrogen-centered radical and its proposed mechanism
在上述工作的基础上, 陈加荣等[17d]还发展了一种使用有机光催化剂将腙底物中N—H键直接转化为氮自由基, 并用于分子内烯烃羟氨基化反应的方法(图 8).在蓝色LED光照射和氧气作用下, 经TEMPO和有机小分子光敏剂Mes-Acr的双重作用, 以K2CO3为碱, β, γ-不饱和腙37经氮自由基反应分别得到38或39.其中, 当37的烯烃2-位被烷基取代时, 经5-exo环化过程得到相应的吡唑衍生物38; 当37的烯烃2-位被芳基取代时, 催化循环过程中新生成的苄基自由基得以稳定, 则发生6-endo氮自由基环化反应, 得到哒嗪衍生物39.该催化体系同样表现出良好的底物普适性, 无论是何种取代类型的底物, 都可以中等以上收率获得相应的芳环(如38a~38e, 38g, 39a~39d)、芳杂环(如39e)或烷基取代(如38f, 39f)产物.值得一提的是, 在该光反应体系中, 氧气兼具了氧化剂和氧源的功能.
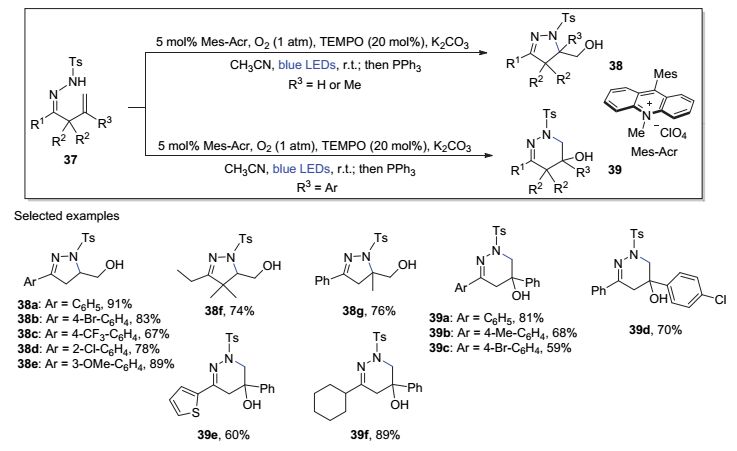 图8
基于腙氮自由基的分子内烯烃羟氨基化反应
Figure8.
Intramolecular alkene oxyamination based on nitrogen-centered radical of hydrazones
图8
基于腙氮自由基的分子内烯烃羟氨基化反应
Figure8.
Intramolecular alkene oxyamination based on nitrogen-centered radical of hydrazones
7 芳(杂)环的脱氢胺化反应
2016年, 俞寿云等[18]报道了可见光催化的非活化磺酰胺对芳杂环的直接氧化酰胺化反应(图 9).研究显示, N-甲基-对甲苯磺酰胺(41)在催化量的光敏剂[Ir(ppy)2(dtbbpy)]PF6及化学计量的氧化剂NaClO存在下, 可有效地对系列芳杂环底物进行直接酰胺化.具体而言, 各种具有芳环取代或不同氮原子取代基团的吲哚核均可获得较高的产率(如42a~42i); 其他芳杂环体系如吡咯和苯并呋喃衍生物等亦可有效地进行直接酰胺化反应转化为相应产物42j~42l.磺酰胺部分带有其他官能团如炔基、烯基、酯基、氨基甲酸酯等时, 该反应也能很好耐受(如42m和42n).另外, 当芳杂环体系本身含有磺酰胺基团时, 该光催化反应过程可以分子内反应的形式进行, 生成相应的杂环衍生物(如42o和42p).该反应可能的机理如图 9所示, 光催化剂IrⅢ在白色LED灯照射下生成激发态的*IrⅢ络合物, 后者被NaClO进一步氧化.磺酰胺底物中的N—H键在IrⅣ和碱的存在下通过去质子化和氧化过程转化为氮自由基, 并与芳杂环进行加成, 所得中间体44再经氧化及去质子化过程生成酰胺化产物42.
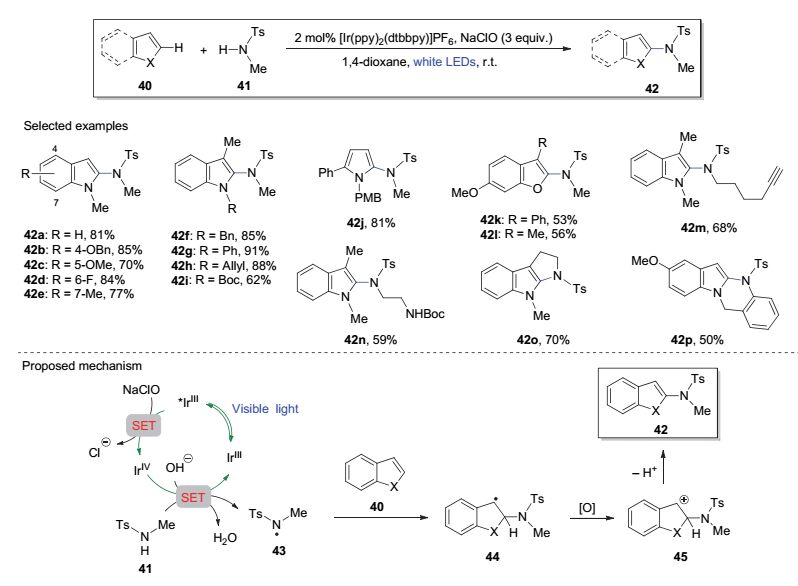 图9
芳杂环体系直接酰胺化反应及其可能的机理
Figure9.
Direct amidation of heteroarenes and its proposed mechanism
图9
芳杂环体系直接酰胺化反应及其可能的机理
Figure9.
Direct amidation of heteroarenes and its proposed mechanism
最近, Murakami, Itami及合作者[19]利用可见光催化条件实现了苯磺酰亚胺底物对芳环或芳杂环体系的直接胺基化.如图 10, 在蓝色LED灯照射下, 以[Ru(bpy)3]Cl2•6H2O为光催化剂, IBB (49)作氧化剂, 功能化的芳香环或芳杂环体系46与苯磺酰亚胺47反应可生成胺基化产物48.苯磺酰亚胺底物的苯环可带有各种给电子或吸电子取代基(如48a~48d).诸多芳环体系, 如萘、菲、苯并噻吩、苯并呋喃、嘧啶及具有高度共轭体系的碗烯和晕苯等均表现出良好的反应性(如48e~48k).该方法为合成一系列有用的芳环磺酰亚胺化产物提供了便利.机理上, 受可见光激发的光敏剂在氧化剂IBB的作用下通过SET生成RuⅢ络合物, 该络合物将苯磺酰亚胺底物(47)中的N—H键转化为活性的氮自由基(50)后回到RuⅡ完成催化循环.氮自由基与芳环体系(如萘)反应, 并进一步经历氧化、去质子化过程生成磺酰亚胺化产物(如48a).
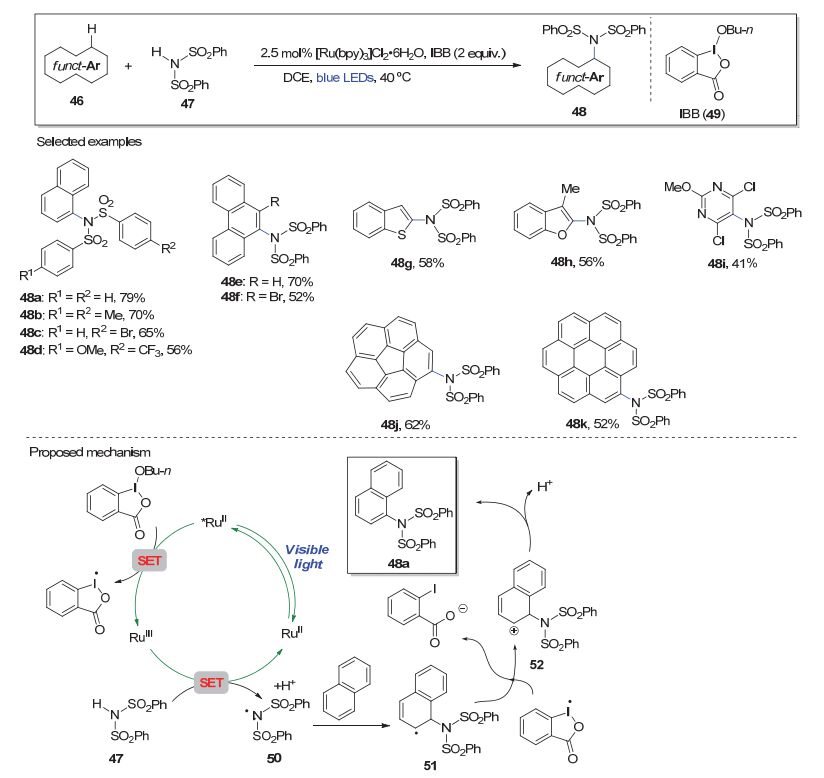 图10
芳(杂)环体系直接磺酰亚胺化反应及其可能的机理
Figure10.
Direct imidation of (hetero)arenes and its proposed mechanism
图10
芳(杂)环体系直接磺酰亚胺化反应及其可能的机理
Figure10.
Direct imidation of (hetero)arenes and its proposed mechanism
夏吾炯小组[20]于近期报道了可见光介导的苯酚与二苯胺类化合物的交叉脱氢胺化反应.如图 11所示, 底物53和54在有机光敏剂55和氧化剂(NH4)2S2O8的存在下, 受蓝色LED灯光照射可在苯酚的邻位发生直接胺化反应生成相应产物56[20b].该方法具有较好的底物普适性, 系列非环或环状的二苯胺底物均能获得中等到优秀的反应收率, 其中芳环上的各种给电子和吸电子取代基均能耐受(如56a~56g).另外, 苯酚部分含有各种基团时也体现出良好的反应性(如56h和56i).值得指出的是, 对于环状的二苯胺底物可在不加入光敏剂的条件下发生反应, 而使用有机光敏剂后反应效率则大大提高(如56j~56m).作者提出了该反应可能的机理, 如图 11所示.简而言之, 在光敏剂、氧化剂存在及可见光照的条件下, 苯酚(53)与二苯胺底物(54)经过系列的SET过程分别生成芳基自由基57和氮自由基58, 两者进行自由基交叉偶联即可得到相应的脱氢胺化产物.
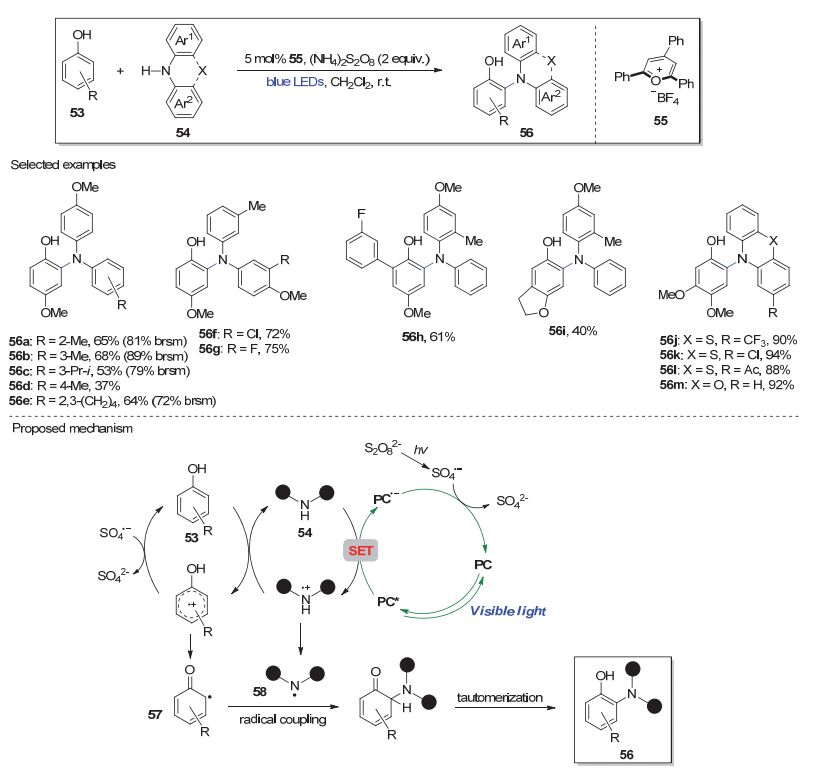 图11
苯酚与二苯胺类化合物的交叉脱氢胺化反应及其可能的机理
Figure11.
Cross-dehydrogenative amination between phenols and diarylamines and its proposed mechanism
图11
苯酚与二苯胺类化合物的交叉脱氢胺化反应及其可能的机理
Figure11.
Cross-dehydrogenative amination between phenols and diarylamines and its proposed mechanism
上述方法几例光催化的脱氢胺化反应直接利用含有未活化C—H键和N—H键的前体物, 在温和的反应条件下构建芳基C—N键实现芳胺化, 具有重要的原子和步骤经济性.
8 氮自由基串联反应构建吲哚生物碱骨架及其全合成
最近, 秦勇课题组[21]借助光催化条件直接活化N—H键产生氮自由基, 并进一步通过串联反应实现了不同复杂吲哚生物碱骨架和天然产物的合成.如图 12所示, 对甲苯磺酰基(Ts)保护的手性苯胺底物59 (或61, 63)在蓝色LED灯照射下, 以Ir(dtbbpy)(ppy)2PF6为光敏剂和KHCO3作碱, 可以成功将苯胺的N—H官能基转化为氮自由基, 缺电子的氮自由基随后分子内加成到富电性的烯胺的β碳原子上, 并进一步引发分子内/分子内、分子内/分子间和分子内/分子间/分子内的三种类型自由基串联反应, 从而一锅多步地高效合成白坚木型60、四氢咔波啉型62和柯楠因型64三种官能化的手性单萜吲哚生物碱骨架.以第一类分子内/分子内自由基串联反应为例, 该反应可能的机理如图 12所示.手性苯胺65在KHCO3碱性条件下首先失去质子, 随后通过SET过程被蓝色LED灯光照射激发的*IrⅢ催化剂氧化成氮自由基66.后者经历自由基串联反应历程转化为碳自由基中间体67. 67被还原态的IrⅡ复合物还原成碳负离子中间体68的同时, IrⅡ自身重新被氧化成IrⅢ催化剂, 完成反应循环过程.经上述循环得到的中间体68进一步获得一个质子即可得到白坚木型衍生物60a.
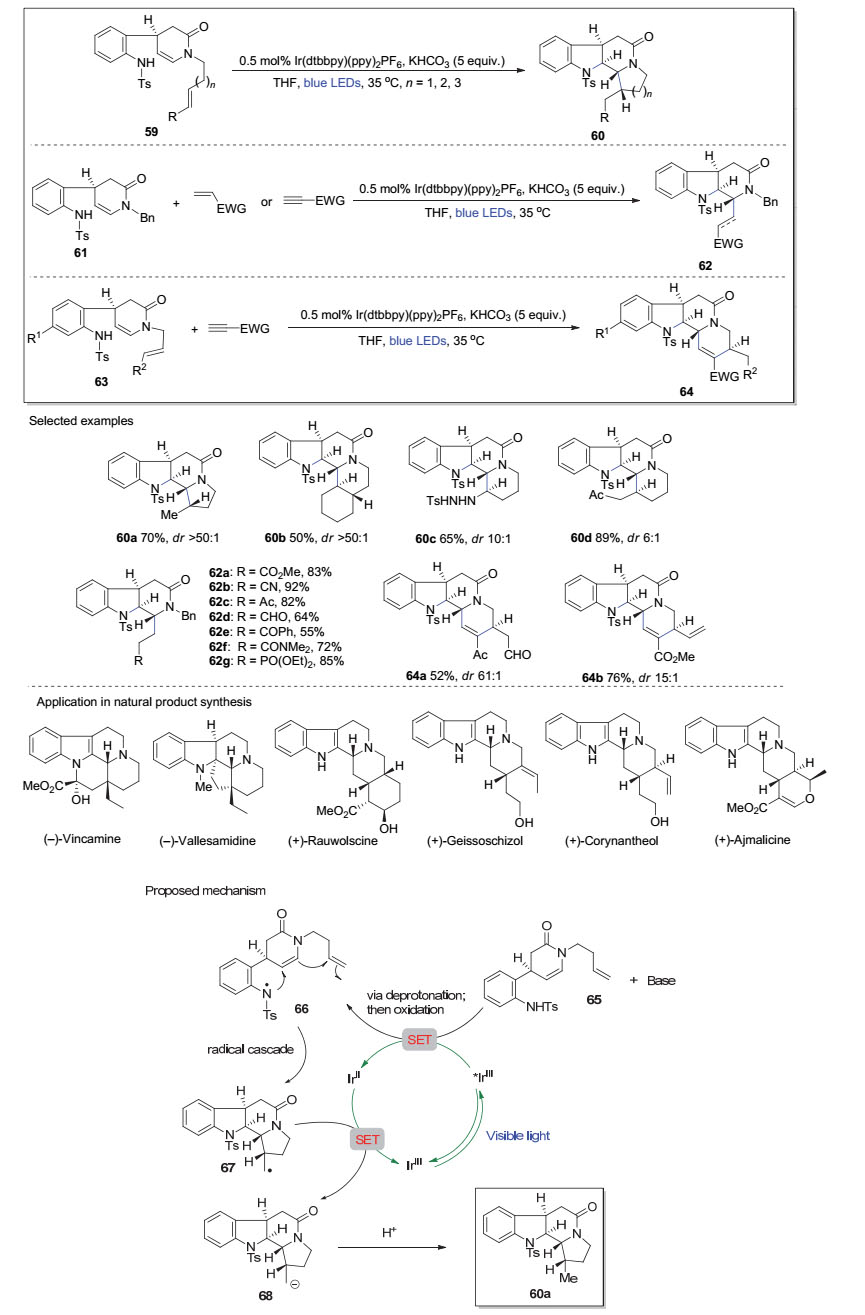 图12
光催化氮自由基串联反应及其可能的反应机理与在天然产物合成中的应用
Figure12.
Photocatalytic nitrogen radical cascade and its proposed mechanism and application in natural product synthesis
图12
光催化氮自由基串联反应及其可能的反应机理与在天然产物合成中的应用
Figure12.
Photocatalytic nitrogen radical cascade and its proposed mechanism and application in natural product synthesis
上述三类自由基串联反应可以中等到优秀的收率同时实现多个C—N键及C—C键的快速构建, 进而形成分子复杂性.更为重要的是, 三类串联反应中新生成的多个手性中心均可实现良好的立体化学控制, 且对酯基、氰基、酮基、醛基等多种官能团具有良好的底物适应性.改变底物的官能团可为合成骨架丰富、稠合方式多样的三种不同类型的吲哚生物碱骨架提供便利(图 12).以该方法为关键反应, 秦勇等进一步实现了(-)-vincamine, (-)-vallesamidine, (+)-rauwolscine, (+)-geissoschizol, (+)-corynantheol及(+)-ajmalicine等33个吲哚生物碱的不对称集群式合成.
9 小结与展望
光催化活化N—H键产生氮自由基的研究在近年来取得了重要的进展, 它对于以前用传统方法不能实现的转化提供了新的思路, 例如本文中介绍的非活化烯烃的氢胺化反应, 芳环及芳杂环的脱氢胺化反应, 以及串联反应构建吲哚生物碱骨架等.该方法具有条件温和、绿色、环保、经济等显著优点.相关的工作再次体现出新型可见光介导的化学转化过程的优势和应用价值.可以预见, 关于该领域进一步的研究重点将聚焦于实现更多样化的反应类型, 为重要药物分子、活性天然产物及功能材料等的合成提供便利.
-
-
[1]
For selected reviews on the N-radical chemistry, see: (a) Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603.(b) Stella, L. In Radicals in Organic Synthesis, Vol. 2, Eds.: Renaud, P.; Sibi, M. P., Wiley, New York, 2001, p. 407.(c) Quiclet-Sire, B.; Zard, S. Z. Beilstein J. Org. Chem. 2013, 9, 557.
-
[2]
For selected examples, see:(a) Lessard, J.; Cote, R.; Mackiewicz, P.; Furstoss, R.; Waegell, B. J. Org. Chem. 1978, 43, 3750.(b) Boivin, J.; Fouquet, E.; Schiano, A.-M.; Zard, S. Z. Tetrahedron 1994, 50, 1769.(c) Gennet, D.; Zard, S. Z.; Zhang, H. Chem. Commun. 2003, 1870.(d) Lu, H.; Li, C. Tetrahedron Lett. 2005, 46, 5983.(e) Guin, J.; Fr hlich, R.; Studer, A. Angew. Chem. Int. Ed. 2008, 47, 779.
-
[3]
For selected examples, see:(a) Sherman, E. S.; Chemler, S. R.; Tan, T. B.; Gerlits, O. Org. Lett. 2004, 6, 1573.(b) Sherman, E. S.; Fuller, P. H.; Kasi, D.; Chemler, S. R. J. Org. Chem. 2007, 72, 3896.(c) Zeng, W.; Chemler, S. R. J. Am. Chem. Soc. 2007, 129, 12948.(d) Zhu, X.; Wang, Y.-F.; Ren, W.; Zhang, F.-L.; Chiba, S. Org. Lett. 2013, 15, 3214.(e) Zhu, M.-K.; Chen, Y.-C.; Loh, T. P. Chem. Eur. J. 2013, 19, 5250.(f) Duan, X.-Y.; Zhou, N.-N.; Fang, R.; Yang, X.-L.; Yu, W.; Han, B. Angew. Chem. Int. Ed. 2014, 53, 3158.(g) Duan, X. Y.; Yang, X. L.; Jia, P. P.; Zhang, M.; Han, B. Org. Lett. 2015, 17, 6022.
-
[4]
For selected reviews on the visible light photocatalysis, see: (a) Xuan, J. ; Xiao, W. -J. Angew. Chem. Int. Ed. 2012, 51, 6828. (b) Shi, L. ; Xia, W. -J. Chem. Soc. Rev. 2012, 41, 7687. (c) Prier, C. K. ; Rankic, D. A. ; Macmillan, D. W. Chem. Rev. 2013, 113, 5322. (d) Xi, Y. -M. ; Yi, H. ; Lei, A. -W. Org. Biomol. Chem. 2013, 11, 2387. (e) Schultz, D. M. ; Yoon, T. P. Science 2014, 343, 985. (f) Xuan, J. ; Zhang, Z. -G. ; Xiao, W. -J. Angew. Chem. Int. Ed. 2015, 54, 15632. (g) Karkas, M. D. ; Porco Jr., J. A. ; Stephenson, C. R. Chem. Rev. 2016, 116, 9683. (h) Romero, N. A. ; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075. (i) Chen, J. -R. ; Hu, X. -Q. ; Lu, L. -Q. ; Xiao, W. -J. Acc. Chem. Res. 2016, 49, 1911. (j) Liu, Y. ; Song, R. ; Li, J. Sci. China Chem. 2016, 59, 161. (k) Zhang, J. ; Chen, Y. Acta Chim. Sinica 2017, 75, 41(in Chinese). (张晶, 陈以昀, 化学学报, 2017, 75, 41. )(l) Chen, J. -R. ; Yan, D. -M. ; Wei, Q. ; Xiao, W. -J. ChemPhotoChem 2017, 1, 148.
-
[5]
For recent reviews, see:(a) Chen, J.-R.; Hu, X. Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044.(b) Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2016, 45, 3069.(c) Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546.(d) Nguyen, L. Q.; Knowles, R. R. ACS Catal. 2016, 6, 2894.(e) K rk s, M. D. ACS Catal. 2017, 7, 4999.
-
[6]
For recent reviews, see:(a) Gensch, T.; Hopkinson, M. N.; Glorius, F.; Wencel-Delord, J. Chem. Soc. Rev. 2016, 45, 2900.(b) Zhu, R.-Y.; Farmer, M. E.; Chen, Y.-Q.; Yu, J.-Q. Angew. Chem. Int. Ed. 2016, 55, 10578.(c) Della Ca', N.; Fontana, M.; Motti, E.; Catellani, M. Acc. Chem. Res. 2016, 49, 1389.(d) He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev. 2017, 117, 8754.
-
[7]
For selected examples, see:(a) Qin, Q.; Yu, S. Org. Lett. 2015, 17, 1894.(b) Hollister, K. A.; Conner, E. S.; Spell, M. L.; Deveaux, K.; Maneval, L.; Beal, M. W.; Ragains, J. R. Angew. Chem. Int. Ed. 2015, 54, 7837.(c) Huang, F.-Q.; Dong, X.; Qi, L.-W.; Zhang, B. Tetrahedron Lett. 2016, 57, 1600.(d) Zhang, J.; Li, Y.; Zhang, F.; Hu, C.; Chen, Y. Angew. Chem. Int. Ed. 2016, 55, 1872.(e) Wang, C.; Harms, K.; Meggers, E. Angew. Chem. Int. Ed. 2016, 55, 13495.(f) Shaaban, S.; Oh, J.; Maulide, N. Org. Lett. 2016, 18, 345.(g) Parasram, M.; Chuentragool, P.; Sarkar, D.; Gevorgyan, V. J. Am. Chem. Soc. 2016, 138, 6340.(h) Hu, X.-Q.; Chen, J.-R.; Xiao, W.-J. Angew. Chem. Int. Ed. 2017, 56, 1960.
-
[8]
Choi, G. J.; Zhu, Q.; Miller, D. C.; Gu, C. J.; Knowles, R. R. Nature 2016, 539, 268. doi: 10.1038/nature19811
-
[9]
Chu, J. C. K.; Rovis, T. Nature 2016, 539, 272. doi: 10.1038/nature19810
-
[10]
Zhang, L.; Meggers, E. Acc. Chem. Res. 2017, 50, 320. doi: 10.1021/acs.accounts.6b00586
-
[11]
Zhou, Z.; Li, Y.; Han, B.; Gong, L.; Meggers, E. Chem. Sci. 2017, 8, 5757. doi: 10.1039/C7SC02031G
-
[12]
Yuan, W.; Zhou, Z.; Gong, L.; Meggers, E. Chem. Commun. 2017, 53, 8964. doi: 10.1039/C7CC04941B
-
[13]
For a recent review, see: Huang, L.; Arndt, M.; Goo en, K.; Heydt, H.; Goo en, L. J. Chem. Rev. 2015, 115, 2596. doi: 10.1021/cr300389u
-
[14]
Musacchio, A. J.; Lainhart, B. C.; Zhang, X.; Naguib, S. G.; Sherwood, T. C.; Knowles, R. R. Science 2017, 355, 727. doi: 10.1126/science.aal3010
-
[15]
For a recent review, see: Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Chem. Soc. Rev. 2015, 44, 5220. doi: 10.1039/C4CS00467A
-
[16]
Shu, W.; Genoux, A.; Li, Z.; Nevado, C. Angew. Chem. Int. Ed. 2017, 56, 10521. doi: 10.1002/anie.v56.35
-
[17]
(a) Hu, X. -Q. ; Chen, J. -R. ; Wei, Q. ; Liu, F. -L. ; Deng, Q. -H. ; Beauchemin, A. M. ; Xiao, W. -J. Angew. Chem. Int. Ed. 2014, 53, 12163. (b) Hu, X. -Q. ; Qi, X. ; Chen, J. -R. ; Zhao, Q. -Q. ; Wei, Q. ; Lan, Y. ; Xiao, W. -J. Nat. Commun. 2016, 7, 11188. (c) Zhao, Q. -Q. ; Hu, X. -Q. ; Yang, M. -N. ; Chen, J. -R. ; Xiao, W. -J. Chem. Commun. 2016, 52, 12749. (d) Hu, X. -Q. ; Chen, J. ; Chen, J. -R. ; Yan, D. -M. ; Xiao, W. -J. Chem. Eur. J. 2016, 22, 14141. (e) Zhao, Q. -Q. ; Chen, J. ; Yan, D. -M. ; Chen, J. -R. ; Xiao, W. -J. Org. Lett. 2017, 19, 3620. (f) Yu, X. ; Zhou, F. ; Chen, J. ; Xiao, W. Acta Chim. Sinica 2017, 75, 86(in Chinese). (余晓叶, 周帆, 陈加荣, 肖文精, 化学学报, 2017, 75, 86. )
-
[18]
Tong, K.; Liu, X.; Zhang, Y.; Yu, S. Chem. Eur. J. 2016, 22, 15669. doi: 10.1002/chem.201604014
-
[19]
Ito, E.; Fukushima, T.; Kawakami, T.; Murakami, K.; Itami, K. Chem 2017, 2, 383. doi: 10.1016/j.chempr.2017.02.006
-
[20]
(a) Zhao, Y.; Huang, B.; Yang, C.; Xia, W. Org. Lett. 2016, 18, 3326. (b) Zhao, Y.; Huang, B.; Yang, C.; Li, B.; Gou, B.; Xia, W. ACS Catal. 2017, 7, 2446.
-
[21]
Wang, X.; Xia, D.; Qin, W.; Zhou, R.; Zhou, X.; Zhou, Q.; Liu, W.; Dai, X.; Wang, H.; Wang, S.; Tan, L.; Zhang, D.; Song, H.; Liu, X.-Y.; Qin, Y. Chem 2017, 2, 803. doi: 10.1016/j.chempr.2017.04.007
-
[1]
-

图 1 基于氮自由基的远端C—H键官能化反应
Figure 1 Remote C—H bond functionalization based on nitrogen-centered radical

图 2 远端C—H键官能化反应的可能机理
Figure 2 The proposed mechanism of remote C—H bond functionalization

图 3 不对称共轭胺化反应及其可能的机理
Figure 3 Asymmetric conjugate amination and its proposed mechanism

图 5 非活化烯烃的氢胺化反应及其可能的机理
Figure 5 Hydroamination of unactivated alkenes and its proposed mechanism

图 6 胺γ-位官能化反应及其可能的机理
Figure 6 γ-Functionalization of amines and its proposed mechanism

图 7 基于氮自由基合成1, 6-二氢哒嗪及其可能的机理
Figure 7 Synthesis of 1, 6-dihydropyridazines based on nitrogen-centered radical and its proposed mechanism

图 8 基于腙氮自由基的分子内烯烃羟氨基化反应
Figure 8 Intramolecular alkene oxyamination based on nitrogen-centered radical of hydrazones

图 9 芳杂环体系直接酰胺化反应及其可能的机理
Figure 9 Direct amidation of heteroarenes and its proposed mechanism

图 10 芳(杂)环体系直接磺酰亚胺化反应及其可能的机理
Figure 10 Direct imidation of (hetero)arenes and its proposed mechanism

图 11 苯酚与二苯胺类化合物的交叉脱氢胺化反应及其可能的机理
Figure 11 Cross-dehydrogenative amination between phenols and diarylamines and its proposed mechanism
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 302
- 文章访问数: 7485
- HTML全文浏览量: 3407
 下载:
下载:
