引用本文:
张盼盼, 吕龙, 沈其龙. 直接三氟甲硫基化试剂及方法的研究进展[J]. 化学学报,
2017, 75(8): 744-769.
doi:
10.6023/A17050202

Citation: Zhang Panpan, Lu Long, Shen Qilong. Recent Progress on Direct Trifluoromethylthiolating Reagents and Methods[J]. Acta Chimica Sinica, 2017, 75(8): 744-769. doi: 10.6023/A17050202
Citation: Zhang Panpan, Lu Long, Shen Qilong. Recent Progress on Direct Trifluoromethylthiolating Reagents and Methods[J]. Acta Chimica Sinica, 2017, 75(8): 744-769. doi: 10.6023/A17050202
直接三氟甲硫基化试剂及方法的研究进展
摘要:
三氟甲硫基脂溶性高、吸电子能力强,将其引入药物分子中可以改变分子药代动力学以及物理化学特性,提高代谢稳定性.含三氟甲硫基的分子在医药、农药、材料以及含氟日用品等领域得到了广泛的应用.因此,如何高效地向分子中引入三氟甲硫基团成为了近些年来有机氟化学领域的一个研究热点.经过几年的研究,新的三氟甲硫基化试剂及方法得到了快速的发展,为药物化学家提供了方便简洁地引入三氟甲硫基团的工具箱.在本篇综述中,将首先简要介绍间接引入三氟甲硫基团的方法,随后将对直接引入三氟甲硫基团的方法,尤其是近些年来发展的过渡金属催化、亲电三氟甲硫基化以及自由基类型的三氟甲硫基化方法做着重的介绍.最后将对三氟甲硫基化领域存在的问题和难点进行简要的展望.
English
Recent Progress on Direct Trifluoromethylthiolating Reagents and Methods
Abstract:
With a significantly high Hansch's hydrophobicity parameter (π=1.44), electron-withdrawing trifluoromethylthio group (CF3S-) has been considered as one of the most lipophilic substituents and privileged fragments that are able to improve drug molecules' pharmacokinetic and physicochemical properties such as lipophilicity and metabolic stability. It is well-known that incorporation of the trifluoromethylthio group into small molecules greatly enhances its ability to cross lipid membranes and in vivo absorpotion rate. In addition, the high electronegativity of the trifluoromethylthio group significantly improves the small molecule's stablity in acidic environments. Not surprisingly, the trifluoromethylthio group has been of special attention not only from the academia but also from pharmaceutical and agrochemical industry for their use in isostere-based drug design. Development of highly efficient methods for the introduction of the trifluoromethylthio group into small molecules, thereafter, has become a subject of recent focus in the field of organic chemistry. In the early 1960s, a few methods for the formation of trifluoromethylthioethers were reported, which typically involved halogen exchange of the trichloromethyl-substituted compounds and trifluoromethylation of thiolated substrates. However, the conditions of these methods were harsh and incompatible with many common functional groups. Since 2008, new reagents and methods that were able to efficiently incorporate the trifluoromethylthio group under mild conditions have emerged, that pave the way for the facile introduction of trifluoromethylthio group into site-specific positions of the target molecules. In this review, we will first briefly introduce the indirect strategies for trifluoromethylthiolation including halogen exchange and trifluoromethylation of thiolated substrates, and then focus on the direct trifluoromethylthiolation strategies including the transition metal-catalyzed trifluoromethylthiolation reactions, electrophilic trifluoromethylthiolation reactions with electrophilic trifluoromethylthiolating reagents and radical trifluoromethylthiolations. These methods represent the most straightforward and promising approaches for the incorporation of the trifluoromethylthio group into small molecules. At the end, we will discuss the remaining problems and challenges in this particular field.
-
Key words:
- fluorine chemistry
- / trifluoromethylthiolation
- / indirect strategy
- / direct method
-
1 引言
2016年10月, 中国科学院科技战略咨询研究院、中国科学院文献情报中心与Clarivate Analytics公司(原汤森路透知识产权与科技事业部)联合发布了《2016研究前沿》报告[1].从该报告的统计数据中我们发现, 在化学与材料科学领域中, 有关三氟甲硫基化反应的研究位列第二!中国科学家在该研究领域发表核心论文多达47篇, 被引频次达到了3168次之多!由此可见, 对于三氟甲硫基化反应的研究是目前有机化学领域的一个热点.
 表 1
2016年化学与材料科学领域热点前沿
Table 1.
Frontiers of chemistry and material science in 2016
表 1
2016年化学与材料科学领域热点前沿
Table 1.
Frontiers of chemistry and material science in 2016
排名 热点前沿 核心论文 被引频次 1 基于非富勒烯受体的有机太阳能电池 41 2249 2 三氟甲硫基化反应 47 3168 3 摩擦纳米发电机 43 2846 4 非贵金属电解水纳米催化剂 26 2427 5 金催化的有机合成 23 2062 6 高效钙钛矿型太阳能电池 30 1647 7 半导体/石墨烯纳米复合物光催化剂 21 3176 8 白光LED用荧光粉 44 4690 9 石墨烯过滤膜 22 3125 10 锂离子电池 4 1998 摘自《2016研究前沿》. 该领域受到研究者们的广泛关注, 其原因在于三氟甲硫基独特的性质以及其重大的应用前景.三氟甲硫基官能团有着较强的吸电子能力, 可以有效地降低底物的电子云密度, 从而提高分子的代谢稳定性; 硫原子有着多种氧化态, 可以进一步氧化成亚砜以及砜基官能团, 从而可以更加灵活地通过电性来对底物的活性进行调控; 脂溶性对生物体对药物分子的吸收、分布以及对药物分子到达靶点的能力有着决定性的影响.而三氟甲硫基官能团在常见的含氟取代基中拥有最高的π系数(1.44) [2], 将其引入分子中可以极大程度地提高分子的脂溶性, 增加细胞膜的穿透性.
如何向分子中引入三氟甲硫基官能团在近些年来得到了长足和快速的发展.关于三氟甲硫基官能团的引入, 相关的总结也有不少[3], 但考虑到该领域快速的发展, 我们认为仍然需要对该领域进行一个归纳.
2 三氟甲硫基的引入
根据底物和三氟甲硫基来源的不同, 我们可以将引入三氟甲硫基的方法分为直接法和间接法.在该领域早期发展的方法大部分都属于间接法, 包括含卤前体的氟卤交换法和含硫底物的亲电、亲核以及自由基类型的三氟甲基化方法(Scheme 1).
 图式1
间接引入三氟甲硫基的方法
图式1.
Indirect strategies for trifluoromethylthiolation
图式1
间接引入三氟甲硫基的方法
图式1.
Indirect strategies for trifluoromethylthiolation
对于氟卤交换法[4]而言, 首先需要预先制备含卤底物.其次需要使用高活性的亲核性氟源如HF、SbF3等进行氟卤交换反应.该方法在实验室操作较为繁琐, 但在工业生产中应用较广.
对于含硫底物的三氟甲基化方法而言, 首先也需要预先制备含硫底物, 如二硫醚、硫氰基底物、硫氯底物或者硫醇、硫酚类底物等.其次采用亲核性[5]、亲电性三氟甲基源[6]或者在紫外光照的条件下[7]对含硫底物进行三氟甲基化.该方法对于简单底物比较适用, 但对于一些复杂的底物分子而言, 制备含硫底物的难度是比较大的, 也就限制了该方法在分子合成设计后期的应用.
而直接向分子中引入三氟甲硫基有着简单、高效的优势, 更有利于在分子合成设计的后期来对底物进行三氟甲硫基化修饰.根据三氟甲硫基的来源, 我们可以将直接三氟甲硫基化法分为亲核型、亲电型以及自由基型三氟甲硫基化法(Scheme 2).
 图式2
直接引入三氟甲硫基的方法
图式2.
Direct strategies for trifluoromethylthiolation
图式2
直接引入三氟甲硫基的方法
图式2.
Direct strategies for trifluoromethylthiolation
2.1 亲核三氟甲硫基化法
2.1.1 亲核三氟甲硫基源
亲核三氟甲硫基化法是采用亲核性三氟甲硫基源与亲电底物反应来实现的.而常用的亲核三氟甲硫基源包括三氟甲硫基金属或非金属试剂.
Muetterties等[8]最早通过HgF2和CS2在250 ℃下制备了首个亲核三氟甲硫基源Hg (SCF3)2.而制备的Hg (SCF3)2可以与Cu粉或者AgNO3反应来进一步制备CuSCF3与AgSCF3, 但在当时其制备的纯度不高.随后MacDuffie等[9]采用与Muetterties类似的方法, 通过AgF和CS2在140 ℃下反应, 以70%~80%的收率制备AgSCF3. Yagupolskii小组[10]从AgSCF3出发, 与CuBr发生一步复分解反应, 可以以当量的收率获得CuSCF3 (Scheme 3).
 图式3
三氟甲硫汞、三氟甲硫银和三氟甲硫铜的制备
图式3.
Preparation of Hg (SCF3)2, AgSCF3 and CuSCF3
图式3
三氟甲硫汞、三氟甲硫银和三氟甲硫铜的制备
图式3.
Preparation of Hg (SCF3)2, AgSCF3 and CuSCF3
随后, 其他一系列的三氟甲硫基物种(K, Cs, NMe4, S (NMe2)3, TDAE)陆续被报道, Yagupolskii小组[11]采用一锅法, 从硫粉和Ruppert-Prakash试剂出发, 可以简单便捷地合成相应的Me4NSCF3、CsSCF3等亲核性三氟甲硫基源(Scheme 4).
 图式4
一锅法制备MSCF3
图式4.
One-pot synthesis of MSCF3
图式4
一锅法制备MSCF3
图式4.
One-pot synthesis of MSCF3
2.1.2 早期直接亲核三氟甲硫基化反应
早期的三氟甲硫基化法主要采用上述各种亲核性的三氟甲硫基源, 与烷基、芳基卤化物等亲电底物在高温条件下实现三氟甲硫基的引入.而为了解决亲核三氟甲硫基源稳定性差的问题, 研究者们也发展了现场生成亲核三氟甲硫基源的方法.如Remy小组[12]利用Hg (SCF3)2与Cu粉现场生成CuSCF3, Yagupolskii小组[13]使用CF3SSCF3与Cu粉现场产生CuSCF3, 上海有机所的陈庆云先生[14]利用CuI, Chen试剂与硫粉现场生成CuSCF3, Clark小组[15]利用CSCl2与KF、NMe4F现场生成KSCF3、NMe4SCF3, 来实现芳基碘化物和溴化物以及活泼氯化物的三氟甲硫基化反应(Scheme 5).
 图式5
早期发展的亲核三氟甲硫基化反应
图式5.
Early development of nucleophilic trifluoromethylation
图式5
早期发展的亲核三氟甲硫基化反应
图式5.
Early development of nucleophilic trifluoromethylation
2.1.3 过渡金属催化三氟甲硫基化反应
从20世纪开始, 三氟甲硫基化的研究走向了“低谷”, 在将近十年期间中, 关于三氟甲硫基化反应的报道与突破相当之少.而由于氟原子和三氟甲基在药物中的应用, 在2010年前后, 三氟甲硫基化反应的研究又重新受到了研究者们的热切关注, 过渡金属催化的三氟甲硫基化反应得到了快速的发展.
2011年, MIT的Buchwald小组[16]首次实现了过渡金属Pd催化下芳基溴化物与AgSCF3的交叉偶联反应, 重新将三氟甲硫基化反应带回了人们的视线.他们小组采用大位阻的BrettPhos作为配体, 成功实现了第一例金属催化的交叉偶联反应(Scheme 6).
 图式6
Pd催化芳基溴化物的三氟甲硫基化反应
图式6.
Palladium-Catalyzed coupling of aryl bromides with AgSCF3
图式6
Pd催化芳基溴化物的三氟甲硫基化反应
图式6.
Palladium-Catalyzed coupling of aryl bromides with AgSCF3
2012年Lehigh University的Vicic小组[17]报道了镍催化下芳基碘化物、溴化物与NMe4SCF3的交叉偶联反应.该反应条件温和, 在室温下就可以以中等到优秀的收率得到相应的三氟甲硫基化产物.当底物中含有吸电子取代基时, 反应的产率会有所降低, 对于活性更低的芳基氯化物, 反应则不能发生(Scheme 7).
 图式7
Ni催化芳基碘化物、溴化物三氟甲硫基化反应
图式7.
Nickel-Catalyzed coupling of ary iodides and bromides with NMe4SCF3
图式7
Ni催化芳基碘化物、溴化物三氟甲硫基化反应
图式7.
Nickel-Catalyzed coupling of ary iodides and bromides with NMe4SCF3
2015年RWTH Aachen University的Schoenebeck小组[18]使用dppf作为配体, 实现了首例镍催化芳基氯化物与NMe4SCF3的交叉偶联反应(Scheme 8).
 图式8
Ni催化芳基氯化物三氟甲硫基化反应
图式8.
Nickel-Catalyzed coupling of ary chlorides with NMe4SCF3
图式8
Ni催化芳基氯化物三氟甲硫基化反应
图式8.
Nickel-Catalyzed coupling of ary chlorides with NMe4SCF3
随后, 该小组合成了双核的一价钯催化剂, 并将其应用到芳基碘化物、溴化物的三氟甲硫基化反应中, 可以获得优秀的反应收率[19] (Scheme 9).
 图式9
双核一价Pd催化芳基碘化物、溴化物三氟甲硫基化反应
图式9.
Dinuclear palladium catalyzed coupling of ary iodides and bromides with NMe4SCF3
图式9
双核一价Pd催化芳基碘化物、溴化物三氟甲硫基化反应
图式9.
Dinuclear palladium catalyzed coupling of ary iodides and bromides with NMe4SCF3
采用芳基氯化物相似的体系, 她们小组使用芳基磺酸酯、烯基磺酸酯等底物也成功实现了Ni催化下的交叉偶联反应[20] (Scheme 10).
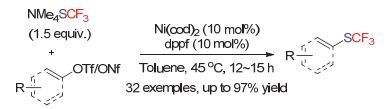 图式10
Ni催化芳基、烯基类卤化物的三氟甲硫基化反应
图式10.
Nickel-Catalyzed coupling of ary, vinyl triflates or nonaflates with NMe4SCF3
图式10
Ni催化芳基、烯基类卤化物的三氟甲硫基化反应
图式10.
Nickel-Catalyzed coupling of ary, vinyl triflates or nonaflates with NMe4SCF3
此外, 通过官能团化导向的策略, 可以更好地对底物中的C—Br键以及C—Cl键进行活化, 从而可以更加高效地实现相应的三氟甲硫基化转化.不列颠哥伦比亚大学的Love小组[21]发现, 使用不同的含氮导向基, 来使金属镍催化剂形成五元环金属中间体, 无需配体和添加剂的加入, 便可以在温和的条件下实现C—Br键、C—Cl键到C—SCF3键的转化(Scheme 11).
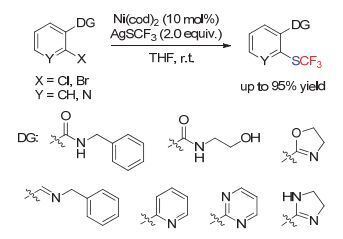 图式11
官能团导向芳基C—Br键、C—Cl键的三氟甲硫基化反应
图式11.
Ligandless nickel-catalyzed ortho-selective directed trifluoromethylthiolation of aryl chlorides and bromides using AgSCF3
图式11
官能团导向芳基C—Br键、C—Cl键的三氟甲硫基化反应
图式11.
Ligandless nickel-catalyzed ortho-selective directed trifluoromethylthiolation of aryl chlorides and bromides using AgSCF3
2.1.4 亲核底物的氧化三氟甲硫基化反应
亲核试剂与另一分子的亲核试剂在氧化性条件下的偶联反应被称为氧化偶联反应.而在三氟甲硫基领域, 上海有机所的卿凤翎小组[22]首先报道了该类转化.他们使用芳基硼酸作为亲核试剂, 在硫粉和Ruppert-Prakash试剂为三氟甲硫基源, 碳酸银作为氧化剂, 硫氰化亚铜作为催化剂的条件实现了该步转化(Scheme 12).
 图式12
铜催化硼酸的氧化三氟甲硫基化反应
图式12.
Copper-Catalyzed oxidative coupling of aryl boronic acid with TMSCF3 and elemental sulfur
图式12
铜催化硼酸的氧化三氟甲硫基化反应
图式12.
Copper-Catalyzed oxidative coupling of aryl boronic acid with TMSCF3 and elemental sulfur
采用同样的策略, 该小组使用炔烃作为亲核试剂也实现了无金属参与下的氧化偶联反应.该反应无须惰性气体保护, 便可以在室温条件下顺利实现转化[23] (Scheme 13).
 图式13
炔烃的氧化三氟甲硫基化反应
图式13.
Metal-free oxidative coupling of terminal alkynes with TMSCF3 and elemental sulfur
图式13
炔烃的氧化三氟甲硫基化反应
图式13.
Metal-free oxidative coupling of terminal alkynes with TMSCF3 and elemental sulfur
2012年, Vicic小组[24]使用NMe4SCF3, 在1 equiv.的一价铜存在下, 也实现了与芳基、烯基硼酸底物的氧化三氟甲硫基化反应.该反应无论对于吸电子或者给电子取代基都可以取得良好到优秀的产率.而对于反式的烯基硼酸底物, 可以得到构型保持的三氟甲硫基化产物(Scheme 14).
 图式14
铜介导的硼酸氧化三氟甲硫基化反应
图式14.
Copper-mediated trifluoromethylthiolation of aryl boronic acids with NMe4SCF3
图式14
铜介导的硼酸氧化三氟甲硫基化反应
图式14.
Copper-mediated trifluoromethylthiolation of aryl boronic acids with NMe4SCF3
2013年, 大连理工大学的段春迎小组[25]改用硫粉和CF3COONa的组合, 采用当量的铜盐和配体, 也可以实现硼酸的氧化偶联反应, 但由于CF3COONa脱羧需要较高的温度, 因此该反应的温度较高. 2017年, 同济大学赵晓明小组[26]使用三氟甲硫基铜络合物, 在氧气条件下实现了硼酸底物的氧化偶联反应.随后, 华东理工大学的曹松小组[27]使用AgSCF3作为三氟甲硫基源并且同时作为氧化剂, 实现铜催化下芳基硼酸的偶联反应(Scheme 15).
 图式15
硼酸氧化三氟甲硫基化反应
图式15.
Oxidative trifluoromethylthiolation of boronic acids
图式15
硼酸氧化三氟甲硫基化反应
图式15.
Oxidative trifluoromethylthiolation of boronic acids
使用亲核性的三氟甲硫基源, 也可以在氧化条件下实现C—H键的三氟甲硫基反应(Scheme 16).卿凤翎小组[28]通过对氧化剂的考察, 发现在一价铜盐的催化下, 使用过氧化物作为氧化剂, 可以实现苄位C—H键无官能团导向的三氟甲硫基化反应.北京大学的黄湧小组[29]报道了官能团导向芳基C—H键的氧化三氟甲硫基化反应.该过程中, 氧化剂的作用可能是将转金属后的Pd (Ⅱ)-SCF3中间体氧化成Pd (Ⅳ)中间体, 从而促进还原消除过程.中山大学的王洪根小组[30]使用吡啶作为导向基, 在空气作为氧化剂的条件下实现了三价钴催化的芳基C—H键氧化三氟甲硫基化反应.
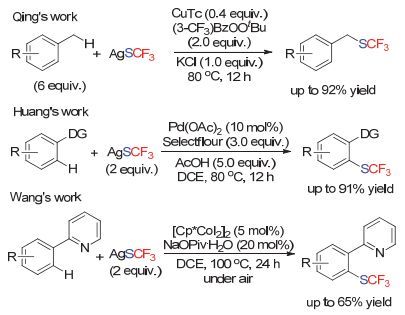 图式16
氧化条件下C—H三氟甲硫基化反应
图式16.
Oxidative trifluoromethylthiolation of C—H bond
图式16
氧化条件下C—H三氟甲硫基化反应
图式16.
Oxidative trifluoromethylthiolation of C—H bond
2.1.5 稳定、易表征的三氟甲硫基金属络合物
合成制备出稳定且容易表征的三氟甲硫基金属络合物一方面可以解决该类三氟甲硫基源稳定性差且难以表征的问题, 另一方面, 制备的络合物会更加有利于研究该类反应的反应机理.
1992年, Munavalli小组[31]使用CF3SSCF3与铜粉在乙腈中反应, 得到了一个晶体化合物, 当时他们认为该结构是1:1的CuSCF3和乙腈的配合物.随后, 他们成功对该络合物的单晶进行培养, 发现该单晶的结构是[CF3SCu]10•8CH3CN, 这是首例可分离得到的三氟甲硫基络合物[32].
2012年, 福州大学的翁志强小组[33]分离得到了双齿氮配体络合的联吡啶单核铜络合物与菲罗琳双核铜络合物.该类络合物在空气和溶液中可以较为稳定的存在.随后他们也通过在体系中加入PPh3, 合成了相应的膦配体络合物[34]. 2015年, Vicic小组[35]使用PMe3与CF3SO2Na反应, 首先观测到了CF3SSCF3的生成.他们随后在反应体系中加入CuCl, 成功以80%的氟谱收率观测到了CuSCF3的生成.通过向反应体系中外加配体, 他们可以成功分离得到与翁志强小组相同的络合物.同样, 通过加入Ph3P和dppf, 他们也分离得到了单齿膦配体和双齿膦配体络合的CuSCF3的络合物(Scheme 17).
 图式17
N, P络合的CuSCF3络合物
图式17.
N, P-ligated CuSCF3 complex
图式17
N, P络合的CuSCF3络合物
图式17.
N, P-ligated CuSCF3 complex
采用该类可分离得到的络合物, 可以实现与各种芳基[36]、烷基(苄基、烯丙基)[34, 37]、烯基[38]、酰卤[39]等一系列底物的亲核三氟甲硫基化反应.
2.1.6 其他一些类型的亲核三氟甲硫基化反应
以上亲核反应中, 反应底物大多局限于卤化物.而在近年来, 其他亲电底物的三氟甲硫基化反应也得到了发展.
芳基重氮盐Sandmeyer类型的三氟甲硫基化反应可以直接通过AgSCF3或CuSCF3的反应来实现. 1975年Kondratenko小组[40]使用AgSCF3与芳基重氮盐反应, 可以以中等的收率得到相应的三氟甲硫基化反应的产物, 这是Sandmeyer类型的三氟甲硫基化反应的首次报道.在2000年, Clark小组[41]使用芳基重氮盐与CuSCF3反应, 实现了相应地转化, 且产率较AgSCF3有所提高.该方法也可以从苯胺底物出发, 通过一锅法实现氨基到三氟甲硫基的转化.
2014年Goossen小组[42]首先通过Sandmeyer类型的硫氰化反应构建C—S键, 再通过Ruppert试剂形成的三氟甲基负离子进攻氰基, 实现了三氟甲硫基化的转化.该方法避免了预先制备相对不稳定的CuSCF3, 在产率和底物兼容性上都有了很大的提升(Scheme 18).
 图式18
Sandmeyer类型的三氟甲硫基化反应
图式18.
Sandmeyer-type trifluoromethylthiolation
图式18
Sandmeyer类型的三氟甲硫基化反应
图式18.
Sandmeyer-type trifluoromethylthiolation
重氮甲酸酯可以在铜、钯等金属盐的存在下形成金属卡宾, 该类卡宾有着很好的亲电性, 可以与各类亲核试剂发生迁移插入反应.
在2014年, 上海有机所的胡金波小组[43a]和北京大学的王剑波小组[43b]几乎同时报道了该类型的反应.他们采用了相同的策略, 首先以AgSCF3和一价铜盐现场生成CuSCF3, 随后与α-重氮甲酸酯反应形成相应的铜卡宾, 经迁移插入和质子淬灭后形成最终产物.随后, Rueping小组[44]发现, 使用预先制备的CuSCF3通过加入水淬灭或者加入另一分子的亲电三氟甲硫基化试剂, 可以实现单/双三氟甲硫基化反应. Goossen小组[45]发现, 无需预先制备CuSCF3, 在CuSCN的催化下, 使用Me4NSCF3为三氟甲硫基源, 也可以实现相应的转化, 并且使用Me4NSeCF3时, 也可以实现三氟甲硒基化反应(Scheme 19).
 图式19
重氮酸酯的三氟甲硫基化反应
图式19.
Trifluoromethylthiolation of diazo compounds through copper carbine migratory insertion
图式19
重氮酸酯的三氟甲硫基化反应
图式19.
Trifluoromethylthiolation of diazo compounds through copper carbine migratory insertion
醇底物的亲核三氟甲硫基化反应由Rueping小组和卿凤翎小组分别于2014年和2015年报道(Scheme 20). Rueping小组[46]使用Lewis活化羟基的策略, 实现了活化羟基底物如苄醇、烯丙醇的脱氧三氟甲硫基化反应.卿凤翎小组[47]发现, 醇羟基可以进攻硫代氟光气而转变为更易离去的硫代氟甲酰氧基团, 使用过量的AgSCF3可以进攻该基团, 从而实现未活化一级、二级醇底物的三氟甲硫基化反应.相比该两个小组的工作, 卿凤翎小组的工作在底物范围上更广, 并且避免了Lewis酸的加入.但是, 由于需要将羟基转变为离去基团, 首先需要消耗等当量的硫代氟光气, 因而该反应一般需要3个当量以上的AgSCF3以及大大过量的四丁基碘化铵.
 图式20
醇底物的亲核三氟甲硫基化反应
图式20.
Dehydrotrifluoromethylthiolation of alcohols
图式20
醇底物的亲核三氟甲硫基化反应
图式20.
Dehydrotrifluoromethylthiolation of alcohols
兰州大学的梁永民小组[48]采用炔丙醇底物, 使用BF3•Et2O为Lewis Acid, 实现了串联关环反应(Scheme 21).作者认为该反应的机理可能是底物在Lewis Acid的活化下首先离去羟基生成联烯三氟甲硫基结构.随后, 在银盐的活化下, 底物中的亲核试剂进攻双键, 最终水解生成最终产物.
 图式21
炔丙醇底物的串联环化三氟甲硫基化反应
图式21.
AgSCF3/BF3•Et2O Mediated trifluoromethylthiolation cascade cyclization of propynols
图式21
炔丙醇底物的串联环化三氟甲硫基化反应
图式21.
AgSCF3/BF3•Et2O Mediated trifluoromethylthiolation cascade cyclization of propynols
将羟基转变为更容易离去的离去基团, 也可以使用亲核三氟甲硫基源来实现相应的三氟甲硫基转化.如上海有机所的游书力小组[49]将羟基转变为碳酸酯, 可以在钌催化下实现端位选择性的三氟甲硫基化反应.在该反应中, 产生的支链产物会进一步转化为热力学稳定的线性产物(Scheme 22).
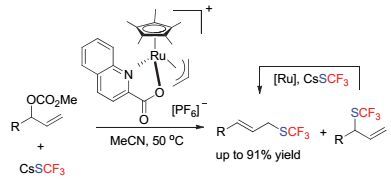 图式22
钌催化区域端位选择性的烯丙基三氟甲硫基化反应
图式22.
Ruthenium-catalyzed regioselective allylic trifluoromethylthiolation reaction
图式22
钌催化区域端位选择性的烯丙基三氟甲硫基化反应
图式22.
Ruthenium-catalyzed regioselective allylic trifluoromethylthiolation reaction
马军安小组和Cahard小组[50]合作, 实现了含三氟甲硫基取代的手性胺以及手性酯类化合物的合成.他们将手性的氨基醇衍生为更容易离去的磺酸酯类化合物, 采用Me4NSCF3为亲核三氟甲硫基源, 通过一锅法的开环、水解反应, 实现了该步转化, 合成了一类含三氟甲硫基取代的结构新颖的化合物(Scheme 23).
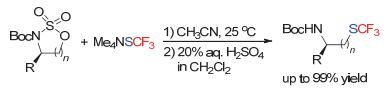 图式23
环状磺酸酯类化合物的三氟甲硫基化反应
图式23.
Trifluoromethylthiolation of cyclic sulfamidates
图式23
环状磺酸酯类化合物的三氟甲硫基化反应
图式23.
Trifluoromethylthiolation of cyclic sulfamidates
二芳基高价碘盐也是一类高活性的亲电试剂, 其相关的亲核三氟甲硫基化反应也有相关报道(Scheme 24). 2016年, Rueping小组[51]使用CuSCF3, 通过与芳基均三甲基苯基高价碘盐实现了区域选择性的三氟甲硫基化反应.张成潘小组[52]使用炔基苯基高价碘盐为亲电试剂, Me4NSCF3为亲核试剂, 实现了炔烃端选择性的三氟甲硫基化反应.此外, 作者采用相应的Me4NSeCF3, 也可以实现三氟甲硒基化反应.
 图式24
高价碘盐的三氟甲硫基化反应
图式24.
Trifluoromethylthiolation of Unsymmetrical λ3-iodane derivatives
图式24
高价碘盐的三氟甲硫基化反应
图式24.
Trifluoromethylthiolation of Unsymmetrical λ3-iodane derivatives
除去AgSCF3中三氟甲硫基的亲核性外, 银也具有一定的Lewis酸性, 而利用该两点, 研究者们也使用AgSCF3来捕捉反应体系中生成的活性中间体. 2013年Lee小组[53]从AgSCF3出发, 一方面利用Ag离子的Lewis酸性来活化三炔底物来形成苯炔中间体, 另一方面利用SCF3阴离子的亲核性来进攻苯炔中间体, 从而实现串联成环反应.最近作者发现, 当炔丙位有氢原子存在时, 该反应会优先发生Alder-ene反应从而形成烯-联烯中间体, 随后受AgSCF3进攻, 并在三氟乙醇存在下通过后续的迁移重排反应来实现串联环化反应[54] (Scheme 25).
 图式25
AgSCF3与苯炔、烯炔中间体的反应
图式25.
Trifluoromethylthiolation of aryne and allene-enyne intermediate
图式25
AgSCF3与苯炔、烯炔中间体的反应
图式25.
Trifluoromethylthiolation of aryne and allene-enyne intermediate
2014年, 复旦大学的吴劼小组[55]从肟底物出发, 使用AgOTf为催化剂, 对甲氧基磺酰氯作为活化剂, 首先形成氧化异喹啉中间体, 随后被AgSCF3捕捉, 在碱促进下形成三氟甲硫基化异喹啉产物. 2016年, 上海有机所的胡金波小组[56]使用AgSCF3为亲核试剂, 炔基碘作为亲电试剂, 实现了苯炔前体的碘化-三氟甲硫基化反应(Scheme 26).
 图式26
AgSCF3与氧化异喹啉、苯炔中间体的反应
图式26.
Trifluoromethylthiolation of N-oxide and aryne intermediate
图式26
AgSCF3与氧化异喹啉、苯炔中间体的反应
图式26.
Trifluoromethylthiolation of N-oxide and aryne intermediate
另外, 在2013年Zard小组[57]设计合成了一类可以现场生成三氟甲硫基阴离子的试剂(Scheme 27).该试剂可以用于高效地制备三氟甲硫基取代的芦竹碱和α-三氟甲硫基取代的酮类化合物.
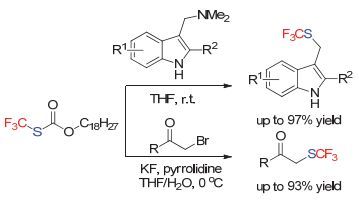 图式27
现场生成三氟甲硫基阴离子的反应
图式27.
In-situ generation and capture of trifluoromethanethiol
图式27
现场生成三氟甲硫基阴离子的反应
图式27.
In-situ generation and capture of trifluoromethanethiol
随后, 上海有机所施敏课题组[58]采用该试剂, 在不同溶剂的条件下实现了MBH酯底物区域选择性的三氟甲硫基化反应.随后, 作者同样采用MBH酯底物, 实现了分子内的成环反应, 并且通过手性三级胺的催化, 实现了不对称二氟甲硫基片段的引入[59] (Scheme 28).该产物的机理可能是由MBH反应的中间体在碱性条件下拔氢, 与三氟甲硫基阴离子现场产生的氟光气反应生成的中间体通过后续的反应形成的.
 图式28
MBH酯的亲核三氟甲硫基化反应
图式28.
Nucleophilic trifluoromethylthiolation of Morita-Baylis-Hillman carbonates
图式28
MBH酯的亲核三氟甲硫基化反应
图式28.
Nucleophilic trifluoromethylthiolation of Morita-Baylis-Hillman carbonates
2.2 亲电三氟甲硫基化反应
相比较于亲核三氟甲硫基化反应, 亲电三氟甲硫基化反应由于早期试剂的匮乏, 其相应的研究报道较少.在20世纪60年代开始, 关于CF3SCl和CF3SSCF3的亲电三氟甲硫基化反应开始被报道.
CF3SCl有着很高的亲电三氟甲硫基化活性, 其反应报道较多, 可以实现一系列富电子底物如富电子芳烃、杂芳烃、活泼亚甲基底物、有机金属试剂、烯醇硅醚、烯胺等亲电三氟甲硫基化反应[60] (Scheme 29).但在上述反应中, 对于底物类型的研究只有有限的个别例子, 并没有进行系统的研究.其次, 由于CF3SCl高的反应活性, 反应的选择性通常较差, 且需要在低温下进行反应操作.更重要的是, CF3SCl有着很高的毒性, 因而限制了其广泛使用.
 图式29
CF3SCl的亲电三氟甲硫基化反应
图式29.
Electrophilic trifluoromethylthiolation using CF3SCl
图式29
CF3SCl的亲电三氟甲硫基化反应
图式29.
Electrophilic trifluoromethylthiolation using CF3SCl
与CF3SCl相比, CF3SSCF3的活性较低, 其相关反应报道较少, 它能与活泼亚甲基类底物、芳基钠等实现相应的三氟甲硫基化反应[61].在2014年, Daugulis小组[62]利用8-氨基喹啉作为导向基, 在醋酸铜的催化下实现了首例C—H活化三氟甲硫基化反应(Scheme 30).
 图式30
8-氨基喹啉导向C—H三氟甲硫基化反应
图式30.
Auxiliary-assisted trifluoromethylthiolation using CF3SSCF3
图式30
8-氨基喹啉导向C—H三氟甲硫基化反应
图式30.
Auxiliary-assisted trifluoromethylthiolation using CF3SSCF3
虽然该两种亲电三氟甲硫基化试剂都有着出色的反应性能, 但是由于其都是高毒性气体[63], 给操作带来了诸多不便, 也因而限制了其应用与发展.因此, 发展新型、高活性且便于操作的亲电三氟甲硫基化试剂成为了该领域迫切需要解决的问题.
从2008年开始, 一系列便于操作、稳定的亲电三氟甲硫基化试剂及方法逐渐被开发并且得到了应用, 并对于各类底物的三氟甲硫基化反应都进行了较为系统的研究.此外, 也有着基于手性骨架的亲电三氟甲硫基试剂的相关报道, 为不对称引入三氟甲硫基团提供了新的策略. Scheme 31列举了文献中已有的亲电三氟甲硫基化试剂及方法, 下面将按照不同的试剂逐一进行介绍.
 图式31
亲电三氟甲硫基试剂种类
图式31.
Different types of electrophilic trifluoromethylthiolation reagents
图式31
亲电三氟甲硫基试剂种类
图式31.
Different types of electrophilic trifluoromethylthiolation reagents
2.2.1 Billard试剂
在2008年, 法国Billard和Langlois等[64]利用二乙胺基三氟化硫(DAST)、Ruppert试剂(TMSCF3)制备了含三氟甲硫基的苯胺类衍生物.该衍生物进一步与碘甲烷发生SN2反应, 可进一步制备含有甲基取代的Billard试剂.该小组通过后续的研究发现, 该类试剂可以在Lewis Acid或者Brønsted Acid的活化下实现烯烃[65]、炔烃[66]、富电子芳烃[67]、胺[68]等一系列底物的亲电三氟甲硫基化反应(Scheme 32).
 图式32
第一代Billard试剂的制备及反应
图式32.
Preparation and reactions of first generation of Billard reagent
图式32
第一代Billard试剂的制备及反应
图式32.
Preparation and reactions of first generation of Billard reagent
同时, 他们也发现, 在合成该苯胺骨架的试剂过程中, 当底物的胺含有吸电子取代基时会优先生成一个亚胺中间体, 该中间体可以在三氟乙酸的水解下, 进一步生成活性更高的第二代Billard试剂[64].在2014年, 他们对该类试剂的反应活性进行了报道, 发现该类试剂相比第一代苯胺结构的试剂有着更为出色的反应活性, 除去上述报道的反应外还可以实现活泼C—H键底物[69]、杂环底物C—H键底物[70]、硼酸底物[71]、杂原子亲核试剂[72] (S, O)、活泼卤化物(类卤化物)极性反转的三氟甲硫基化反应[73]、炔烃的单、双三氟甲硫基化反应[74]以及富电子芳烃、杂芳烃底物的C—H键三氟甲硫基化反应[75](Scheme 33).
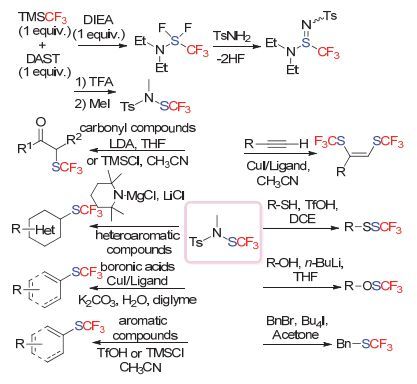 图式33
第二代Billard试剂的制备及反应
图式33.
Preparation and reactions of Second generation of Billard reagent
图式33
第二代Billard试剂的制备及反应
图式33.
Preparation and reactions of Second generation of Billard reagent
最近Billard小组[76]也发现, 采用第二代活性更高的试剂, 可以在超强酸的作用下对该试剂进行进一步的活化, 从而可以在低温下对一系列芳胺底物实现芳环上的三氟甲硫基化反应(Scheme 34).作者也通过核磁手段, 观察到了活性中间体的产生.作者认为, 采用该方法, 可以在合成设计的后期方便地在芳环上引入三氟甲硫基官能团.
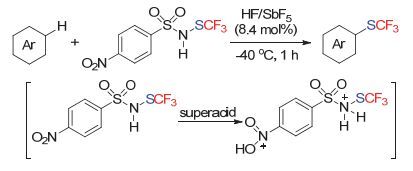 图式34
超强酸催化下芳胺底物的三氟甲硫基化反应
图式34.
Superacid-catalyzed trifluoromethylthiolation of aromatic amines
图式34
超强酸催化下芳胺底物的三氟甲硫基化反应
图式34.
Superacid-catalyzed trifluoromethylthiolation of aromatic amines
在Billard小组工作的基础上, 其他课题组采用不同的酸活化剂, 实现了不同类型的亲电三氟甲硫基化反应. 2013年上海有机所的卿凤翎小组[77]使用色胺底物, 分别在Lewis Acid和Brønsted Acid活化的条件下实现了吲哚2位三氟甲硫基化以及串联关环三氟甲硫基化反应.随后, 他们采用乙酰氯作为Lewis Acid, 实现了烯丙基硅试剂的三氟甲硫基化反应[78a].而同样采用酰氯活化的策略, 上海有机所的刘国生小组[78b]实现了钯催化条件下吡啶导向的C—H键三氟甲硫基化反应.华东理工大学曹松小组[78c]采用乙酰氯活化的策略, 发现对于酮类化合物, 可以很好地实现羰基α位的三氟甲硫基化反应(Scheme 35).
 图式35
酸和酰氯活化下的三氟甲硫基化反应
图式35.
Trifluoromethylthiolation under the activation of acid or acyl chloride using the first generation of Billard reagent
图式35
酸和酰氯活化下的三氟甲硫基化反应
图式35.
Trifluoromethylthiolation under the activation of acid or acyl chloride using the first generation of Billard reagent
复旦大学的吴劼小组发现BiCl3可以作为Lewis Acid来高效地对该试剂进行活化, 从而实现一系列串联关环反应, 成功构建了苯并[1, 2]噻嗪1, 1-二氧化物[79]、吲哚[80]、苯并呋喃[81]、苯并噻吩[81]以及苯并吡喃类衍生物[82].此外, 炔基硅底物[83]也可以在该条件下, 实现相应的三氟甲硫基化转化(Scheme 36).
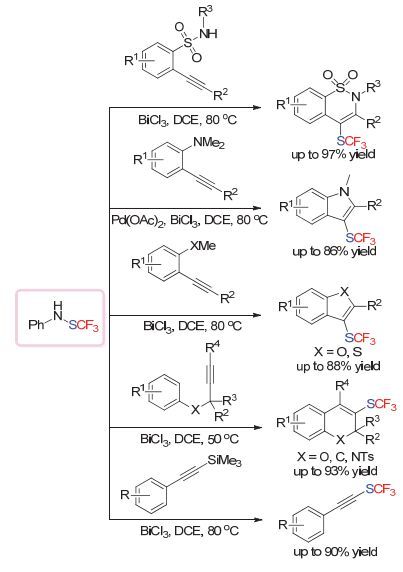 图式36
吴劼小组的工作
图式36.
Wu's Work using the first generation of Billard reagent
图式36
吴劼小组的工作
图式36.
Wu's Work using the first generation of Billard reagent
嘉兴学院的邱观音生小组与上海海洋大学的盛洁小组[84]合作, 从磺酰氯出发, 现场生成亚磺酸钠盐后, 使用对甲苯磺酸为酸添加剂对试剂进行活化, 可以实现砜基三氟甲硫基产物的合成(Scheme 37).
 图式37
磺酰氯的一锅法三氟甲硫基化转化
图式37.
One-pot reaction of sulfonyl chloride and Billard reagent
图式37
磺酰氯的一锅法三氟甲硫基化转化
图式37.
One-pot reaction of sulfonyl chloride and Billard reagent
2.2.2 Lu and Shen试剂
在2013年, 上海有机所的吕龙和沈其龙小组[85]在试图发展基于Togni试剂类型的亲电三氟甲硫基试剂过程中, 合成了一类新颖结构的亲电三氟甲硫基化试剂. 2014年, MIT的Buchwald小组[86]通过对该类底物的衍生化, 合成了固体状态的衍生物, 发现在该衍生物的结构中, 三氟甲硫基是与氧原子相连.随后他们通过MOF技术, 将该试剂吸附并通过X单晶射线衍射, 确认该结构并不是类似于Togni试剂而是三氟甲基次磺酸酯的结构(Scheme 38).
 图式38
Lu and Shen试剂的合成及结构修正
图式38.
Preparation and modify of Lu and Shen reagent
图式38
Lu and Shen试剂的合成及结构修正
图式38.
Preparation and modify of Lu and Shen reagent
采用该试剂, 吕龙和沈其龙小组[85]首先实现了β-酮酸酯、芳基硼酸、炔烃以及羰基α位C—H键的三氟甲硫基化反应(Scheme 39).在芳基硼酸的反应基础上, 通过提高反应温度, 他们可以实现烷基硼酸的三氟甲硫基化反应[87].随后, 他们也发现, 该试剂可以在Brønsted Acid的活化下实现吲哚底物的三氟甲硫基化反应[88].此外, 他们小组对该类试剂的结构活性相关做了比较, 发现该试剂中的碘原子在大多数反应情况下对反应并没有产生明显的差别.采用不带碘的试剂, 他们在醋酸作为反应溶剂的条件下, 实现苯基亚磺酸钠底物的三氟甲硫基化反应[89].此外, 在β-酮酸酯的反应基础上, 作者在奎宁作为碱或奎宁定作为相转移催化剂的条件下实现了β-酮酸酯、吲哚酮底物的不对称三氟甲硫基化反应[90] (Scheme 40).
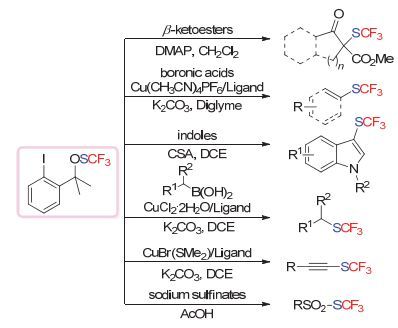 图式39
Lu and Shen试剂的反应
图式39.
Reactions of Lu and Shen reagent
图式39
Lu and Shen试剂的反应
图式39.
Reactions of Lu and Shen reagent
 图式40
不对称三氟甲硫基化反应
图式40.
Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent
图式40
不对称三氟甲硫基化反应
图式40.
Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent
2014年, Gade小组[91]采用该试剂使用Boxmi作为配体, 在铜催化下实现了β-酮酸酯的反应(Scheme 41).该反应体系无论对于5元环还是6元环都可以适用, 且不需要大位阻的金刚烷基取代基.
 图式41
Cu (OTf)2/boxmi体系下不对称三氟甲硫基化反应
图式41.
Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent and copper-boxmi complexes
图式41
Cu (OTf)2/boxmi体系下不对称三氟甲硫基化反应
图式41.
Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent and copper-boxmi complexes
浙江师范大学朱向明小组[92]在硫糖苷的活化中, 使用该亲电三氟甲硫基化试剂活化糖苷键, 实现了醇的糖苷化反应(Scheme 42).在该反应中, 作者认为, 该试剂在三氟磺酸三甲基硅酯作为Lewis Acid的活化下, 接受硫原子进攻, 可以形成更易离去的硫鎓盐中间体, 最后实现糖苷化反应.
 图式42
糖苷键的活化
图式42.
Thioperoxide-mediated activation of thioglycoside donors
图式42
糖苷键的活化
图式42.
Thioperoxide-mediated activation of thioglycoside donors
普渡大学的戴明骥小组[93]采用环丙醇底物, 在铜催化条件下通过开环三氟甲硫基化反应, 构建了β位三氟甲硫基取代的羰基化合物(Scheme 43).作者认为该反应的机理可能是生成的重排后的烷基铜物种与亲电试剂发生偶联进而生成最终产物.
 图式43
铜催化环丙醇亲电开环三氟甲硫基化反应
图式43.
Copper-catalyzed electrophilic ring-opening cross-coupling of cyclopropanols
图式43
铜催化环丙醇亲电开环三氟甲硫基化反应
图式43.
Copper-catalyzed electrophilic ring-opening cross-coupling of cyclopropanols
通过吕龙和沈其龙小组的研究还发现, 该试剂除去实现亲电三氟甲硫基化反应以外, 还可以作为体系中自由基的淬灭剂, 实现自由基类型的三氟甲硫基化反应, 我们将在自由基反应中进行介绍.
2.2.3 Shibata试剂
2013年, 日本的Shibata小组[94]发展了一种基于高价碘叶立德类型的亲电三氟甲硫基化试剂.该试剂的合成较为廉价, 从三氟亚磺酸钠出发与溴代苯乙酮反应, 随后在等当量的醋酸碘苯的存在下, 可以以当量的收率获得该试剂(Scheme 44).
 图式44
Shibata试剂的制备
图式44.
Preparation of Shibata reagent
图式44
Shibata试剂的制备
图式44.
Preparation of Shibata reagent
采用该试剂, 他们小组首先实现了β-酮酸酯、吲哚衍生物、烯胺类底物在铜催化下的反应[94].随后, 他们利用该试剂, 也实现了铜催化下硼酸[95]、芳胺[96]、吡咯[97]、烯丙基硅[98]、烯醇硅醚[98]的亲电三氟甲硫基化反应.对于烯丙醇的底物, 他们也同样采用该试剂, 可以实现重排反应[95] (Scheme 45).
 图式45
Shibata试剂的反应
图式45.
Reactions of Shibata reagent
图式45
Shibata试剂的反应
图式45.
Reactions of Shibata reagent
该试剂参与的反应一般都需要铜盐的催化才能发生, 其反应机理可能是该卡宾前体在铜盐作用下首先形成卡宾中间体, 随后发生重排来生成活性中间体, 最后与亲核底物发生三氟甲硫基化反应(Scheme 46).
 图式46
Shibata试剂的反应机理
图式46.
Proposed mechanism for Cu (Ⅰ)-catalyzed trifluoromethylthiolation with Shibata reagent
图式46
Shibata试剂的反应机理
图式46.
Proposed mechanism for Cu (Ⅰ)-catalyzed trifluoromethylthiolation with Shibata reagent
在2016年, 他们课题组采用相似的策略合成了偶氮类型的试剂, 该试剂除去实现以上大部分反应外, 还可以实现碘苯的三氟甲硫基化反应, 但是效率偏低[99] (Scheme 47).
 图式47
偶氮类型的Shibata试剂
图式47.
Diazo-type of Shibata reagent
图式47
偶氮类型的Shibata试剂
图式47.
Diazo-type of Shibata reagent
2.2.4 N-三氟甲硫基二酰亚胺试剂
该类结构的酰胺试剂目前有两种, 包括N-三氟甲硫基丁二酰亚胺以及N-三氟甲硫基邻苯二甲酰亚胺.该两种试剂的首次合成分别由Haas小组[100]和Munavalli小组[101]于1996年和2000年报道(Scheme 48).
 图式48
二酰亚胺N-SCF3试剂的早期制备
图式48.
Early preparation of N-SCF3 reagent
图式48
二酰亚胺N-SCF3试剂的早期制备
图式48.
Early preparation of N-SCF3 reagent
Munavalli小组[101]首先使用N-三氟甲硫基邻苯二甲酰亚胺尝试了烯胺底物的亲电反应, 发现可以以88%的收率获得相应羰基α位三氟甲硫基化反应的产物, 初步验证了该化合物作为一种亲电三氟甲硫基化试剂的可能性(Scheme 49).但当他们采用该试剂尝试其他类型亲核试剂的反应时, 却没有获得成功.
 图式49
Munavalli小组的初步尝试
图式49.
Preliminary trifluoromethylthiolation attempts by Munavalli group
图式49
Munavalli小组的初步尝试
图式49.
Preliminary trifluoromethylthiolation attempts by Munavalli group
该类试剂在往后的十多年内, 似乎已被研究人员所“遗忘”.直到2013年德国的Rueping小组[102]和上海有机所的沈其龙小组[103]几乎同时报道了该试剂的合成以及反应, 让该试剂重新回到人们的视线.他们在合成中均避开了高毒性的CF3SCl, 分别采用CuSCF3和AgSCF3与N-卤化合物发生复分解反应, 来高效、安全地制备该试剂(Scheme 50).
 图式50
N-三氟甲硫基邻苯二甲酰亚胺试剂合成方法的改良
图式50.
Modified preparation of N-(trifluoromethylthio) phthalimide by Rueping and shen group
图式50
N-三氟甲硫基邻苯二甲酰亚胺试剂合成方法的改良
图式50.
Modified preparation of N-(trifluoromethylthio) phthalimide by Rueping and shen group
2013年, Rueping小组[104]使用该试剂, 首先报道了在奎宁定作为有机碱条件下β-酮酸酯的不对称三氟甲硫基化反应.该反应无论对于苯并五元、六元或者是非苯并的β-酮酸酯类底物都可以获得优秀的产率和对映选择性, 并且底物中的酯基取代基对对映选择性的影响不大.随后, 他们又在金鸡纳碱(DHQD)2Pyr作为有机碱的条件下实现了2-吲哚酮底物的不对称三氟甲硫基化反应[105] (Scheme 51).该体系与沈其龙小组的反应体系形成了互补, 当底物3位上的取代基是芳基时可以获得优秀的产率和对映选择性.
 图式51
N-三氟甲硫基邻苯二甲酰亚胺试剂参与的不对称反应
图式51.
Asymmetric trifluoromethylthiolation using N-(trifluoro-methylthio) phthalimide
图式51
N-三氟甲硫基邻苯二甲酰亚胺试剂参与的不对称反应
图式51.
Asymmetric trifluoromethylthiolation using N-(trifluoro-methylthio) phthalimide
2014年, Rueping小组[102]和沈其龙小组[103]几乎同时报道了硼酸与该试剂在铜催化下的偶联反应, 反应条件非常相似, 有着优秀的底物兼容性(Scheme 52).采用该试剂, Rueping小组[102]也实现了铜催化下炔烃的偶联反应.另外, 通过他们后续的研究发现, 采用该试剂, 可以在使用三氟甲苯作为反应溶剂的条件下, 实现硫醇、硫酚以及胺类底物的三氟甲硫基化反应[106] (Scheme 53).
 图式52
N-三氟甲硫基邻苯二甲酰亚胺试剂与硼酸的偶联反应
图式52.
Cross-coupling reaction between boronic acids and N-(trifluoromethylthio) phthalimide
图式52
N-三氟甲硫基邻苯二甲酰亚胺试剂与硼酸的偶联反应
图式52.
Cross-coupling reaction between boronic acids and N-(trifluoromethylthio) phthalimide
 图式53
Rueping小组其它的工作
图式53.
Other works by Rueping group
图式53
Rueping小组其它的工作
图式53.
Other works by Rueping group
2015年, 南方科技大学的汪珺小组[107]采用该试剂实现了活化烯烃底物的双官能团化反应.同时他们发现, 采用硝基取代的试剂, 可以进一步提高试剂的反应活性, 从而可以进一步提高产率(Scheme 54).
 图式54
碱促进下不饱和羰基化合物的胺化三氟甲硫基化反应
图式54.
DABCO-promoted aminotrifluoromethylthiolation of α, β-unsaturated carbonyl compounds
图式54
碱促进下不饱和羰基化合物的胺化三氟甲硫基化反应
图式54.
DABCO-promoted aminotrifluoromethylthiolation of α, β-unsaturated carbonyl compounds
Glorius小组[108]采用该试剂也做出了漂亮的工作. 2015年, 他们采用NaCl为添加剂, 实现了吡咯、吲哚、氮杂吲哚等底物的三氟甲硫基化反应(Scheme 55).他们认为, 在该反应体系中, 加入的NaCl可能与试剂反应现场生成了高活性的CF3SCl, 但是他们并未检测到该中间体的存在, 因此NaCl所起的作用还有待考证.此外, 他们采用该试剂在光催化条件下也实现了一系列自由基类型的三氟甲硫基化反应, 我们将在后续进行介绍.
 图式55
NaCl催化下含氮杂环的三氟甲硫基化反应
图式55.
NaCl-catalyzed trifluoromethylthiolation of N-heteroarenes
图式55
NaCl催化下含氮杂环的三氟甲硫基化反应
图式55.
NaCl-catalyzed trifluoromethylthiolation of N-heteroarenes
N-三氟甲硫基丁二酰亚胺试剂随后也由沈其龙小组[109]采用相似的策略合成.他们采用该试剂, 首次实现了吡啶导向的芳烃C—H键三氟甲硫基化反应(Scheme 56).
 图式56
Pd催化芳基C—H三氟甲硫基化反应
图式56.
Palladium-catalyzed trifluoromethylthiolation of Aryl C—H bonds
图式56
Pd催化芳基C—H三氟甲硫基化反应
图式56.
Palladium-catalyzed trifluoromethylthiolation of Aryl C—H bonds
采用该丁二酰亚胺试剂, 北京理工大学的杜大明小组[110]在双方酰化氨基酸配体的催化下, 实现了一锅法的不对称三氟甲硫基化和Michael/aldol串联反应, 以优秀的对映选择性和良好的非对映选择性得到了串联环化的产物(Scheme 57).
 图式57
双方酰化氨基酸配体催化下不对称三氟甲硫基化反应
图式57.
Enantioselective squaramide-catalyzed trifluoromethylthiolation-sulfur-Michael/adol cascade reaction
图式57
双方酰化氨基酸配体催化下不对称三氟甲硫基化反应
图式57.
Enantioselective squaramide-catalyzed trifluoromethylthiolation-sulfur-Michael/adol cascade reaction
2.2.5 Shen试剂
在2014年, 上海有机所沈其龙小组[111]在发展N-三氟甲硫基邻苯二甲酰亚胺试剂的基础上, 进一步报道合成了基于糖精骨架的亲电三氟甲硫基化试剂(Scheme 58).该试剂的合成同样极为方便, 且有着更为出色的反应性能.通过他们小组的研究发现, 采用该试剂可以在温和条件下实现醇、胺、硫醇、硫酚、富电子芳烃、羰基α位C—H键、酮酸酯、炔烃等一系列底物的三氟甲硫基化反应.而当采用TMSCl作为Lewis Acid时, 他们也报道了首例分子内羧酸、酰胺与双键的双官能团化三氟甲硫基化反应[112] (Scheme 59).
 图式58
Shen试剂的合成
图式58.
Preparation of N-trifluoromethylthiosaccharin
图式58
Shen试剂的合成
图式58.
Preparation of N-trifluoromethylthiosaccharin
 图式59
Shen试剂的反应
图式59.
Reactions of N-trifluoromethylthiosaccharin
图式59
Shen试剂的反应
图式59.
Reactions of N-trifluoromethylthiosaccharin
除此之外, 其他小组采用糖精试剂, 也实现了其他转化.如2015年大连化物所的李兴伟小组[113]采用FeCl3或AuCl3, 实现了富电子芳烃底物的三氟甲硫基化反应.另外, 他们采用官能团导向的策略实现了Rh (Ⅲ)催化吲哚2位C—H键的三氟甲硫基化反应[114] (Scheme 60).
 图式60
李兴伟小组的工作
图式60.
Works by Li group
图式60
李兴伟小组的工作
图式60.
Works by Li group
Shibata小组与Cahard小组[115]合作, 采用该试剂与烯丙醇底物反应, 发现可以经过重排反应生成烯丙基三氟亚砜的产物(Scheme 61).在该反应中, 二级烯丙醇可以获得热力学稳定的反式选择性的产物.而对于一级、三级烯丙醇类底物, 作者并没有进行相关地考察.
中山大学的赵晓丹小组[116]使用二芳基硒作为催化剂, 在三氟磺酸作为添加剂的条件下, 实现了苯乙烯以及烯烃衍生物的双官能团化三氟甲硫基化反应.他们认为, 二芳基硒与糖精类型的试剂在三氟甲磺酸的存在下可以生成活性的[Se-SCF3]中间体, 接受烯烃的进攻后形成的阳离子可进一步被体系中的亲核试剂捕获, 从而生成双官能团化产物.随后, 他们采用相似的策略, 发现对于炔烃底物, 同样可以实现相应的双官能团化反应, 并且有着优秀的顺反选择性[117].他们也对该体系下的手性硫醚、硒醚催化剂进行了考察.而通过一系列的筛选发现, 茚胺醇衍生的手性硫醚催化剂, 可以很好地实现烯烃内酯化三氟甲硫基化反应.当采用糖精亲电三氟甲硫基化试剂在此条件下反应时, 可以获得92%的收率和83%的对映选择性[118](Scheme 62).
 图式62
赵晓丹小组的工作
图式62.
Zhao's works
图式62
赵晓丹小组的工作
图式62.
Zhao's works
北京大学叶新山小组[119]发现, 使用烯糖与糖精试剂在TMSCl作为Lewis Acid的条件下反应, 会生成氯化三氟甲硫基化的双官能团化产物, 该产物在DBU作为碱的条件下, 可以顺利地发生消除反应, 从而实现双键C—H上的三氟甲硫基化反应(Scheme 63).作者考察多种烯糖类底物, 发现都可以以优秀的收率得到相应的产物.
 图式63
烯糖C—H键三氟甲硫基化反应
图式63.
2-Trifluoromethylthiolation of glycals
图式63
烯糖C—H键三氟甲硫基化反应
图式63.
2-Trifluoromethylthiolation of glycals
湖北科技学院的吴鸣虎和孙绍发小组[120]利用烯胺中间体的亲核性, 通过对二级胺的筛选, 发现吡咯烷可以有效地催化醛底物和糖精试剂的反应.作者通过硼氢化钠现场对羰基进行还原, 实现了一级醇α位三氟甲硫基取代产物的构建(Scheme 64).
 图式64
醛α位三氟甲硫基化反应
图式64.
α-Trifluoromethylthiolation of aldehydes
图式64
醛α位三氟甲硫基化反应
图式64.
α-Trifluoromethylthiolation of aldehydes
Glorius小组[121]采用之前提到过的NaCl活化的策略, 采用糖精试剂, 实现了呋喃2位C—H键的三氟甲硫基化反应(Scheme 65).作者认为该反应可能也是生成了高活性的CF3SCl中间体.但是该反应采用N-三氟甲硫基邻苯二甲酰亚胺却不能反应, 因此反应体系的真正活性中间体是否是CF3SCl还只是个推测.
 图式65
呋喃衍生物的三氟甲硫基化反应
图式65.
NaCl-Catalyzed trifluoromethylthiolation of furans
图式65
呋喃衍生物的三氟甲硫基化反应
图式65.
NaCl-Catalyzed trifluoromethylthiolation of furans
山东大学徐政虎小组[122]首先通过三组分的Click反应构建了5-锡基三氮唑化合物, 随后在AlCl3作为Lewis Acid的条件下与糖精试剂反应, 两步构建了5-三氟甲硫基取代的三氮唑化合物(Scheme 66).
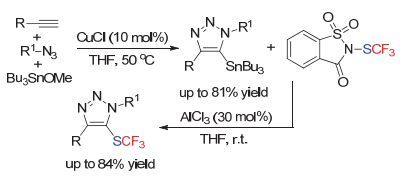 图式66
5-锡基三氮唑的三氟甲硫基化反应
图式66.
5-Stannyl triazole towards trifluoromethylthiotriazoles
图式66
5-锡基三氮唑的三氟甲硫基化反应
图式66.
5-Stannyl triazole towards trifluoromethylthiotriazoles
2.2.6 N-三氟甲硫基双苯磺酰亚胺试剂
2016年上海有机所的吕龙和沈其龙小组[123]进一步报道了基于双苯磺酰亚胺类型的新型亲电三氟甲硫基试剂(Scheme 67).他们对该试剂的反应活性进行了细致地研究, 发现并支持了该试剂高的亲电三氟甲硫基活性.
 图式67
N-三氟甲硫基双苯磺酰亚胺亲电三氟甲硫基试剂的制备
图式67.
Preparation of N-trifluoromethylthio-dibenzenesulfonimide
图式67
N-三氟甲硫基双苯磺酰亚胺亲电三氟甲硫基试剂的制备
图式67.
Preparation of N-trifluoromethylthio-dibenzenesulfonimide
他们与南开大学薛晓松小组合作, 对该试剂的亲电三氟甲硫基化活性进行了理论计算[123, 124] (Scheme 68).计算结果以Tt+DA, 即给出三氟甲硫基阳离子的能力给出.结果表明, 该试剂在目前具有最高的亲电三氟甲硫基化活性, 其活性甚至高过了CF3SCl.基于该理论计算的结果, 他们对该试剂的反应活性进行了较为系统的考察.
 图式68
亲电三氟甲硫基试剂理论计算结果
图式68.
Tt+DA of electrophilic trifluoromethylthiolating reagents
图式68
亲电三氟甲硫基试剂理论计算结果
图式68.
Tt+DA of electrophilic trifluoromethylthiolating reagents
采用该试剂, 他们首先实现了无添加剂存在条件下富电子芳烃、杂芳烃底物的三氟甲硫基化反应.随后, 他们使用2-萘酚衍生物发现了去芳构化反应, 并在三氟磺酸钪和双恶唑啉配体的催化下, 实现了首例去芳构化三氟甲硫基化反应(Scheme 69).
 图式69
N-三氟甲硫基双苯磺酰亚胺的反应
图式69.
Reactions between heteroarenes and naphthol derivatives with N-trifluoromethylthio-dibenzenesulfonimide
图式69
N-三氟甲硫基双苯磺酰亚胺的反应
图式69.
Reactions between heteroarenes and naphthol derivatives with N-trifluoromethylthio-dibenzenesulfonimide
此外, 采用该试剂, 作者发现, 同样无需添加剂的活化, 便可以实现苯乙烯衍生物C—H键反式选择性的三氟甲硫基化反应.作者对该反应的机理进行了研究, 发现该过程经历了碳正离子过程.苯乙烯衍生物在常温下会和该试剂反应从而生成碳正离子中间体或者DMF进攻碳正离子中间体所形成的亚胺离子中间体.中间体在加热条件下发生会发生消除反应, 从而生成烯烃产物(Scheme 70).
 图式70
苯乙烯衍生物C—H键三氟甲硫基化反应及机理
图式70.
Reaction and mechanism for direct trifluoromethylthiolation of styrene derivatives
图式70
苯乙烯衍生物C—H键三氟甲硫基化反应及机理
图式70.
Reaction and mechanism for direct trifluoromethylthiolation of styrene derivatives
随后, 作者在该机理研究的基础上, 实现了一系列双官能团化反应(Scheme 71).作者通过改变反应的溶剂, 实现了烯烃衍生物的甲酰氧化、乙酰氧化、羟化以及胺化三氟甲硫基化反应.
 图式71
烯烃衍生物的双官能团化三氟甲硫基化反应
图式71.
Difunctionalized trifluoromethylthiolation
图式71
烯烃衍生物的双官能团化三氟甲硫基化反应
图式71.
Difunctionalized trifluoromethylthiolation
他们同时也尝试了一系列碳亲核试剂以及杂原子亲核试剂的反应, 发现都可以以优秀的收率得到相应的产物(Scheme 72).
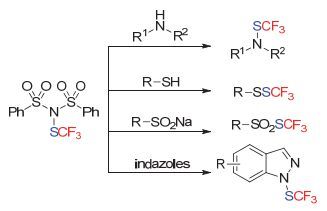 图式72
杂原子亲核试剂的三氟甲硫基化反应
图式72.
Trifluoromethylthiolation of heteroatom-nucleophiles
图式72
杂原子亲核试剂的三氟甲硫基化反应
图式72.
Trifluoromethylthiolation of heteroatom-nucleophiles
前面我们提到中山大学的赵晓丹小组[118]实现了分子内的内酯化反应(Scheme 73).他们在该工作中也对该双苯磺酰亚胺试剂进行了合成.相比较于糖精结构的试剂, 该试剂可以获得更优秀的对映选择性.但是在他们的工作中, 对于该试剂的分离纯化问题没有得到很好地解决.
 图式73
手性硫醚催化下的不对称内酯化三氟甲硫基化反应
图式73.
Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane-based chiral sulfide
图式73
手性硫醚催化下的不对称内酯化三氟甲硫基化反应
图式73.
Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane-based chiral sulfide
2.2.7 手性亲电三氟甲硫基试剂
在2017年年初, 上海有机所的沈其龙小组[125]报道了第一例手性骨架的亲电三氟甲硫基试剂.他们从樟脑磺酰胺出发, 与次氯酸叔丁酯生成N-Cl前体后与AgSCF3发生复分解反应, 可以以优秀的收率得到相应的手性亲电三氟甲硫基试剂(Scheme 74).采用相同的方法, 他们也合成了基于恶唑烷酮骨架的手性亲电三氟甲硫基试剂.
 图式74
手性亲电三氟甲硫基试剂的合成
图式74.
Preparation of optically pure trifluoromethylthiolation reagents
图式74
手性亲电三氟甲硫基试剂的合成
图式74.
Preparation of optically pure trifluoromethylthiolation reagents
采用该类手性试剂, 作者发现, 使用二甲氧基取代的手性磺酰胺试剂, 可以很好地实现β-酮酸酯底物、2-吲哚酮以及苯并呋喃酮底物的不对称三氟甲硫基化反应, 实现试剂手性向底物手性的传递[125].并且对于该类反应, 只需要加入催化量的碱, 便可以顺利地实现转化(Scheme 75).
 图式75
β-酮酸酯、苯并呋喃酮及氧化吲哚底物的不对称三氟甲硫基化反应
图式75.
Trifluoromethylthiolation of β-ketoesters, oxindoles and benzofuranones
图式75
β-酮酸酯、苯并呋喃酮及氧化吲哚底物的不对称三氟甲硫基化反应
图式75.
Trifluoromethylthiolation of β-ketoesters, oxindoles and benzofuranones
2.2.8 现场生成亲电三氟甲硫基试剂
利用稳定且易于制备的亲电三氟甲硫基试剂, 可以高效地在分子中引入三氟甲硫基团.而通过现场生成亲电试剂的策略, 则无需对亲电试剂进行预先制备和分离, 进一步简化了反应操作.
2014年, 南方科技大学的谭斌和刘心元小组[126]通过考察一系列金鸡钠碱催化剂, 实现了3-芳基-2-吲哚酮底物的不对称三氟甲硫基化(Scheme 76).他们通过AgSCF3和TCCA现场生成亲电三氟甲硫基物种的策略实现了该步转化.而在他们初步的机理研究中, 发现CF3SSCF3是该反应的活性物种之一, 此外, 可能存在的其他亲电三氟甲硫基物种尚未被确定.
 图式76
AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现氧化吲哚的不对称三氟甲硫基化反应
图式76.
In situ generation of electrophilic trifluoromethylthio reagents for enantioselective trifluoromethylthiolation of oxindoles
图式76
AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现氧化吲哚的不对称三氟甲硫基化反应
图式76.
In situ generation of electrophilic trifluoromethylthio reagents for enantioselective trifluoromethylthiolation of oxindoles
上海药物所的杨春皓小组[127]采用同样的策略, 在温和的条件下对不同取代基取代的色酮进行了合成(Scheme 77).该反应的机理可能是该推拉电子的底物首先发生分子内的迈克尔加成反应, 随后生成的中间体与现场产生的亲电三氟甲硫基物种反应, 最后脱除二甲基胺生成最终产物.
 图式77
AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现色酮的合成
图式77.
In situ generation of electrophilic trifluoromethylthio reagents for the synthesis of 3-((trifluoromethyl) thio)-4H-chromen-4-on
图式77
AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现色酮的合成
图式77.
In situ generation of electrophilic trifluoromethylthio reagents for the synthesis of 3-((trifluoromethyl) thio)-4H-chromen-4-on
卿凤翎小组[128]从末端炔烃出发, 使用AgSCF3和NCS, 在磷酸钾作碱的条件下, 实现了末端炔烃的三氟甲硫基化反应(Scheme 78).有意思的是, 对于该反应, 先预先合成亲电三氟甲硫基化试剂, 反应是不能发生的.因此, 反应体系中存在的银盐可能对该反应起到了关键性的作用!
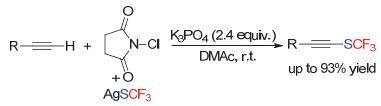 图式78
AgSCF3/NCS下实现末端炔烃的三氟甲硫基化反应
图式78.
Trifluoromethylthiolation of terminal alkynes using AgSCF3 and NCS
图式78
AgSCF3/NCS下实现末端炔烃的三氟甲硫基化反应
图式78.
Trifluoromethylthiolation of terminal alkynes using AgSCF3 and NCS
而在之前我们所提到的亲电三氟甲硫基化试剂的制备大多都是采用亲核性的三氟甲硫基源来完成的. 2015年, 南京理工大学的易文斌与UMass Boston的张玮课题组[129]合作, 使用廉价的三氟亚磺酸钠(CF3SO2Na, Langlois reagent, 常被用来作为三氟甲基自由基试剂)作为三氟甲硫基源, 在亚磷酸酯作为还原剂, DMSO与CuCl作为氧化剂和促进剂的条件下, 实现了吲哚、吡咯以及富电子烯胺类底物的直接三氟甲硫基化反应(Scheme 79).作者对该反应的机理进行了初步的考察, 通过氟谱跟踪和质谱检测, 作者认为, 在该反应中, 首先是三氟亚磺酸钠被亚磷酸酯还原生成活性的CF3S (=O) H, 随后发生重排生成CF3S-OH.该物种可以直接作为亲电试剂与富电子底物发生反应或者是在DMSO与CuCl的存在下生成CF3SSCF3, 从而进一步与底物发生反应.
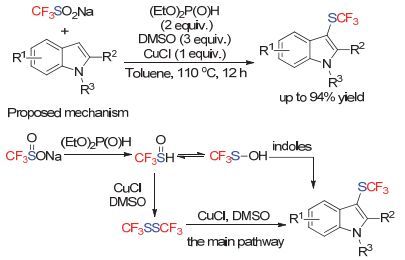 图式79
CF3SO2Na作为三氟甲硫基源实现吲哚的三氟甲硫基化反应
图式79.
Direct trifluoromethylthiolation of indoles with CF3SO2Na
图式79
CF3SO2Na作为三氟甲硫基源实现吲哚的三氟甲硫基化反应
图式79.
Direct trifluoromethylthiolation of indoles with CF3SO2Na
此前我们也提到, Vicic小组[35]使用PPh3与CF3SO2Na反应, 同样观测到了CF3SSCF3的生成.随后, 他们在反应体系中加入CuCl, 可以以80%的氟谱收率, 观测到CuSCF3的生成.
在2016年, Shibata小组与Cahard小组[130]合作, 使用CF3SO2Cl作为三氟甲硫基源, 在PMe3作为还原剂的条件下实现了吲哚、吡咯、烯胺等富电子底物的亲电三氟甲硫基化反应.他们对反应机理进行了初步的考察, 发现体系中的CF3SCl是该反应的活性物种, 而CF3SSCF3不具有反应活性.天津科技大学的赵霞小组[131]使用相似的策略, 发现在体系中加入催化量的碘化钠可以更有效地促进该反应的发生.易文斌小组[132]采用亚磷酸酯为还原剂, 在他们已有工作的基础上, 也可以实现吲哚底物的三氟甲硫基化反应.并且在此条件下, 吡咯衍生物、苯甲醚衍生物以及硫酚衍生物也可以很好地实现三氟甲硫基化转化(Scheme 80).
 图式80
CF3SO2Cl作为三氟甲硫基源实现吲哚三氟甲硫基化反应
图式80.
Direct trifluoromethylthiolation of indoles with CF3SO2Cl
图式80
CF3SO2Cl作为三氟甲硫基源实现吲哚三氟甲硫基化反应
图式80.
Direct trifluoromethylthiolation of indoles with CF3SO2Cl
南京理工大学的蔡春小组[133]使用CF3SO2Na作为三氟甲硫基源, PPh3作为还原剂, N-氯代邻苯二甲酰亚胺作为氯源, 通过现场生成CF3SCl的策略, 同样实现了吲哚、吡咯、烯胺等富电子底物的三氟甲硫基化反应(Scheme 81).
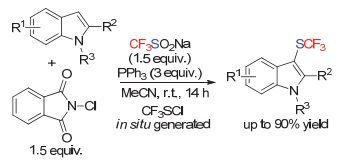 图式81
CF3SO2Na作为三氟甲硫基源实现吲哚三氟甲硫基化反应
图式81.
Direct trifluoromethylthiolation of indoles with CF3SO2Na
图式81
CF3SO2Na作为三氟甲硫基源实现吲哚三氟甲硫基化反应
图式81.
Direct trifluoromethylthiolation of indoles with CF3SO2Na
2.2.9 亲电三氟甲硫基试剂的理论计算结果
在2016年, 南开大学薛小松小组[123, 124]对已有的亲电三氟甲硫基试剂的亲电三氟甲硫基化活性进行了理论计算.他们采用阴离子交换三氟甲硫基正离子的模型, 在二氯甲烷作为反应溶剂的条件下, 对46种亲电三氟甲硫基试剂的亲电活性进行了理论计算(Scheme 82).
 图式82
亲电三氟甲硫基化反应活性及酰胺类型亲电三氟甲硫基化试剂的活性比较
图式82.
Reactivity of electrophilic trifluoromethylthiolating reagents and comparison of amide-type electrophilic trifluoromethylthiolating reagents
图式82
亲电三氟甲硫基化反应活性及酰胺类型亲电三氟甲硫基化试剂的活性比较
图式82.
Reactivity of electrophilic trifluoromethylthiolating reagents and comparison of amide-type electrophilic trifluoromethylthiolating reagents
他们以CF3COOSCF3为基准, 以给出三氟甲硫基正离子的能力(Tt+DA)为指标, 对已有的亲电试剂的活性进行了排序.理论计算结果表明, 该类试剂的反应活性与试剂所对应的酸根阴离子的共轭酸的pKa有着很好的相关性, 共轭酸的pKa越低, 其亲电三氟甲硫基化活性越高.如邻苯二甲酰亚胺、糖精及双磺酰亚胺的pKa分别为10.06, 1.8和1.45, 其所对应的试剂的Tt+DA值为33.0, 17.9和9.8.该9.8的Tt+DA值甚至高过了高活性的CF3SCl (Tt+DA值为11.1), 因而也说明, 基于双苯磺酰亚胺的亲电三氟甲硫基试剂在目前已知的亲电试剂中有着最高的反应活性.
2.2.10 其他类型的亲电三氟甲硫基化反应
前面我们提到, Billard小组[64]利用DAST和TMSCF3合成了其苯胺骨架的亲电三氟甲硫基试剂, 该反应的中间体是CF3-DAST. Shibata小组[134]通过相同的方法对该中间体进行了合成, 并发现该中间体可以与环状酮酸酯类底物或者只含有一个活泼氢的开链酮酸酯底物发生反应, 通过C—C键的断裂来实现一类新颖的重排三氟甲硫基化反应.随后他们发现当底物中含有两个活泼氢时, 则会首先通过烯醇异构化, 进攻CF3-DAST, 并且通过后续的重排反应, 来实现活泼C—H键的三氟甲硫基化反应[135](Scheme 83).值得一提的是, 对于该两类反应, 都可以通过合成不同类型的DAST类似物, 从而实现不同类型的硫氟烷基化反应.
 图式83
CF3-DAST作为三氟甲硫基源的重排反应
图式83.
CF3-DAST as trifluoromethylthiol source
图式83
CF3-DAST作为三氟甲硫基源的重排反应
图式83.
CF3-DAST as trifluoromethylthiol source
2.3 自由基类型三氟甲硫基化反应
自由基类型的亲电三氟甲硫基化反应主要有以下两种反应模式: (1) 体系中先生成三氟甲硫基自由基再与底物反应; (2) 体系中先生成自由基再与亲电三氟甲硫基试剂反应(Scheme 84).
 图式84
两种自由基类型的三氟甲硫基反应
图式84.
Two types of radical trifluoromethylthiolating reactions
图式84
两种自由基类型的三氟甲硫基反应
图式84.
Two types of radical trifluoromethylthiolating reactions
最早的自由基类型三氟甲硫基化反应是使用CF3SH[136]、CF3SCl[137]、CF3SSCF3[138]等在紫外或者X射线的条件下产生三氟甲硫基自由基, 进而与C—H键、双键等发生反应.但是该类反应的条件较为苛刻, 且底物的选择性差, 因而限制了其发展.
2.3.1 三氟甲硫基自由基的反应
在2014年, 中国科技大学的王细胜小组[139]发展了AgSCF3/K2S2O8体系, 实现了活化烯烃的串联关环反应(Scheme 85).作者通过自由基关环、开环以及动力学同位素实验支持了自由基的反应历程.作者认为在该过程中, 三氟甲硫基自由基的产生可能有以下两种途径:一是AgSCF3被氧化成Ag (Ⅱ) SCF3后, 产生三氟甲硫基自由基; 二是体系中的CF3SSCF3可能会与银盐作用产生三氟甲硫基自由基.
 图式85
活化烯烃AgSCF3/K2S2O8体系下自由基三氟甲硫基化反应
图式85.
Activated alkenes radical trifluoromethylthiolating reaction using AgSCF3/K2S2O8
图式85
活化烯烃AgSCF3/K2S2O8体系下自由基三氟甲硫基化反应
图式85.
Activated alkenes radical trifluoromethylthiolating reaction using AgSCF3/K2S2O8
卿凤翎小组[140]发现, 在AgSCF3/K2S2O8体系下, 加入当量铜盐可以实现末端烯烃的三氟甲硫基化反应, 从而构建烯丙基三氟甲硫基化产物.而由于产生的中间体在消除时选择性较差, 产物的E/Z选择性不高, 一般在6:1左右.采用相似的策略, 作者在铜参与下也实现了苯醌衍生物C—H键的三氟甲硫基化反应[141] (Scheme 86).
 图式86
AgSCF3/K2S2O8在铜盐氧化下的自由基三氟甲硫基化反应
图式86.
Copper-mediated oxidative trifluoromethylthiolation of alkenes and quinones
图式86
AgSCF3/K2S2O8在铜盐氧化下的自由基三氟甲硫基化反应
图式86.
Copper-mediated oxidative trifluoromethylthiolation of alkenes and quinones
瑞士苏黎世大学的Nevado小组[142]采用预先构建的底物, 通过活化的双键来捕获三氟甲硫基自由基发生自由基串联关环反应后脱除二氧化硫, 成功地构建了四环产物(Scheme 87).
 图式87
AgSCF3/K2S2O8体系下串联环化反应
图式87.
Cyclization cascades under AgSCF3/K2S2O8system
图式87
AgSCF3/K2S2O8体系下串联环化反应
图式87.
Cyclization cascades under AgSCF3/K2S2O8system
兰州大学梁永民小组[143]使用1, 6-烯炔底物, 在该体系下通过串联环化反应来构建并环骨架.随后, 他们发现, 环化反应的自由基中间体可以被过氧自由基所捕获, 并随后脱除叔丁醇生成氧化去芳构产物[144] (Scheme 88).
 图式88
1, 6-烯炔底物在AgSCF3/K2S2O8体系下串联环化反应
图式88.
Radical cascade cyclization of 1, 6-enynes under AgSCF3/ K2S2O8system
图式88
1, 6-烯炔底物在AgSCF3/K2S2O8体系下串联环化反应
图式88.
Radical cascade cyclization of 1, 6-enynes under AgSCF3/ K2S2O8system
中山大学王洪根小组[145]采用炔酸苯酯类衍生物, 在该体系下通过自由基串联环化反应成功构建了三氟甲硫基取代的香豆素类衍生物(Scheme 89).而采用硫氰酸银也可以实现硫氰基取代的香豆素衍生物的合成.
 图式89
炔酸苯酯类衍生物在AgSCF3/K2S2O8体系下串联环化反应
图式89.
Radical trifluoromethylthiolation of aryl alkynoate esters under AgSCF3/K2S2O8system
图式89
炔酸苯酯类衍生物在AgSCF3/K2S2O8体系下串联环化反应
图式89.
Radical trifluoromethylthiolation of aryl alkynoate esters under AgSCF3/K2S2O8system
上海有机所卿凤翎小组[146]从肉桂酸出发, 在外加铜盐和氧化剂的条件下实现了氧化脱酸三氟甲硫基化反应.对于该反应, 可以得到反式为主的产物, 顺反选择性最高可达11:1.而当采用铁盐为氧化剂, 使用乙腈和水为混合溶剂时, 可以进一步对自由基氧化后所形成碳正离子进行水和, 并在氧气的氛围下进一步氧化成羰基结构(Scheme 90).
 图式90
AgSCF3/K2S2O8体系下肉桂酸衍生物脱羧三氟甲硫基化反应
图式90.
Decarboxylative trifluoromethylthiolation of cinnamic acid under AgSCF3/K2S2O8system
图式90
AgSCF3/K2S2O8体系下肉桂酸衍生物脱羧三氟甲硫基化反应
图式90.
Decarboxylative trifluoromethylthiolation of cinnamic acid under AgSCF3/K2S2O8system
华东理工大学的曹松小组[147]从炔烃出发, 通过自由基类型的三氟甲硫化反应, 可以实现马氏与反马氏选择性的氢-三氟甲硫基化反应(Scheme 91).对于马氏选择性反应, 底物中需要连有烷氧基等给电子基团.而对于马氏选择性反应, 存在着顺反选择性问题, 其比例接近于1:1.但是该体系对于非苯乙烯类的烯烃底物, 反应并不适用.
 图式91
AgSCF3/K2S2O8体系下炔烃马氏、反马氏氢/三氟甲硫基化
图式91.
Anti-Markovnikov and Markovnikov-selective hydrotrifluoromethylthiolation of terminal alkyne under AgSCF3/K2S2O8system
图式91
AgSCF3/K2S2O8体系下炔烃马氏、反马氏氢/三氟甲硫基化
图式91.
Anti-Markovnikov and Markovnikov-selective hydrotrifluoromethylthiolation of terminal alkyne under AgSCF3/K2S2O8system
上海有机所施敏小组[148]使用亚甲基环丙烷底物, 在该体系下通过三元环开环和随后的自由基成环反应, 构建了烯基三氟甲硫基化产物.该产物在过硫酸钠作为氧化剂的条件下, 可以进一步生成芳构化的三氟甲硫基化产物(Scheme 92).
 图式92
AgSCF3/K2S2O8体系下亚甲基环丙烷底物的开环串联反应
图式92.
Trifluoromethylthiolation of methylenecyclopropane under AgSCF3/K2S2O8system
图式92
AgSCF3/K2S2O8体系下亚甲基环丙烷底物的开环串联反应
图式92.
Trifluoromethylthiolation of methylenecyclopropane under AgSCF3/K2S2O8system
美国西弗吉尼亚大学的Hoover小组[149]发现, 对于香豆素类羧酸衍生物, 可以在该体系下以优秀的收率实现氧化脱羧三氟甲硫基化反应(Scheme 93).
 图式93
AgSCF3/K2S2O8体系下香豆素类羧酸脱羧三氟甲硫基化反应
图式93.
Decarboxylative trifluoromethylthiolation of coumarin-3-carboxylic acid under AgSCF3/K2S2O8system
图式93
AgSCF3/K2S2O8体系下香豆素类羧酸脱羧三氟甲硫基化反应
图式93.
Decarboxylative trifluoromethylthiolation of coumarin-3-carboxylic acid under AgSCF3/K2S2O8system
华东理工大学的刘培念小组[150]报道了烯丙醇底物在自由基条件下的半频哪醇重排反应(Scheme 94).在该反应中, 吡啶的加入对产率的提高至关重要, 其在反应体系中起着缚酸剂的作用.
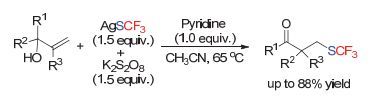 图式94
AgSCF3/K2S2O8体系下半频哪醇重排三氟甲硫基化反应
图式94.
Trifluoromethylthiolation of diaryl allylic alcohols via radical neophyl rearrangement under AgSCF3/K2S2O8system
图式94
AgSCF3/K2S2O8体系下半频哪醇重排三氟甲硫基化反应
图式94.
Trifluoromethylthiolation of diaryl allylic alcohols via radical neophyl rearrangement under AgSCF3/K2S2O8system
Glorius小组采用光催化的策略, 使用邻苯二甲酰亚胺试剂, 同样可以产生三氟甲硫基自由基.使用该策略, 作者实现了烯烃[151], 烯丙醇三氟磺酸酯[151]以及丙烯酰胺[151]等底物的三氟甲硫基化反应(Scheme 95).作者在反应体系中加入了当量的四丁基卤化铵, 其作用可能是生成活性更高的CF3S-X中间体.
 图式95
N-三氟甲硫基邻苯二甲酰亚胺在光催化条件下的自由基三氟甲硫基化反应
图式95.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) phthalimide as SCF3 source
图式95
N-三氟甲硫基邻苯二甲酰亚胺在光催化条件下的自由基三氟甲硫基化反应
图式95.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) phthalimide as SCF3 source
东京工业大学Akita小组[152]在光催化的条件下, 采用N-三氟甲硫基丁二酰亚胺试剂实现了双官能团化反应(Scheme 96).该反应首先经过单电子还原产生三氟甲硫基自由基, 随后进攻苯乙烯衍生物, 生成的苄基自由基经过单电子氧化后接受水的进攻从而实现双官能团化反应.此外, 当使用二氯甲烷和醇为混合溶剂时, 可以实现烷氧基化三氟甲硫基双官能团化反应.
 图式96
N-三氟甲硫基丁二酰亚胺在光催化条件下的自由基三氟甲硫基化反应
图式96.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) succinimide as SCF3 source
图式96
N-三氟甲硫基丁二酰亚胺在光催化条件下的自由基三氟甲硫基化反应
图式96.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) succinimide as SCF3 source
法国的Magnier小组与Billard小组[153]合作, 发现使用糖精试剂在光催化条件下可以产生三氟甲硫基自由基, 并且通过电子顺磁共振检测到了三氟甲硫基自由基的存在(Scheme 97).采用该体系, 作者实现了分子内的串联环化反应以及苯乙烯衍生物的双官能团化反应.
 图式97
Shen试剂在光催化条件下的自由基三氟甲硫基化反应
图式97.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) saccharin as SCF3 source
图式97
Shen试剂在光催化条件下的自由基三氟甲硫基化反应
图式97.
Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) saccharin as SCF3 source
2.3.2 自由基与亲电三氟甲硫基试剂的反应
自由基类型的三氟甲硫基化反应的另外一种反应是体系中首先产生一个活性的自由基中间体, 随后亲电型的三氟甲硫基试剂来作为自由基淬灭剂, 实现三氟甲硫基化反应.
上海有机所的吕龙和沈其龙小组[154]首先报道了该类反应.他们采用烷基羧酸以及烷基硼酸钾盐, 在水相、银催化条件下实现了脱羧三氟甲硫基化反应.在该反应中, SDS的加入至关重要, 作者推测该反应可能是在胶束中进行的, 生成的烷基自由基随后被亲电三氟甲硫基试剂淬灭, 生成三氟甲硫基产物.在此工作的基础上, 作者随后采用Fe盐和硼烷产生[Fe-H]物种后, 对烯烃进行加成, 生成烷基自由基后与次磺酸酯试剂反应, 实现了未活化烯烃马氏选择性的氢化三氟甲硫基化反应[155] (Scheme 98).
 图式98
Lu and Shen试剂作为自由基淬灭剂的反应
图式98.
Lu and Shen reagent as radical quencher
图式98
Lu and Shen试剂作为自由基淬灭剂的反应
图式98.
Lu and Shen reagent as radical quencher
Glorius小组[156]利用光催化的策略, 在金属铱或者有机染料作为光敏剂的条件下, 实现烷基羧酸的脱羧三氟甲硫基化反应(Scheme 99).在此条件下, 烷基自由基的形成通过光氧化过程实现, 因而不用外加的氧化剂便可以产生烷基自由基, 从而与亲电三氟甲硫基试剂进行反应.对于一级羧酸底物, 也可以很好地发生转化.
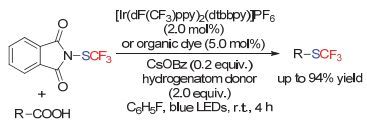 图式99
光催化条件下的脱羧三氟甲硫基化反应
图式99.
Visible light-promoted decarboxylative trifluoromethylthiolation of alkyl carboxylic acids
图式99
光催化条件下的脱羧三氟甲硫基化反应
图式99.
Visible light-promoted decarboxylative trifluoromethylthiolation of alkyl carboxylic acids
上海有机所的陈庆云小组[157]发现, 使用选择性氟化试剂(selectfluor)也可以作为体系中的氧化剂, 从而实现脱羧三氟甲硫基化反应(Scheme 100).该反应的机理可能是AgSCF3被选择性氟化试剂氧化生成Ag (Ⅱ) SCF3.该物种作为氧化剂对羧酸进行氧化脱羧并淬灭, 形成最终的脱羧三氟甲硫基产物.
 图式100
氧化条件下的脱羧三氟甲硫基化反应
图式100.
Oxidative decarboxylative trifluoromethylthiolation of alkyl carboxylic acids
图式100
氧化条件下的脱羧三氟甲硫基化反应
图式100.
Oxidative decarboxylative trifluoromethylthiolation of alkyl carboxylic acids
上海有机所的陈庆云小组[158a]与南开大学的汤平平小组[158b]几乎同时报道了首例烷基C—H键自由基类型的三氟甲硫基化反应(Scheme 101).该类反应的机理可能是体系中产生的硫酸根自由基首先攫取烷基C—H形成烷基自由基, 随后与体系中的Ag (Ⅱ) SCF3、CF3SSCF3或者是[SCF3•]自由基发生反应从而形成最终的C—H键三氟甲硫基化产物.
 图式101
1非活化烷基C—H键自由基三氟甲硫基化反应
图式101.
Unactivated C—H bond trifluoromethylthiolation under radical conditions
图式101
1非活化烷基C—H键自由基三氟甲硫基化反应
图式101.
Unactivated C—H bond trifluoromethylthiolation under radical conditions
Glorius小组[159]采用光催化的策略, 使用苯甲酸钠为碱, 实现了烷基C—H的三氟甲硫基化反应(Scheme 102).相比较于陈庆云小组和汤平平小组的工作, 该工作的反应条件更加温和, 并且有着更优秀的三级C—H键的区域选择性.
 图式102
光催化条件下非活化烷基C—H键三氟甲硫基化反应
图式102.
Radical trifluoromethylthiolation of unactivated C—H bond under photocatalytic conditions
图式102
光催化条件下非活化烷基C—H键三氟甲硫基化反应
图式102.
Radical trifluoromethylthiolation of unactivated C—H bond under photocatalytic conditions
最近, 山东大学的徐政虎小组[160]采用金催化/光催化协同催化的策略, 采用三氟甲硫基苯基砜结构的试剂, 实现了苯乙烯衍生物的双官能团化反应(Scheme 103).该反应的机理可能是在光催化条件下, 活性的阳离子一价金物种IPrAu (Ⅰ)首先被还原成零价金IPrAu (0).该零价金被三氟甲硫基苯基砜还原, 生成IPrAuSCF3与砜基自由基.砜基自由基对苯乙烯底物进行自由基加成, 生成的烷基自由基与亲电型的IPrAuSCF3结合生成二价金中间体, 随后被三价Ru催化剂氧化生成三价金中间体.该三价金中间体发生还原消除生成最终产物, 同时生成一价金物种继续催化循环.该反应有着高的原子经济性.除了三氟甲硫基以外, 硫烷基以及硫苯基类型的试剂也可以很好地实现相应的双官能团化反应.
 图式103
金催化/光催化协同催化下烯烃的砜基三氟甲硫基化反应
图式103.
Dual gold and photoredox catalysis: visible light-mediated intermolecular atom transfer trifluoromethylthiosulfonylation of alkene
图式103
金催化/光催化协同催化下烯烃的砜基三氟甲硫基化反应
图式103.
Dual gold and photoredox catalysis: visible light-mediated intermolecular atom transfer trifluoromethylthiosulfonylation of alkene
3 总结与展望
从以上总结中, 我们可以看到, 在近十年来的时间里, 三氟甲硫基化反应得到了快速的发展, 其中包括过渡金属催化的反应、自由基类型的反应以及不同类型的亲电三氟甲硫基试剂的开发.这些都为化学工作者以及药物化学工作者方便简洁地引入三氟甲硫基团提供了一个较为全面的工具箱.
但与此同时, 该领域仍然存在着一些问题与挑战需要我们去解决.首先, 对于直接三氟甲硫基化反应而言, 通常都需要用到三氟甲硫基金属化合物如AgSCF3和CuSCF3, 这在成本是比较昂贵的.其次, 不对称三氟甲硫基化研究还处于起步阶段, 其相关报道基本都集中于酮酸酯和吲哚酮底物, 对于其他底物如羰基α位的不对称三氟甲硫基化反应或许会成为一个重要的研究课题.
最后, 我们也认为, 对于目前已经存在的含三氟甲硫基的药物、农药分子, 对其相关的代谢途径的研究可能会为我们更好地设计和合成含三氟甲硫基的化合物提供更进一步的理论支持.
-
-
[1]
《2016研究前沿》.
-
[2]
(a) Leo, A.; Hansch, C.; Elkins, D. Chem. Rev. 1971, 71, 525. (b) Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165.
-
[3]
(a) Boiko, V. N. Beilstein J. Org. Chem. 2010, 6, 880. (b) Liang, T. ; Neumann, C. N. ; Ritter, T. Angew. Chem. Int. Ed. 2013, 52, 8214. (c) He, W. -M. ; Weng, Z. -Q. Prog. Chem. 2013, 25, 1071 (in Chinese). (何伟明, 翁志强, 化学进展, 2013, 25, 1071). (d) Fang, L. Huaxue Tongbao, 2014, 77, 1058 (in Chinese). (方玲, 化学通报, 2014, 77, 1058). (e) Liu, Z. -J. ; Liu, P. ; Wu, F. -H. J. Shanghai Instit. of Tech. (Nat. Sci. ). 2014, 14, 93. (f) Toulgoat, F. ; Alazet, S. ; Billard, T. Eur. J. Org. Chem. 2014, 2415. (g) Chu, L. -L. ; Qing, F. -L. Acc. Chem. Res. 2014, 47, 1513. (h) Shao, X. -X. ; Xu, C. -F. ; Lu, L. ; Shen, Q. -L. Acc. Chem. Res. 2015, 48, 1227. (i) Xu, X. H. ; Matsuzaki, K. ; Shibata, N. Chem. Rev. 2015, 115, 731. (j) Zhang, K. ; Xu, X. -H. ; Qing, F. -L. Chin. J. Org. Chem. 2015, 35, 556 (in Chinese). (张柯, 徐修华, 卿凤翎, 有机化学, 2015, 35, 556). (k) Yang, X. -Y. ; Wu, Tao. ; Phipps, R. J. ; Toste, F. D. Chem. Rev. 2015, 115, 826. (l) Lin, J. -H. ; Ji, Y. -L. ; Xiao, J. -C. Current Org. Chem. 2015, 19, 1541. (m) Chachignon, H. ; Cahard, D. Chin. J. Chem. 2016, 34, 445. (n) Zheng, H. -D. ; Huang, Y. -J. ; Weng, Z. -Q. Tetrahedron Lett. 2016, 57, 1397. (o) Guo, Y. ; Huang, M. -W. ; Fu, X. -L. ; Liu, C. ; Chen, Q. -Y. ; Zhao, Z. -G. ; Zeng, B. -Z. ; Chen, J. Chin. Chem. Lett. 2017, 28, 719. (p) Li, G. -M. ; Sun, D. -Q. Chin. J. Org. Chem. 2016, 36, 1715 (in Chinese). (李恭铭, 孙德群, 有机化学, 2016, 36, 1715).
-
[4]
(a) Fluorverbindungen, U. Angew. Chem. 1939, 52, 457. (b) Yagupolski, L. M.; Marenets, M. S. J. Gen. Chem. U.S.S.R. 1954, 24, 885.
-
[5]
(a) Ruppert, I.; Schlich, K.; Volbach, W. Tetrahedron Lett. 1984, 25, 2195. (b) Kolomeitsev, A. A.; Movchun, V. N.; Kondranenko, N. V.; Yagupolski, Y. L. Synthesis 1990, 1151. (c) Shono, T.; IShifune, M.; Okada, T.; Kashimura, S. J. Org. Chem. 1991, 56, 2. (d) Billard, T.; Large, S.; Langlois, B. R. Tetrahedron Lett. 1997, 38, 65. (e) Singh, R. P.; Cao, G.; Kirchmeier, R. L.; Shreeve, J. J. Org. Chem. 1999, 64, 2873. (f) Folleas, B.; Marek, I.; Normant, J. F.; Saint-Jalmes, L. Tetrahedron 2000, 56, 275. (g) Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848. (h) Billard, T.; Langlois, B. R.; Blond, G. Eur. J. Org. Chem. 2001, 1467. (i) Steensma, R. W.; Galabi, S.; Tagat, J. R.; McCombie, S. W. Tetrahedron Lett. 2001, 42, 2281. (j) Potash, S.; Rozen, S. J. Fluorine Chem. 2014, 168, 173.
-
[6]
(a) Yagupolski, L. M.; Kondranenko, N. V.; Timofeeva, G. N. Zh. Org. Khim. 1984, 20, 115. (b) Umemoto, T.Chem. Rev. 1996, 96, 1757. (c) Yang, J. J.; Kirchmeier, R. L.; Shreeve, J. J. Org. Chem. 1998, 63, 2656. (d) Eisenberger, P.; Gischig, S.; Togni, A. Chem. Eur. J. 2006, 12, 2579. (e) Kieltsch, I.; Eisenberger, P.; Togni, A. Angew. Chem. Int. Ed. 2007, 46, 754. (f) Koller, R.; Stanek, K.; Stolz, D.; Ardoom, R.; Niedermann, K.; Togni, A. Angew. Chem. Int. Ed. 2009, 48, 4332.
-
[7]
(a) Boiko, V. N.; Shchupak, G. M.; Yagupolski, L. M. Zh. Org. Khim. 1977, 13, 1057. (b) Popov, I.; Boiko, V. N.; Kondranenko, N. V.; Sambur, V. P.; Yagupolski, L. M. 1977, 13, 2135. (c) Boiko, V. N.; Dashevskaya, T. A.; Shchupak, G. M.; Yagupolski, L. M. Zh. Org. Khim. 1979, 15, 396.
-
[8]
Man, E. H.; Coffman, D. D.; Muetterties, E. L. J. Am. Chem. Soc. 1959, 81, 3575. doi: 10.1021/ja01523a023
-
[9]
Emeleus, H. J.; MacDuffie, D. E. J. Am. Chem. Soc. (Resumed) 1961, 83, 2572.
-
[10]
Yagupolskii, L. M.; Kondratenko, N. V.; Sambur, V. P. Synthesis 1975, 1975, 721. doi: 10.1055/s-1975-23905
-
[11]
Tyrra, W.; Naumann, D.; Hoge, B.; Yagupolskii, Y. L. J. Fluorine Chem. 2003, 119, 101. doi: 10.1016/S0022-1139(02)00276-2
-
[12]
Remy, D. C.; Rittle, K. E.; Hunt, C. A.; Freedman, M. B. J. Org. Chem. 1976, 41, 1644. doi: 10.1021/jo00871a037
-
[13]
Kondratenko, N. V.; Kolomeytsev, A. A.; Popov, V. I.; Yagupolskii, L. M. Synthesis 1985, 667.
-
[14]
Chen, Q.-Y.; Duan, J.-X. J. Chem. Soc., Chem. Commun. 1993, 918.
-
[15]
Clark, J. H.; Tavener, S. J. J. Fluorine Chem. 1997, 85, 169. doi: 10.1016/S0022-1139(97)00057-2
-
[16]
Teverovskiy, G.; Surry, D. S.; Buchwald, S. L. Angew. Chem. Int. Ed. 2011, 50, 7312. doi: 10.1002/anie.v50.32
-
[17]
Zhang, C.-P.; Vicic, D. A. J. Am. Chem. Soc. 2012, 134, 183. doi: 10.1021/ja210364r
-
[18]
Yin, G.; Kalvet, I.; Englert, U.; Schoenebeck, F. J. Am. Chem. Soc. 2015, 137, 4164. doi: 10.1021/jacs.5b00538
-
[19]
Yin, G.; Kalvet, I.; Schoenebeck, F. Angew. Chem. Int. Ed. 2015, 54, 6809. doi: 10.1002/anie.201501617
-
[20]
Dürr, A. B.; Yin, G.; Kalvet, I.; Napoly, F.; Schoenebeck, F. Chem. Sci. 2016, 7, 1076. doi: 10.1039/C5SC03359D
-
[21]
Nguyen, T.; Chiu, W.-L.; Wang, X.-Y.; Sattler, M. O.; Love, J. A. Org. Lett. 2016, 18, 5492. doi: 10.1021/acs.orglett.6b02689
-
[22]
Chen, C.; Xie, Y.; Chu, L.-L.; Wang, R.-W.; Zhang, X.; Qing, F. -L. Angew. Chem. Int. Ed. 2012, 51, 2492. doi: 10.1002/anie.v51.10
-
[23]
Chen, C.; Chu, L.-L.; Qing, F.-L. J. Am. Chem. Soc. 2012, 134, 12454. doi: 10.1021/ja305801m
-
[24]
Zhang, C.-P.; Vicic, D. A. Chem. Asian J. 2012, 7, 1756. doi: 10.1002/asia.v7.8
-
[25]
Zhai, L.-J.; Li, Y.-M.; Yin, J.; Jin, K.; Zhang, R.; Fu, X.-M.; Duan, C.-Y. Tetrahedron 2013, 69, 10262. doi: 10.1016/j.tet.2013.10.028
-
[26]
Zhao, M.-Z.; Zhao, X.-M.; Zheng, P.-R.; Tian, Y.-W. J. Fluorine Chem. 2017, 194, 73. doi: 10.1016/j.jfluchem.2017.01.007
-
[27]
Wu, W.; Wang, B.-Y.; Ji, X.-F.; Cao, S. Org. Chem. Front. 2017, DOI: 10.1039/c7qo00198c.
-
[28]
Chen, C.; Xu, X.-H.; Yang, B.; Qing, F.-L. Org. Lett. 2014, 16, 3372. doi: 10.1021/ol501400u
-
[29]
Yin, W.; Wang, Z.; Huang, Y. Adv. Synth. Catal. 2014, 356, 2998. doi: 10.1002/adsc.201400362
-
[30]
Liu, X.-G.; Li, Q.-J.; Wang, H.-G. Adv. Synth. Catal. 2017, 359, 1942. doi: 10.1002/adsc.v359.11
-
[31]
Munavalli, S.; Rossman, D. I.; Rohrbaugh, D. K.; Ferguson, C. P.; Hsu, F. L. Heteroat. Chem. 1992, 3, 189. doi: 10.1002/(ISSN)1098-1071
-
[32]
Rheingold, A. L.; Munavalli, S.; Rossman, D. I.; Ferguson, C. P. In-org. Chem. 1994, 33, 1723.
-
[33]
Weng, Z.-Q.; He, W.; Chen, C.; Lee, R.; Tan, D.; Lai, Z.; Kong, D.; Yuan, Y.; Huang, K.-W. Angew. Chem. Int. Ed. 2013, 52, 1548. doi: 10.1002/anie.201208432
-
[34]
Wang, Z.; Tu, Q.; Weng, Z.-Q. J. Organomet. Chem. 2014, 751, 830. doi: 10.1016/j.jorganchem.2013.08.008
-
[35]
Yang, Y.; Xu, L.; Yu, S.; Liu, X.; Zhang, Y.; Vicic, D. A. Chem. Eur. J. 2016, 22, 858. doi: 10.1002/chem.201504790
-
[36]
Zhang, M.; Weng, Z.-Q. Adv. Synth. Catal. 2016, 358, 386. doi: 10.1002/adsc.201500575
-
[37]
(a) Kong, D.; Jiang, Z.; Xin, S.; Bai, Z.; Yuan, Y.; Weng, Z.-Q. Tetrahedron 2013, 69, 6046. (b) Huang, Y.; He, X.; Li, H.; Weng, Z.-Q. Eur. J. Org. Chem. 2014, 2014, 7324. (c) Lin, Q.; Chen, L.; Huang, Y.; Rong, M.; Yuan, Y.; Weng, Z.-Q. Org. Biomol. Chem. 2014, 12, 5500.
-
[38]
(a) Zhu, P.; He, X.; Chen, X.; You, Y.; Yuan, Y.; Weng, Z.-Q. Tetrahedron 2014, 70, 672. (b) Huang, Y.; Ding, J.; Wu, C.; Zheng, H.; Weng, Z.-Q. J. Org. Chem. 2015, 80, 2912.
-
[39]
Zhang, M.; Chen, J.; Chen, Z.; Weng, Z.-Q. Tetrahedron 2016, 72, 3525. doi: 10.1016/j.tet.2016.04.081
-
[40]
Kondratenko, N. V.; Sambur, V. P. Ukr. Khim. Zh. (Russ. Ed.) 1975, 41, 516.
-
[41]
Adams, D. J.; Goddard, A.; Clark, J. H.; Macquarrie, D. J. Chem. Commun. 2000, 46, 987.
-
[42]
Danoun, G.; Bayarmagnai, B.; Gruenberg, M. F.; Goossen, L. J. Chem. Sci. 2014, 5, 1312. doi: 10.1039/c3sc53076k
-
[43]
(a) Hu, M.; Rong, J.; Miao, W.; Ni, C.; Han, Y.; Hu, J.-B. Org. Lett. 2014, 16, 2030. (b) Wang, X.; Zhou, Y.; Ji, G.; Wu, G.; Li, M.; Zhang, Y.; Wang, J.-B. Eur. J. Org. Chem. 2014, 3093.
-
[44]
Lefebvre, Q.; Fava, E.; Nikolaienko, P.; Rueping, M. Chem. Commun. 2014, 50, 6617. doi: 10.1039/c4cc02060j
-
[45]
Matheis, C.; Krause, T.; Bragoni, V.; Goossen, L. J. Chem. Eur. J. 2016, 22, 12270. doi: 10.1002/chem.v22.35
-
[46]
Nikolaienko, P.; Pluta, R.; Rueping, M. Chem. Eur. J. 2014, 20, 9867. doi: 10.1002/chem.201402679
-
[47]
Liu, J.-B.; Xu, X.-H.; Chen, Z.-H.; Qing, F.-L. Angew. Chem. Int. Ed. 2015, 54, 897. doi: 10.1002/anie.201409983
-
[48]
Qiu, Y.-F.; Song, X.-R.; Li, M.; Zhu, X.-Y.; Wang, A.-Q.; Yang, F.; Han, Y.-P.; Zhang, H.-R.; Jin, D.-P.; Li, Y.-X.; Liang, Y.-M. Org. Lett. 2016, 18, 1514. doi: 10.1021/acs.orglett.6b00065
-
[49]
Ye, K.-Y.; Zhang, X.; Dai, L.-X.; You, S.-L. J. Org. Chem. 2014, 79, 12106. doi: 10.1021/jo5019393
-
[50]
Zeng, J.-L.; Chachignon, H.; Ma, J.-A.; Cahard, D. Org. Lett. 2017, 19, 1974. doi: 10.1021/acs.orglett.7b00501
-
[51]
Nikolaienko, P.; Yildiz, T.; Rueping, M. Eur. J. Org. Chem. 2016, 1091.
-
[52]
Fang, W.-Y.; Dong, T.; Han, J.-B.; Zha, G.-F.; Zhang, C.-P. Org. Biomol. Chem. 2016, 14, 11502. doi: 10.1039/C6OB02107G
-
[53]
Wang, K.-P.; Yun, S.-Y.; Mamidipalli, P.; Lee, D. Chem. Sci. 2013, 4, 3205. doi: 10.1039/c3sc50992c
-
[54]
Karmakar, R.; Mamidipalli, P.; Salzman, R. M.; Hong, S.; Yun, S. -Y.; Guo, W.; Xia, Y.; Lee, D. Org. Lett. 2016, 18, 3530. doi: 10.1021/acs.orglett.6b01443
-
[55]
Xiao, Q.; Sheng, J.; Ding, Q.; Wu, J. Eur. J. Org. Chem. 2014, 217.
-
[56]
Zeng, Y. W.; Hu, J.-B. Org. Lett. 2016, 18, 856. doi: 10.1021/acs.orglett.6b00142
-
[57]
Li, S.-G.; Zard, S. Z. Org. Lett. 2013, 15, 5898. doi: 10.1021/ol403038f
-
[58]
Yang, H.-B.; Fan, X.; Wei, Y.; Shi, M. Org. Chem. Front. 2015, 2, 1088. doi: 10.1039/C5QO00198F
-
[59]
Fan, X.; Yang, H.; Shi, M. Adv. Synth.Catal. 2017, 359, 49. doi: 10.1002/adsc.v359.1
-
[60]
(a) Andreades, S.; Harris, J. F.; Sheppard, W. A. J. Org. Chem. 1964, 29, 898. (b) Sheppard, W. A. J. Org. Chem. 1964, 29, 895; (c) Scribner, R. M. J. Org. Chem. 1966, 31, 3671. (d) Bayreuther, H.; Haas, A. Chem. Ber. 1973, 106, 1418. (e) Croft, T. S.; McBrady, J. J. J. Heterocycl. Chem. 1975, 12, 845. (f) Haas, A.; hellwig, V. Chem. Ber. 1976, 109, 2475. (g) Haas, A.; Niemann, U. Chem. Ber. 1977, 110, 67. (h) Popov, V. I.; Kondranenko, N. V.; Haas, A. UKr. Khim. Zh. 1983, 49, 861. (i) Haas, A.; Lieb, M.; Zhang, Y. J. Fluorine Chem. 1985, 29, 311; (j) Bogdanowicz-Szwed, K.; Kawalek, B.; Lieb, M. J. Fluorine Chem. 1987, 35, 317. (k) Rossman, D. I.; Muller, A. J.; Lewis, E. O. J. Fluorine Chem. 1991, 55, 221.
-
[61]
(a) Sharpe, T. R.; Cherkofsky, S. C.; Hewes, W. E.; Smith, D. H.; Gregory, W. A.; Haber, S. B.; Leadbetter, M. R.; Whitney, J. G. J. Med. Chem. 1985, 28, 1188. (b) South, M. S.; Van Sant, K. A. J. Heterocycl. Chem 1991, 28, 1017. (c) Boese, R.; Haas, A.; Lieb, M.; Roeske, U. Chem. Ber. 1994, 127, 449.
-
[62]
Tran, L. D.; Popov, I.; Daugulis, O. J. Am. Chem. Soc. 2012, 134, 18237. doi: 10.1021/ja3092278
-
[63]
CF3SCl was reported to have an L(ct)50 of between 440 and 880 ppm/min and CF3SSCF3 was reported to have an L(ct)50 of about 200 ppm/min. Chem. Eng. News 1967, 45(51), 44.
-
[64]
Ferry, A.; Billard, T.; Langlois, B. R.; Bacque, E. J. Org. Chem. 2008, 73, 9362. doi: 10.1021/jo8018544
-
[65]
Ferry, A.; Billard, T.; Langlois, B. R.; Bacque, E. Angew. Chem. Int. Ed. 2009, 48, 8551. doi: 10.1002/anie.v48:45
-
[66]
Alazet, S.; Zimmer, L.; Billard, T. Angew. Chem. Int. Ed. 2013, 52, 10814. doi: 10.1002/anie.201305179
-
[67]
Ferry, A.; Billard, T.; Bacqué, E.; Langlois, B. R. J. Fluorine Chem. 2012, 134, 160. doi: 10.1016/j.jfluchem.2011.02.005
-
[68]
Alazet, S.; Ollivier, K.; Billard, T. Beilstein J. Org. Chem. 2013, 9, 2354.
-
[69]
(a) Alazet, S.; Zimmer, L.; Billard, T. Chem. Eur. J. 2014, 20, 8589. (b) Alazet, S.; Ismalaj, E.; Glenadel, Q.; Le Bars, D.; Billard, T. Eur. J. Org. Chem. 2015, 4607.
-
[70]
Alazet, S.; Zimmer, L.; Billard, T. J. Fluorine Chem. 2015, 171, 78. doi: 10.1016/j.jfluchem.2014.09.009
-
[71]
Glenadel, Q.; Alazet, S.; Tlili, A.; Billard, T. Chem. Eur. J. 2015, 21, 14694. doi: 10.1002/chem.201502338
-
[72]
Glenadel, Q.; Billard, T. Chin. J. Chem. 2016, 34, 455.
-
[73]
Glenadel, Q.; Bordy, M.; Alazet, S.; Tlili, A.; Billard, T. Asian J. Org. Chem. 2016, 5, 428.
-
[74]
Tlili, A.; Alazet, S.; Glenadel, Q.; Billard, T. Chem. Eur. J. 2016, 22, 10230. doi: 10.1002/chem.201601338
-
[75]
Alazet, S.; Billard, T. Synlett 2015, 26, 76. doi: 10.1055/s-00000083
-
[76]
Bonazaba Milandou, L. J. C.; Carreyre, H.; Alazet, S.; Greco, G.; Martin-Mingot, A.; Nkounkou Loumpangou, C.; Ouamba, J. M.; Bouazza, F.; Billard, T.; Thibaudeau, S. Angew. Chem. Int. Ed. 2017, 56, 169. doi: 10.1002/anie.v56.1
-
[77]
Yang, Y.; Jiang, X.; Qing, F.-L. J. Org. Chem. 2012, 77, 7538. doi: 10.1021/jo3013385
-
[78]
(a) Liu, J. -B. ; Chu, L. -L. ; Qing, F. -L. Org. Lett. 2013, 15, 894. (b) Xu, J. -B. ; Chen, P. -H. ; Ye, J. -X. ; Liu, G. -S. Acta Chim. Sinica 2015, 73, 1294 (in Chinese). (徐佳斌, 陈品红, 叶金星, 刘国生, 化学学报, 2015, 73, 1294). (c) Wu, W. ; Zhang, X. -X. ; Liang, F. ; Cao, S. Org. Biomol. Chem. 2015, 13, 6992.
-
[79]
Xiao, Q.; Sheng, J.; Chen, Z.; Wu, J. Chem. Commun. 2013, 49, 8647. doi: 10.1039/c3cc44263b
-
[80]
Sheng, J.; Fan, C.; Wu, J. Chem. Commun. 2014, 50, 5494. doi: 10.1039/c4cc01904k
-
[81]
Sheng, J.; Li, S.; Wu, J. Chem. Commun. 2014, 50, 578. doi: 10.1039/C3CC45958F
-
[82]
Liu, T.; Qiu, G.-Y.-S.; Ding, Q.-P.; Wu, J. Tetrahedron 2016, 72, 1472. doi: 10.1016/j.tet.2016.01.053
-
[83]
Sheng, J.; Wu, J. Org. Biomol. Chem. 2014, 12, 7629. doi: 10.1039/C4OB01451K
-
[84]
Liu, Y.-W.; Qiu, G.-Y.-S.; Wang, H.-L.; Sheng, J. Tetrahedron Lett. 2017, 58, 690. doi: 10.1016/j.tetlet.2017.01.018
-
[85]
Shao, X.-X.; Wang, X.-Q.; Yang, T.; Lu, L.; Shen, Q.-L. Angew. Chem. Int. Ed. 2013, 52, 3457. doi: 10.1002/anie.v52.12
-
[86]
Vinogradova, E. V.; Muller, P.; Buchwald, S. L. Angew. Chem. Int. Ed. 2014, 53, 3125. doi: 10.1002/anie.201310897
-
[87]
Shao, X.-X.; Liu, T.-F.; Lu, L.; Shen, Q.-L. Org. Lett. 2014, 16, 4738. doi: 10.1021/ol502132j
-
[88]
Ma, B.-Q.; Shao, X.-X.; Shen, Q.-L. J. Fluorine Chem. 2015, 171, 73. doi: 10.1016/j.jfluchem.2014.09.011
-
[89]
Shao, X.-X.; Liu, T.-F.; Lu, L.; Shen, Q.-L. Org. Lett. 2015, 80, 3012.
-
[90]
(a) Wang, X.-Q.; Yang, T.; Cheng, X.; Shen, Q.-L. Angew. Chem. Int. Ed. 2013, 52, 12860. (b) Yang, T.; Shen, Q.-L.; Lu, L. Chin. J. Chem. 2014, 32, 678.
-
[91]
Deng, Q. H.; Rettenmeier, C.; Wadepohl, H.; Gade, L. H. Chem. Eur. J. 2014, 20, 93. doi: 10.1002/chem.201303641
-
[92]
He, H.; Zhu, X. Org. Lett. 2014, 16, 3102. doi: 10.1021/ol501211z
-
[93]
Li, Y.; Ye, Z.; Bellman, T. M.; Chi, T.; Dai, M. Org. Lett. 2015, 17, 2186. doi: 10.1021/acs.orglett.5b00782
-
[94]
Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. J. Am. Chem. Soc. 2013, 135, 8782. doi: 10.1021/ja402455f
-
[95]
Arimori, S.; Takada, M.; Shibata, N. Dalton Trans. 2015, 44, 19456. doi: 10.1039/C5DT02214B
-
[96]
Huang, Z.; Yang, Y.-D.; Tokunaga, E.; Shibata, N. Asian J. Org. Chem. 2015, 4, 525.
-
[97]
Huang, Z.; Yang, Y.-D.; Tokunaga, E.; Shibata, N. Org. Lett. 2015, 17, 1094. doi: 10.1021/ol503616y
-
[98]
Arimori, S.; Takada, M.; Shibata, N. Org. Lett. 2015, 17, 1063. doi: 10.1021/acs.orglett.5b00057
-
[99]
Huang, Z.; Okuyama, K.; Wang, C.; Tokunaga, E.; Li, X.; Shibata, N. ChemistryOpen 2016, 5, 188. doi: 10.1002/open.201500225
-
[100]
Haas, A.; Möller, G. Chemische Berichte 1996, 129, 1383. doi: 10.1002/(ISSN)1099-0682
-
[101]
Munavalli, S.; Rohrbaugh, D. K.; Rossman, D. I.; Berg, F. J.; Wagner, G. W.; Durst, H. D. Synth. Commun. 2000, 30, 2847. doi: 10.1080/00397910008087435
-
[102]
Pluta, R.; Nikolaienko, P.; Rueping, M. Angew. Chem. Int. Ed. 2014, 53, 1650. doi: 10.1002/anie.201307484
-
[103]
Kang, K.; Xu, C.-F.; Shen, Q.-L. Org. Chem. Front. 2014, 1, 294. doi: 10.1039/c3qo00068k
-
[104]
Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. Angew. Chem. Int. Ed. 2013, 52, 12856. doi: 10.1002/anie.201304957
-
[105]
Rueping, M.; Liu, X.; Bootwicha, T.; Pluta, R.; Merkens, C. Chem. Commun. 2014, 50, 2508. doi: 10.1039/c3cc49877h
-
[106]
Pluta, R.; Rueping, M. Chem. Eur. J. 2014, 20, 17315. doi: 10.1002/chem.201405654
-
[107]
Xiao, Q.; He, Q.; Li, J.; Wang, J. Org. Lett. 2015, 17, 6090. doi: 10.1021/acs.orglett.5b03116
-
[108]
Honeker, R.; Ernst, J. B.; Glorius, F. Chem. Eur. J. 2015, 21, 8047. doi: 10.1002/chem.201500957
-
[109]
Xu, C.-F.; Shen, Q.-L. Org. Lett. 2014, 16, 2046. doi: 10.1021/ol5006533
-
[110]
Zhao, B.-L.; Du, D.-M. Org. Lett. 2017, 19, 1036. doi: 10.1021/acs.orglett.6b03846
-
[111]
Xu, C.-F.; Ma, B.-Q.; Shen, Q.-L. Angew. Chem. Int. Ed. 2014, 53, 9316. doi: 10.1002/anie.201403983
-
[112]
Xu, C.-F.; Shen, Q.-L. Org. Lett. 2015, 17, 4561. doi: 10.1021/acs.orglett.5b02315
-
[113]
Wang, Q.; Qi, Z.; Xie, F.; Li, X.-W. Adv. Synth. Catal. 2015, 357, 355. doi: 10.1002/adsc.201400717
-
[114]
Wang, Q.; Xie, F.; Li, X.-W. J. Org. Chem. 2015, 80, 8361. doi: 10.1021/acs.joc.5b00940
-
[115]
Maeno, M.; Shibata, N.; Cahard, D. Org. Lett. 2015, 17, 1990. doi: 10.1021/acs.orglett.5b00750
-
[116]
Luo, J.; Zhu, Z.; Liu, Y.; Zhao, X.-D. Org. Lett. 2015, 17, 3620. doi: 10.1021/acs.orglett.5b01727
-
[117]
Wu, J.-J.; Xu, J.; Zhao, X.-D. Chem. Eur. J. 2016, 22, 15265. doi: 10.1002/chem.v22.43
-
[118]
Liu, X.; An, R.; Zhang, X.; Luo, J.; Zhao, X.-D. Angew. Chem. Int. Ed. 2016, 55, 5846. doi: 10.1002/anie.201601713
-
[119]
Yu, Y.; Xiong, D.-C.; Ye, X.-S. Org. Biomol. Chem. 2016, 14, 6403. doi: 10.1039/C6OB01001F
-
[120]
Hu, L.-Q.; Wu, M.-H.; Wan, H.-X.; Wang, J.; Wang, G.-Q.; Guo, H.-B.; Sun, S.-F. New J. Chem. 2016, 40, 6550.
-
[121]
Ernst, J. B.; Rakers, L.; Glorius, F. Synthesis 2017, 49, 260.
-
[122]
Wei, F.; Zhou, T.; Ma, Y.; Tung, C.-H.; Xu, Z.-H. Org. Lett. 2017, 19, 2098. doi: 10.1021/acs.orglett.7b00701
-
[123]
Zhang, P.-P.; Li, M.; Xue, X.-S.; Xu, C.-F.; Zhao, Q.-C.; Liu, Y.-F.; Wang, H.-Y.; Guo, Y.-L.; Lu, L.; Shen, Q.-L. J. Org. Chem. 2016, 81, 7486. doi: 10.1021/acs.joc.6b01178
-
[124]
Li, M.; Guo, J.; Xue, X.-S.; Cheng, J.-P. Org. Lett. 2016, 18, 264. doi: 10.1021/acs.orglett.5b03433
-
[125]
Zhang, H.; Leng, X.-B.; Wan, X.-L.; Shen, Q.-L. Org. Chem. Front. 2017, 4, 1051. doi: 10.1039/C7QO00042A
-
[126]
Zhu, X.-L.; Xu, J.-H.; Cheng, D.-J.; Zhao, L.-J.; Liu, X.-Y.; Tan, B. Org. Lett. 2014, 16, 2192. doi: 10.1021/ol5006888
-
[127]
Xiang, H.; Yang, C.-H. Org. Lett. 2014, 16, 5686. doi: 10.1021/ol502751k
-
[128]
Zhu, S.-Q.; Xu, X.-H.; Qing, F.-L. Eur J. Org. Chem. 2014, 4453.
-
[129]
Jiang, L.; Qian, J.; Yi, W.; Lu, G.; Cai, C.; Zhang, W. Angew. Chem. Int. Ed. 2015, 54, 14965. doi: 10.1002/anie.201508495
-
[130]
Chachignon, H.; Maeno, M.; Kondo, H.; Shibata, N.; Cahard, D. Org. Lett. 2016, 18, 2467. doi: 10.1021/acs.orglett.6b01026
-
[131]
Lu, K.; Deng, Z.-J.; Li, M.; Li, T.-J.; Zhao, X. Org. Biomol. Chem. 2017, 15, 1254. doi: 10.1039/C6OB02465C
-
[132]
Jiang, L.-Q.; Yi, W.-B.; Liu, Q.-R. Adv. Synth. Catal. 2016, 358, 3700. doi: 10.1002/adsc.201600651
-
[133]
Bu, M. J.; Lu, G. P.; Cai, C. Org. Chem. Front. 2017, 4, 266. doi: 10.1039/C6QO00622A
-
[134]
Saidalimu, I.; Suzuki, S.; Tokunaga, E.; Shibata, N. Chem. Sci. 2016, 7, 2106. doi: 10.1039/C5SC04208A
-
[135]
Saidalimu, I.; Suzuki, S.; Yoshioka, T.; Tokunaga, E.; Shibata, N. Org. Lett. 2016, 18, 6404. doi: 10.1021/acs.orglett.6b03301
-
[136]
Harris, J. F.; Stacey, F. W. J. Am. Chem. Soc. 1961, 83, 840. doi: 10.1021/ja01465a026
-
[137]
(a) Harris, J. F. J. Am. Chem. Soc. 1962, 84, 3148. (b) Harris, J. F. J. Org. Chem. 1966, 31, 931.
-
[138]
Haran, G.; Sharp, D. W. A. J. Chem. Soc., Perkin Trans. 11972, 34.
-
[139]
Yin, F.; Wang, X.-S. Org. Lett. 2014, 16, 1128. doi: 10.1021/ol403739w
-
[140]
Zhang, K.; Liu, J.-B.; Qing, F.-L. Chem. Commun. 2014, 50, 14157. doi: 10.1039/C4CC07062C
-
[141]
Li, C.; Zhang, K.; Xu, X.-H.; Qing, F.-L. Tetrahedron Lett. 2015, 56, 6273. doi: 10.1016/j.tetlet.2015.09.117
-
[142]
Fuentes, N.; Kong, W.; Fernandez-Sanchez, L.; Merino, E.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 964. doi: 10.1021/ja5115858
-
[143]
Qiu, Y.-F.; Zhu, X.-Y.; Li, Y.-X.; He, Y.-T.; Yang, F.; Wang, J.; Hua, H.-L.; Zheng, L.; Wang, L.-C.; Liu, X.-Y.; Liang, Y.-M. Org. Lett. 2015, 17, 3694. doi: 10.1021/acs.orglett.5b01657
-
[144]
Jin, D.-P.; Gao, P.; Chen, D.-Q.; Chen, S.; Wang, J.; Liu, X.-Y.; Liang, Y.-M. Org. Lett. 2016, 18, 3486. doi: 10.1021/acs.orglett.6b01702
-
[145]
Zeng, Y.-F.; Tan, D.-H.; Chen, Y.; Lu, W.-X.; Liu, X.-G.; Li, Q.; Wang, H.-G. Org. Chem. Front. 2015, 2, 1511. doi: 10.1039/C5QO00271K
-
[146]
Pan, S.; Huang, Y.; Qing, F.-L. Chem. Asian J. 2016, 11, 2854. doi: 10.1002/asia.201601098
-
[147]
Wu, W.; Dai, W.; Ji, X.; Cao, S. Org. Lett. 2016, 18, 2918. doi: 10.1021/acs.orglett.6b01286
-
[148]
Chen, M.-T.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2017, 4, 86. doi: 10.1039/C6QO00536E
-
[149]
Li, M.; Petersen, J. L.; Hoover, J. M. Org. Lett. 2017, 19, 638. doi: 10.1021/acs.orglett.6b03806
-
[150]
Liu, K.; Jin, Q.; Chen, S.; Liu, P.-N. RSC Adv. 2017, 7, 1546. doi: 10.1039/C6RA25378D
-
[151]
Honeker, R.; Garza-Sanchez, R. A.; Hopkinson, M. N.; Glorius, F. Chem. Eur. J. 2016, 22, 4395. doi: 10.1002/chem.201600190
-
[152]
Li, Y.; Koike, T.; Akita, M. Asian J. Org. Chem. 2017, 6, 445.
-
[153]
Dagousset, G.; Simon, C.; Anselmi, E.; Tuccio, B.; Billard, T.; Magnier, E. Chem. Eur. J. 2017, 23, 4282. doi: 10.1002/chem.201700734
-
[154]
Hu, F.; Shao, X.-X.; Zhu, D.-H.; Lu, L.; Shen, Q.-L. Angew. Chem. Int. Ed. 2014, 53, 6105. doi: 10.1002/anie.201402573
-
[155]
Yang, T.; Lu, L.; Shen, Q.-L. Chem. Commun. 2015, 51, 5479. doi: 10.1039/C4CC08655D
-
[156]
Candish, L.; Pitzer, L.; Gomez-Suarez, A.; Glorius, F. Chem. Eur. J. 2016, 22, 4753. doi: 10.1002/chem.201600421
-
[157]
He, B.; Xiao, Z.-W.; Wu, H.; Guo, Y.; Chen, Q.-Y.; Liu, C. RSC Adv. 2017, 7, 880. doi: 10.1039/C6RA26133G
-
[158]
(a) Wu, H.; Xiao, Z.; Wu, J.; Guo, Y.; Xiao, J.-C.; Liu, C.; Chen, Q.-Y. Angew. Chem. Int. Ed. 2015, 54, 4070. (b) Guo, S.; Zhang, X.; Tang, P.-P. Angew. Chem. Int. Ed. 2015, 54, 4065.
-
[159]
Mukherjee, S.; Maji, B.; Tlahuext-Aca, A.; Glorius, F. J. Am. Chem. Soc. 2016, 138, 16200. doi: 10.1021/jacs.6b09970
-
[160]
Li, H.; Shan, C.; Tung, C.-H.; Xu, Z.-H. Chem. Sci. 2017, 8, 2610. doi: 10.1039/C6SC05093J
-
[1]
-

图式6 Pd催化芳基溴化物的三氟甲硫基化反应
Scheme 6 Palladium-Catalyzed coupling of aryl bromides with AgSCF3

图式7 Ni催化芳基碘化物、溴化物三氟甲硫基化反应
Scheme 7 Nickel-Catalyzed coupling of ary iodides and bromides with NMe4SCF3

图式8 Ni催化芳基氯化物三氟甲硫基化反应
Scheme 8 Nickel-Catalyzed coupling of ary chlorides with NMe4SCF3

图式9 双核一价Pd催化芳基碘化物、溴化物三氟甲硫基化反应
Scheme 9 Dinuclear palladium catalyzed coupling of ary iodides and bromides with NMe4SCF3

图式10 Ni催化芳基、烯基类卤化物的三氟甲硫基化反应
Scheme 10 Nickel-Catalyzed coupling of ary, vinyl triflates or nonaflates with NMe4SCF3

图式11 官能团导向芳基C—Br键、C—Cl键的三氟甲硫基化反应
Scheme 11 Ligandless nickel-catalyzed ortho-selective directed trifluoromethylthiolation of aryl chlorides and bromides using AgSCF3

图式12 铜催化硼酸的氧化三氟甲硫基化反应
Scheme 12 Copper-Catalyzed oxidative coupling of aryl boronic acid with TMSCF3 and elemental sulfur

图式13 炔烃的氧化三氟甲硫基化反应
Scheme 13 Metal-free oxidative coupling of terminal alkynes with TMSCF3 and elemental sulfur

图式14 铜介导的硼酸氧化三氟甲硫基化反应
Scheme 14 Copper-mediated trifluoromethylthiolation of aryl boronic acids with NMe4SCF3

图式19 重氮酸酯的三氟甲硫基化反应
Scheme 19 Trifluoromethylthiolation of diazo compounds through copper carbine migratory insertion

图式21 炔丙醇底物的串联环化三氟甲硫基化反应
Scheme 21 AgSCF3/BF3•Et2O Mediated trifluoromethylthiolation cascade cyclization of propynols

图式22 钌催化区域端位选择性的烯丙基三氟甲硫基化反应
Scheme 22 Ruthenium-catalyzed regioselective allylic trifluoromethylthiolation reaction

图式24 高价碘盐的三氟甲硫基化反应
Scheme 24 Trifluoromethylthiolation of Unsymmetrical λ3-iodane derivatives

图式25 AgSCF3与苯炔、烯炔中间体的反应
Scheme 25 Trifluoromethylthiolation of aryne and allene-enyne intermediate

图式26 AgSCF3与氧化异喹啉、苯炔中间体的反应
Scheme 26 Trifluoromethylthiolation of N-oxide and aryne intermediate

图式27 现场生成三氟甲硫基阴离子的反应
Scheme 27 In-situ generation and capture of trifluoromethanethiol

图式28 MBH酯的亲核三氟甲硫基化反应
Scheme 28 Nucleophilic trifluoromethylthiolation of Morita-Baylis-Hillman carbonates

图式30 8-氨基喹啉导向C—H三氟甲硫基化反应
Scheme 30 Auxiliary-assisted trifluoromethylthiolation using CF3SSCF3

图式31 亲电三氟甲硫基试剂种类
Scheme 31 Different types of electrophilic trifluoromethylthiolation reagents

图式32 第一代Billard试剂的制备及反应
Scheme 32 Preparation and reactions of first generation of Billard reagent

图式33 第二代Billard试剂的制备及反应
Scheme 33 Preparation and reactions of Second generation of Billard reagent

图式34 超强酸催化下芳胺底物的三氟甲硫基化反应
Scheme 34 Superacid-catalyzed trifluoromethylthiolation of aromatic amines

图式35 酸和酰氯活化下的三氟甲硫基化反应
Scheme 35 Trifluoromethylthiolation under the activation of acid or acyl chloride using the first generation of Billard reagent

图式37 磺酰氯的一锅法三氟甲硫基化转化
Scheme 37 One-pot reaction of sulfonyl chloride and Billard reagent

图式40 不对称三氟甲硫基化反应
Scheme 40 Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent

图式41 Cu (OTf)2/boxmi体系下不对称三氟甲硫基化反应
Scheme 41 Enantioselective electrophilic trifluoromethylthiolation using Lu and Shen reagent and copper-boxmi complexes

图式43 铜催化环丙醇亲电开环三氟甲硫基化反应
Scheme 43 Copper-catalyzed electrophilic ring-opening cross-coupling of cyclopropanols

图式46 Shibata试剂的反应机理
Scheme 46 Proposed mechanism for Cu (Ⅰ)-catalyzed trifluoromethylthiolation with Shibata reagent

图式49 Munavalli小组的初步尝试
Scheme 49 Preliminary trifluoromethylthiolation attempts by Munavalli group

图式50 N-三氟甲硫基邻苯二甲酰亚胺试剂合成方法的改良
Scheme 50 Modified preparation of N-(trifluoromethylthio) phthalimide by Rueping and shen group

图式51 N-三氟甲硫基邻苯二甲酰亚胺试剂参与的不对称反应
Scheme 51 Asymmetric trifluoromethylthiolation using N-(trifluoro-methylthio) phthalimide

图式52 N-三氟甲硫基邻苯二甲酰亚胺试剂与硼酸的偶联反应
Scheme 52 Cross-coupling reaction between boronic acids and N-(trifluoromethylthio) phthalimide

图式54 碱促进下不饱和羰基化合物的胺化三氟甲硫基化反应
Scheme 54 DABCO-promoted aminotrifluoromethylthiolation of α, β-unsaturated carbonyl compounds

图式55 NaCl催化下含氮杂环的三氟甲硫基化反应
Scheme 55 NaCl-catalyzed trifluoromethylthiolation of N-heteroarenes

图式56 Pd催化芳基C—H三氟甲硫基化反应
Scheme 56 Palladium-catalyzed trifluoromethylthiolation of Aryl C—H bonds

图式57 双方酰化氨基酸配体催化下不对称三氟甲硫基化反应
Scheme 57 Enantioselective squaramide-catalyzed trifluoromethylthiolation-sulfur-Michael/adol cascade reaction


图式66 5-锡基三氮唑的三氟甲硫基化反应
Scheme 66 5-Stannyl triazole towards trifluoromethylthiotriazoles

图式67 N-三氟甲硫基双苯磺酰亚胺亲电三氟甲硫基试剂的制备
Scheme 67 Preparation of N-trifluoromethylthio-dibenzenesulfonimide

图式68 亲电三氟甲硫基试剂理论计算结果
Scheme 68 Tt+DA of electrophilic trifluoromethylthiolating reagents

图式69 N-三氟甲硫基双苯磺酰亚胺的反应
Scheme 69 Reactions between heteroarenes and naphthol derivatives with N-trifluoromethylthio-dibenzenesulfonimide

图式70 苯乙烯衍生物C—H键三氟甲硫基化反应及机理
Scheme 70 Reaction and mechanism for direct trifluoromethylthiolation of styrene derivatives

图式73 手性硫醚催化下的不对称内酯化三氟甲硫基化反应
Scheme 73 Enantioselective trifluoromethylthiolating lactonization catalyzed by an indane-based chiral sulfide

图式74 手性亲电三氟甲硫基试剂的合成
Scheme 74 Preparation of optically pure trifluoromethylthiolation reagents

图式75 β-酮酸酯、苯并呋喃酮及氧化吲哚底物的不对称三氟甲硫基化反应
Scheme 75 Trifluoromethylthiolation of β-ketoesters, oxindoles and benzofuranones

图式76 AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现氧化吲哚的不对称三氟甲硫基化反应
Scheme 76 In situ generation of electrophilic trifluoromethylthio reagents for enantioselective trifluoromethylthiolation of oxindoles

图式77 AgSCF3/TCCA现场生成亲电三氟甲硫基试剂实现色酮的合成
Scheme 77 In situ generation of electrophilic trifluoromethylthio reagents for the synthesis of 3-((trifluoromethyl) thio)-4H-chromen-4-on

图式78 AgSCF3/NCS下实现末端炔烃的三氟甲硫基化反应
Scheme 78 Trifluoromethylthiolation of terminal alkynes using AgSCF3 and NCS

图式79 CF3SO2Na作为三氟甲硫基源实现吲哚的三氟甲硫基化反应
Scheme 79 Direct trifluoromethylthiolation of indoles with CF3SO2Na

图式80 CF3SO2Cl作为三氟甲硫基源实现吲哚三氟甲硫基化反应
Scheme 80 Direct trifluoromethylthiolation of indoles with CF3SO2Cl

图式81 CF3SO2Na作为三氟甲硫基源实现吲哚三氟甲硫基化反应
Scheme 81 Direct trifluoromethylthiolation of indoles with CF3SO2Na

图式82 亲电三氟甲硫基化反应活性及酰胺类型亲电三氟甲硫基化试剂的活性比较
Scheme 82 Reactivity of electrophilic trifluoromethylthiolating reagents and comparison of amide-type electrophilic trifluoromethylthiolating reagents

图式84 两种自由基类型的三氟甲硫基反应
Scheme 84 Two types of radical trifluoromethylthiolating reactions

图式85 活化烯烃AgSCF3/K2S2O8体系下自由基三氟甲硫基化反应
Scheme 85 Activated alkenes radical trifluoromethylthiolating reaction using AgSCF3/K2S2O8

图式86 AgSCF3/K2S2O8在铜盐氧化下的自由基三氟甲硫基化反应
Scheme 86 Copper-mediated oxidative trifluoromethylthiolation of alkenes and quinones

图式88 1, 6-烯炔底物在AgSCF3/K2S2O8体系下串联环化反应
Scheme 88 Radical cascade cyclization of 1, 6-enynes under AgSCF3/ K2S2O8system

图式89 炔酸苯酯类衍生物在AgSCF3/K2S2O8体系下串联环化反应
Scheme 89 Radical trifluoromethylthiolation of aryl alkynoate esters under AgSCF3/K2S2O8system

图式90 AgSCF3/K2S2O8体系下肉桂酸衍生物脱羧三氟甲硫基化反应
Scheme 90 Decarboxylative trifluoromethylthiolation of cinnamic acid under AgSCF3/K2S2O8system

图式91 AgSCF3/K2S2O8体系下炔烃马氏、反马氏氢/三氟甲硫基化
Scheme 91 Anti-Markovnikov and Markovnikov-selective hydrotrifluoromethylthiolation of terminal alkyne under AgSCF3/K2S2O8system

图式92 AgSCF3/K2S2O8体系下亚甲基环丙烷底物的开环串联反应
Scheme 92 Trifluoromethylthiolation of methylenecyclopropane under AgSCF3/K2S2O8system

图式93 AgSCF3/K2S2O8体系下香豆素类羧酸脱羧三氟甲硫基化反应
Scheme 93 Decarboxylative trifluoromethylthiolation of coumarin-3-carboxylic acid under AgSCF3/K2S2O8system

图式94 AgSCF3/K2S2O8体系下半频哪醇重排三氟甲硫基化反应
Scheme 94 Trifluoromethylthiolation of diaryl allylic alcohols via radical neophyl rearrangement under AgSCF3/K2S2O8system

图式95 N-三氟甲硫基邻苯二甲酰亚胺在光催化条件下的自由基三氟甲硫基化反应
Scheme 95 Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) phthalimide as SCF3 source

图式96 N-三氟甲硫基丁二酰亚胺在光催化条件下的自由基三氟甲硫基化反应
Scheme 96 Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) succinimide as SCF3 source

图式97 Shen试剂在光催化条件下的自由基三氟甲硫基化反应
Scheme 97 Radical trifluoromethylthiolation under photocatalytic conditions using N-(trifluoromethylthio) saccharin as SCF3 source

图式99 光催化条件下的脱羧三氟甲硫基化反应
Scheme 99 Visible light-promoted decarboxylative trifluoromethylthiolation of alkyl carboxylic acids

图式100 氧化条件下的脱羧三氟甲硫基化反应
Scheme 100 Oxidative decarboxylative trifluoromethylthiolation of alkyl carboxylic acids

图式101 1非活化烷基C—H键自由基三氟甲硫基化反应
Scheme 101 Unactivated C—H bond trifluoromethylthiolation under radical conditions

图式102 光催化条件下非活化烷基C—H键三氟甲硫基化反应
Scheme 102 Radical trifluoromethylthiolation of unactivated C—H bond under photocatalytic conditions

图式103 金催化/光催化协同催化下烯烃的砜基三氟甲硫基化反应
Scheme 103 Dual gold and photoredox catalysis: visible light-mediated intermolecular atom transfer trifluoromethylthiosulfonylation of alkene
表 1 2016年化学与材料科学领域热点前沿
Table 1. Frontiers of chemistry and material science in 2016
排名 热点前沿 核心论文 被引频次 1 基于非富勒烯受体的有机太阳能电池 41 2249 2 三氟甲硫基化反应 47 3168 3 摩擦纳米发电机 43 2846 4 非贵金属电解水纳米催化剂 26 2427 5 金催化的有机合成 23 2062 6 高效钙钛矿型太阳能电池 30 1647 7 半导体/石墨烯纳米复合物光催化剂 21 3176 8 白光LED用荧光粉 44 4690 9 石墨烯过滤膜 22 3125 10 锂离子电池 4 1998 摘自《2016研究前沿》.  下载: 导出CSV
下载: 导出CSV
-









































































































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 166
- 文章访问数: 18814
- HTML全文浏览量: 2621
 下载:
下载:
