 图 1
木质素结构和木质素模型物
Figure 1.
Lignin structure and lignin model compound
图 1
木质素结构和木质素模型物
Figure 1.
Lignin structure and lignin model compound
引用本文:
刘新鑫, 严龙, 傅尧. 酸性体系V催化木质素β-O-4模型物C-C键高选择性切断[J]. 化学学报,
2017, 75(8): 788-793.
doi:
10.6023/A17050199

Citation: Liu Xinxin, Yan Long, Fu Yao. Lignin C-C Bond's Cleavage by Vanadium Catalyzed with High Selectivity in Acid Environment[J]. Acta Chimica Sinica, 2017, 75(8): 788-793. doi: 10.6023/A17050199
Citation: Liu Xinxin, Yan Long, Fu Yao. Lignin C-C Bond's Cleavage by Vanadium Catalyzed with High Selectivity in Acid Environment[J]. Acta Chimica Sinica, 2017, 75(8): 788-793. doi: 10.6023/A17050199
酸性体系V催化木质素β-O-4模型物C-C键高选择性切断
摘要:
研究了酸性体系下NH4VO3催化木质素模型物2-(苯氧基)-1-苯乙酮(1a)的C-C键氧化切断过程.通过优选反应溶剂,在温和条件下(100℃,101 kPa O2)于DMSO-HOAc(V:V=3:1)溶剂中高选择性地得到了苯甲酸和苯酚(产率分别为82.1%和88.1%),并通过对反应过程的监测和催化剂的研究提出了该反应可能的反应路径.反应过程存在两条可能的途径,一是1a先发生C-O键断裂生成苯酚和2-羟基苯乙酮,再催化2-羟基苯乙酮C-C键氧化断裂生成苯甲酸;二是1a直接发生C-C键氧化断裂生成苯甲酸和苯酚.同时,催化剂表征结果表明,+5价钒氧离子是催化活性物种.钒催化剂在反应过程中通过+4和+5价循环完成催化过程.
English
Lignin C-C Bond's Cleavage by Vanadium Catalyzed with High Selectivity in Acid Environment
Abstract:
Lignin is a potential resources of aromatic compound that can be obtained from renewable biomass. There are many ongoing research efforts to utilize lignin as a sustainable alternative to petroleum derived aromatic compounds. Because of the complex three-dimensional structure, the depolymerization of lignin into monomer molecule became a core challenge for the utilization of lignin. The β-O-4 structure is the most abundant linkage in lignin. Owing to its abundance, the β-O-4 structure has been representatively studied in many aspects of scientific research on lignin degradation. Among the different reported strategies for the cleavage of β-O-4 ether bonds, C-C bond cleavage is one of the most important approaches to depolymerizing lignin. In this study, we accomplished the oxidative C-C bond cleavage of the β-O-4 structure by the catalysis of NH4VO3 using the pre-oxidized 2-phenoxy-1-phenylethanone (1a) as a model compound of lignin. In the DMSO-HOAc solvent system, benzoic acid and phenol were produced in a moderate condition, the yeild of benzoic acid and phenol were 82.1% and 88.1%, respectively. The reaction process was investigated via 1H NMR and X-ray photoelectron spectra (XPS) characterizations and the possible reaction pathway was further proposed. As the results shown, two possible reaction routes existed in this catalytic system. Pathway one:the 2-hydroxyacetophenone and phenol formed after the C-O bond cleavage of 1a in the acidic system, then, the intermediate 2-hydroxyacetophenone was converted to benzoic via the cleavage of C-C bond. Pathway two:benzoic acid and phenol yielded by the C-C bond of 1a cleaved directly over the catalyst. In addition, the catalyst characterization results confirmed that the oxovanadium(V) directly catalyzed the depolymerization of the β-O-4 structure and generated oxovanadium(Ⅳ), then oxovanadium(Ⅳ) was oxidized by O2 and finish the catalytic cycle. All reactions were carried out by the following general procedure. This reaction was carried out in glass tube and heated by oil bath. 0.5 mmol of 1a was added into 2 mL of DMSO-HOAc (V:V=3:1) with 30 mol% NH4VO3 (17.5 mg) under an oxygen atmosphere (101 kPa, in balloon). The reactor was heated to 100℃ with a powerful stirring. After 8 h, the reaction was cooled to room temperature, then 5 mL of ethyl acetate was added into the mixtures. Ash black precipitate was removed by filtration and the liquid mixture was detected by GC.
-
Key words:
- lignin
- / depolymerization
- / C-C bond cleavage
- / V-catalyzed reaction
- / oxidation reaction
-
1 引言
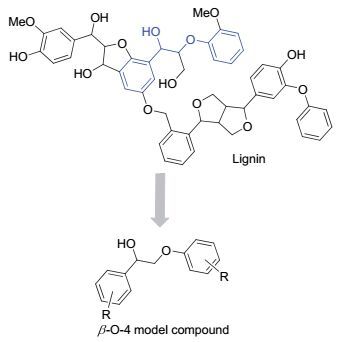
随着现代工业的不断发展, 支撑现代社会运行的不可再生化石燃料消耗持续快速增长, 由此造成的资源危机和环境问题对现代社会的影响日益严重[1~3].为解决这些问题, 开发新型可再生、环境友好的绿色资源成为当今学术界研究的重大课题之一.木质纤维素作为一种自然界广泛分布的可再生资源, 以其巨大的储量、快速再生性和非食用性的特点被研究者认为是具有巨大发展潜力的新型绿色资源并逐渐进入了人们的视野中[4~6].木质纤维素主要由纤维素、半纤维素和木质素三种聚合物组成, 具有分子聚合度较高和含氧官能团丰富的结构特点[7].因此, 与石油精炼过程不同, 高效解聚和去官能化处理是工业应用木质纤维素生产化学品的核心问题[8].如今, 关于纤维素与半纤维素的工业应用较为广泛, 而木质素的利用仍相对较少, 大量的木质素在造纸工业中作为废渣被丢弃, 造成了严重的环境污染和极大的资源浪费[9].因含有丰富的苯环结构, 木质素被认为是一种获得天然芳香类化合物的潜在原料[10].研究发现, 木质素是主要由酚类及其衍生物通过C—C, C—O键连接而形成的无定型聚合物, 其中β-O-4型连接结构占比超过40%[11].因此, 研究者们合成了β-O-4型连接结构的木质素模型物用于木质素的解聚体系的开发和反应过程的研究[10](图 1).
图 1
木质素结构和木质素模型物
Figure 1.
Lignin structure and lignin model compound
由于木质素具有通过C—C/C—O键连接的结构特点, 催化C—O/C—C键的选择性切断因而成为木质素解聚研究的核心科学问题.目前, 基于过渡金属催化剂(如Ni, Fe, Re, Ru, Pd等)催化C—O键切断解聚木质素的研究已取得了一定的进展[6, 12~17].而有关通过切断C—C键降解木质素的催化体系的报道仍相对较少.近来, 基于V, Cu等催化剂催化木质素C—C键断裂的解聚体系陆续被报道[18~21]. Hanson课题组[18]报道了一系列钒配合物均相催化剂的木质素模型物解聚工作, 并对不同配体结构的钒均相催化剂催化木质素C—C键切断过程进行了细致研究.他们发现通过改变钒催化剂配体可以实现对反应产物的选择性进行调控.随后, 徐杰课题组[19]发现, 在钒催化剂和特定的溶剂体系共同作用下可以完成木质素模型物的C—C键断裂解聚, 并对可能的反应路径进行了研究.总的来说, 虽然木质素β-O-4模型物的解聚研究取得了一定的成果, 但仍存在反应体系复杂、配体试剂昂贵、氧化体系产物选择性不高等问题.因此, 温和条件下高效高选择性的木质素催化降解体系仍需进一步开发研究.
2013年, Stahl课题组[22]提出氧化-还原两步法降解木质素新策略.研究发现, 通过模型物α-位醇羟基的选择性氧化有效地活化了β-O-4结构, 使反应在更温和的条件下取得了更高的产物选择性.但反应体系中需要添加三倍量的甲酸钠, 降低了反应的原子经济性.为解决以上问题, 他们提出了氧化-氧化体系, 利用双氧水作为氧化剂完成β-O-4模型物C—C键的切断过程.但由于在该体系中酚类产物不稳定, 选择性有待提高.在此之后, 预氧化的处理过程也被越来越多的科研工作者研究[23], 预氧化处理的木质素模型物降解研究也有诸多报道[24~26].王峰课题组[24a, 24b]发展了一系列基于Cu催化剂的催化体系, 完成了预氧化模型物C—C键的氧化切断降解.但该反应体系需要的配体和试剂存在成本较高的问题, 不利于大规模生产应用.随后, 他们[24c]报道了一种Cu (NO3)2催化氧化木质素的简单高效体系, 但酚类产物的选择性还有待提高.本文中, 我们报道了一种基于钒化合物高效催化氧化降解木质素β-O-4模型物的反应体系(图式1).该体系中通过酸性混合溶剂的促进作用, 通过NH4VO3催化完成了模型物1a的高效C—C键切断过程, 并高选择性地得到苯甲酸与苯酚两种基础化学品.通过机理研究, 本文提出了不同于前人的反应机制.
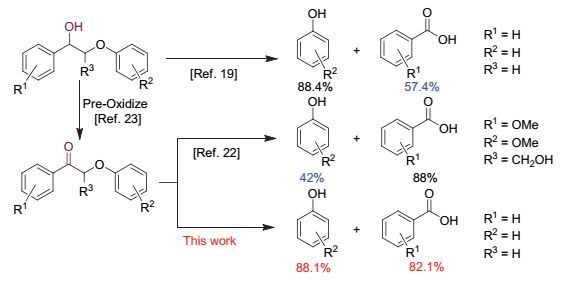 图式1
本文主要研究工作
图式1.
The main reaction in this research
图式1
本文主要研究工作
图式1.
The main reaction in this research
2 结果与讨论
2.1 催化剂的筛选
Stahl课题组[21]提出了一种预氧化处理促进木质素降解策略后, 预氧化处理模型物1a被广泛应用于木质素降解的研究[21, 23, 24].因此, 化合物1a被选为木质素β-O-4模型物用于本研究工作.与此同时, 钒金属化合物在前人研究中显示出良好的C—C键断键活性[18~20, 27], 是一类高效的氧化催化剂.本工作选取了几类常见的钒化合物作为氧化催化剂进行催化氧化研究, 不同钒催化剂催化木质素模型物1a氧化反应的结果列于表 1.从结果可以看出, 在DMSO溶剂中、氧气氛围下, NH4VO3展现出了较强的C—C键切断能力. 100 ℃条件下反应8 h, 1a的转化率达到了35.7%, 产物苯甲酸和苯酚的产率分别为17.2%和24.0%(表 1, Entry 1).当加入的催化剂为VOSO4或VOPO4时, 木质素模型物的转化率分别为21.3%和10.0%(表 1, Entries 2, 3), 低于NH4VO3催化效果.当以不同价态钒氧化物作为催化剂时, VO2只得到只有4.0%的1a转化率, 产物苯甲酸和苯酚产率分别为3.1%和2.7%(表 1, Entry 4), 而在V2O5催化下, 未检测到目标产物苯甲酸和苯酚(表 1, Entry 5).根据Marsh课题组[28]的研究报道, VO2+阳离子可能是酸性C—C键氧化体系(NaVO3-H2SO4)的催化核心.因此, 提高反应体系中VO2+含量可能是提高反应效率的关键.为提高溶液中钒氧离子浓度, 我们考察了不同反应溶剂对反应效果的影响.
 表 1
反应催化剂的筛选a
Table 1.
Catalysts screening of the model reaction
表 1
反应催化剂的筛选a
Table 1.
Catalysts screening of the model reaction

Entry Catalyst Conversionb/% Yieldb/% 1b 1c 1 NH4VO3 35.7 17.2 24.0 2 VOSO4 21.3 13.1 10.4 3 VOPO4 10.0 8.0 8.2 4 VO2 4.0 3.1 2.7 5 V2O5 <5 — — aReaction condition: 1a (0.5 mmol), 30 mol% catalysts, added into DMSO (2 mL), keep in 100 ℃ for 8 h at 101 kPa O2 atmosphere; bConversions and yields were confirmed by GC. 2.2 溶剂对反应结果的影响
根据不同钒催化剂测试结果, NH4VO3被选为模型反应催化剂, 为进一步提高催化效率, 本工作对不同的反应溶剂进行了筛选, 结果列于表 2中.值得注意的是, 使用乙酸作为反应溶剂反应8 h后催化效率明显提高, 1a的转化率达到了93%, 并得到了62.6%的苯甲酸和55.3%的苯酚(表 2, Entry 2).然而, 在纯水和乙腈溶剂的反应当中, 模型物1a没有转化.在此基础上.不同的乙酸混合溶剂被用于催化反应的测试.结果显示, 二甲基亚砜稀释乙酸形成的混合溶剂能够有效地提高反应产物苯酚和苯甲酸的产率和选择性.在DMSO-HOAc (V:V=1:1) 溶剂中, 底物1a完全转化, 苯甲酸和苯酚的产率分别为80.5%和68.5%(表 2, Entry 5), 产物收率有明显提高. H2O-HOAc与CH3CN-HOAc混合溶剂中反应的底物转化率均低于DMSO-HOAc溶剂中的反应, 分别为81.4%和67.6%(表 2, Entries 6, 7).
表 2
溶剂对反应结果的影响a
Table 2.
Solvent influence for the reaction
Entry Solventb (V:V) Conversionc/% Yieldc/% 1b 1c 1 DMSO 35.7 17.2 24.0 2 HOAc 93.0 62.6 55.3 3 CH3CN — — — 4 H2O — — — 5 DMSO/HOAc (1:1) 100 80.5 68.5 6 H2O/HOAc (1:1) 81.4 57.9 63.2 7 CH3CN/HOAc (1:1) 67.6 36.2 36.2 8 DMSO/HOAc (1:3) 100 73.8 67.2 9d DMSO/HOAc (3:1) 96.4 82.1 88.1 10 DMSO/H3PO4 (1:1) 100 76.1 55.0 11e DMSO/HOAc (3:1) 61.2 34.1 33.5 12f DMSO/HOAc (3:1) 100 — 48.1 13g DMSO/HOAc (3:1) 100 — 41.4 a0.5 mmol of 1a, 30 mol% NH4VO3 were added into the reaction under 101 kPa O2 atmosphere and kept 100 ℃ for 8 h; bThe ratio of acids and organic solvents is based on volume; cConversions and yields were confirmed by GC; d The isolated yeilds of 1b and 1c are 75.2% and 71.4%, respectively; e0.5 mmol 2-phenoxy-1-phenylethanol was used as substrate; f The initial O2 pressure is 0.5 MPa; g The initial O2 pressure is 1 MPa. 随后, 我们进一步测试不同浓度的乙酸对反应体系催化效果影响.结果显示, 当使用DMSO-HOAc (V: V=1:3) 溶剂进行反应时, 反应转化率达到100%, 苯甲酸和苯酚产率分别为73.8%和67.2%(表 2, Entry 8).降低乙酸浓度后, DMSO-HOAc (V:V=3:1) 溶剂中反应的转化率降低为96.4%.然而, 该溶剂条件下产物产率达到最高, 苯甲酸、苯酚的气相产率分别达到82.1%和88.1%, 分离收率也分别达到了75.2%和71.4%(表 2, Entry 9).而在DMSO-H3PO4混合溶剂中, 底物的转化率虽然达到了100%, 但苯甲酸和苯酚的产率分别为76.1%和55.0%(表 2, Entry 10), 低于DMSO-HOAc混合溶剂中的产率.前人研究显示, 过高的酸性可能导致木质素结焦, 从而降低产物产率[29].事实上, 在最优反应条件下, 酸性体系对VOSO4与VOPO4的催化活性也表现出一定的提升作用, 但仍低于相同条件下NH4VO3的催化活性.测试结果显示, VOSO4催化下苯甲酸和苯酚的产率分别为46.7%和68.2%, VOPO4催化下苯甲酸和苯酚的产率分别为53.1%和64.2%(表S1).除此之外, 在最优反应条件下, 当以未进行预氧化处理的模型物2-(苯氧基)-1-苯乙醇作为反应底物时, 反应转化率和产物选择性显著降低, 底物转化率为62.1%, 苯甲酸、苯酚产率分别为34.1%和33.5%(表 2, Entry 11).以上结果证实, 经预氧化处理后, 木质素模型物的反应活性明显提高.反应氧气压力影响的测试结果显示, 较高氧气压力降低了产物的收率.当反应氧气压力分别提高至0.5与1 MPa时, 测得反应底物1a转化率均为100%, 然而, 反应体系中未检测到苯甲酸产物.同时, 产物苯酚的产率明显低于常压反应结果, 分别为48.1%和41.4%(表 2, Entries 12, 13).
2.3 反应过程的研究
在前人报道的文献基础上[19, 24], 我们提出了三条可能的反应路径(图式2).路径一: C—O键先通过酸解过程断裂生成苯酚和2-羟基苯乙酮, 2-羟基苯乙酮随后在钒氧离子催化下C—C键氧化断裂产生苯甲酸; 路径二: C—C键被钒氧离子氧化切断生成苯酚, 产生中间体苯甲醛并随后氧化生成苯甲酸; 路径三: C—C键在钒氧离子催化下直接断裂生成苯甲酸与苯酚.对反应体系1H NMR监测分析显示(图 2), 反应2 h, 底物1a、产物苯酚和苯甲酸的核磁特征氢化学位移均可被检测到.然而, 可能的中间体苯甲醛(δ 10.0) 和2-羟基苯乙酮(δ 4.85) 的特征峰在混合体系的核磁检测中并未被发现.随着反应时间延长, 1a的量逐渐减少, 产物酚和苯甲酸的量逐渐增加, 但体系中始终未检测到苯甲醛与2-羟基苯乙酮特征化学位移(图S4).
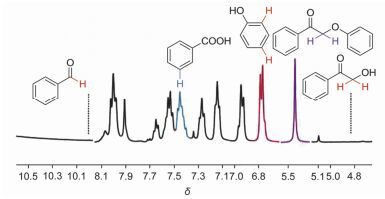 图 2
反应2 h后的反应混合物1H NMR谱图
Figure 2.
1H NMR spectrum of the mixtures after reaction for 2 h
图 2
反应2 h后的反应混合物1H NMR谱图
Figure 2.
1H NMR spectrum of the mixtures after reaction for 2 h
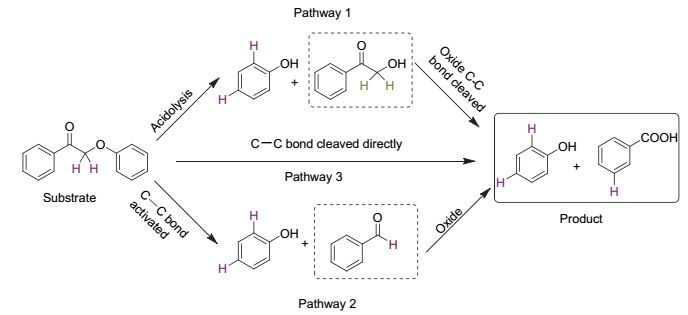 图式2
三条可能的C—C键切断路径
图式2.
Three possible pathways of C—C cleavage
图式2
三条可能的C—C键切断路径
图式2.
Three possible pathways of C—C cleavage
随后, 苯甲醛和2-羟基苯乙酮在模型体系中的反应性被分别进行了测试(图式3).苯甲醛在该体系中稳定, 经过8 h反应后转化率低于5%, 证实苯甲醛在该体系中难以进一步氧化.然而, 2-羟基苯乙酮则显示较高的反应性, 经过8 h反应后完全转化, 苯甲酸产率达到91.4%.由此推测, 模型反应中可能的反应历程为: (1) 酸性条件下C—O键切断后生成苯酚和2-羟基苯乙酮, 2-羟基苯乙酮再进一步氧化C—C键, 生成苯甲酸; (2) 模型物1a在NH4VO3催化作用下直接发生C—C键氧化断裂, 生成苯甲酸和苯酚.根据前人研究发现, 醛的产生可能会导致聚合副反应的发生[30].本文催化反应途径中不经过苯甲醛中间体的过程, 与徐杰课题组[19]报道的反应路径有所不同.由于避免了醛类中间体的生成, 体系中苯甲酸产物的产率因而得到了有效提高.
 图式3
可能中间体在模型反应条件下的结果
图式3.
Reactions of possible intermediates as substrate
图式3
可能中间体在模型反应条件下的结果
图式3.
Reactions of possible intermediates as substrate
为进一步研究催化剂的催化过程, 我们利用X射线光电子能谱技术(XPS)表征对催化剂中钒元素的化学价态进行了分析.首先, 对V2O5和VO2标准品的表征结果显示, 位于516.3, 517.6 eV位置峰分别属于+4和+5价V物种特征峰.随后, 对反应后的催化剂的XPS分析显示, 催化剂中的钒元素以+4和+5价两种价态形式存在(图S3).当VO2作为催化剂在模型反应体系中反应后, 检测到存在的钒元素价态与NH4VO3反应后结果类似.此外, 在Ar气氛下, 当4.0 equiv.(相对于1a) VO2作为氧化剂投入反应体系中, 反应不能进行.但在该条件下用V2O5替换VO2作为氧化剂后, 反应物1a完全转化并得到苯酚和苯甲酸产物, 产率分别为68.9%和69.4%.上述结果证明了+5价钒物种是氧化反应进行的关键, 直接参与催化氧化的过程, 而+4价钒不能直接进行催化氧化过程, 区别于报道的自由基氧化切断C—C键反应过程[25a, 31].
在参考前人报道的文献基础上[18, 19, 24, 25, 31, 32], 我们提出可能的反应机理应为(图式4) :在酸性混合溶剂中, 催化剂首先溶解并产生+5价钒氧离子, 随后+5价钒氧离子对底物1a进行氧化, 切断C—C键生成苯酚和苯甲酸, 同时, 钒氧离子被还原至+4价. +4价的钒氧离子在氧气氛围下被重新氧化至+5价, 并进入下一个催化循环过程.反应决速步方面, 反应过程核磁监测结果中未发现2-羟基苯乙酮的生成(图S4), 但对2-羟基苯乙酮的氧化测试显示, 该催化体系能够将2-羟基苯乙酮高效氧化为苯甲酸(图式3).由此推测, 反应路径一(图式2, Pathway 1) 中模型物1a至中间体2-羟基苯乙酮的过程可能是反应的决速步.另一方面, 针对反应过程氧插入机理的探讨, 本研究中发现, V2O5可作为氧化剂直接完成木质素模型物的C—C键切断(表 3, Entry 3), 区别于韩布兴等[25]提出的[BnMIm][NTf2]离子液氧化体系单电子自由基氧化过程.对于催化过程氧插入切断C—C键的具体机理我们将通过理论计算和实验验证结合的方法进行进一步的研究.
表 3
不同氧化剂对反应的影响a
Table 3.
The study of different oxidants
Entry Oxidant Atmosphere Conversion/% Yieldb/% 1b 1c 1 VO2 (0.3 equiv.) O2 71.4 55.1 53.8 2 VO2 (4.0 equiv.) Ar — — — 3 V2O5 (4.0 equiv.) Ar 100 69.4 68.9 aReaction condition: 1a 0.5 mmol, 30 mol% catalysts, added into 2 mL of DMSO/HOAc (V:V=3:1), keep in 100 ℃ for 8 h at 101 kPa O2 atmosphere; bConversions and yields were confirmed by GC. 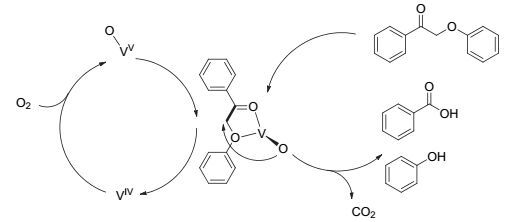 图式4
可能的钒催化氧化解聚过程
图式4.
The possible process of V catalyzed oxidation
图式4
可能的钒催化氧化解聚过程
图式4.
The possible process of V catalyzed oxidation
3 结论
综上所述, 在DMSO/HOAc混合溶剂中, NH4VO3呈现出对预氧化木质素β-O-4模型物优异的催化解聚活性, 能够在101 kPa氧气、100 ℃条件下高效高选择性地完成模型物1a的C—C键氧化切断, 获得了88.1%的苯酚和82.1%的苯甲酸.对反应过程的监测和对反应催化剂的表征分析显示, 反应路径可能为: (1) C—O键断裂生成苯酚和2-羟基苯乙酮, 2-羟基苯乙酮再经过C—C键催化氧化断裂生成苯甲酸; (2) 1a模型物直接发生C—C键氧化断裂生成苯甲酸和苯酚.反应过程中钒催化剂存在+4价和+5价循环过程, 催化剂中+5价钒氧离子是催化的活性中心.相对于已报道的体系而言, 该体系具有廉价、高效、高选择性等优点, 具有较大的工业应用潜力.
-
-
[1]
Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411. doi: 10.1021/cr050989d
-
[2]
(a) Delidovich, I. ; Hausoul, P. J. C. ; Deng, L. ; Pfuzenreuter, R. ; Rose, M. ; Palkovits, R. Chem. Rev. 2016, 116, 1540. (b) Yang, Z. ; Fu, Y. ; Guo, Q. X. Chin. J. Org. Chem. 2015, 35, 273 (in Chinese). (杨珍, 傅尧, 郭庆祥, 有机化学, 2016, 35, 273. )
-
[3]
(a) Ragauskas, A. J. ; Williams, C. K. ; Davison, B. H. ; Britovsek, G. ; Cairney, J. ; Eckert, C. A. ; Hallett, J. P. ; Leak, D. J. ; Liotta, C. L. ; Mielenz, J. R. ; Murphy, R. ; Templer, R. ; Tschaplinski. T. Science 2006, 311, 484. (b) Regalbuto, J. R. Science 2009, 325, 822. (c) Holm, M. S. ; Saravanamurugan, S. ; Taarning, E. Science 2010, 328, 602. (d) Luo, C. ; Wang, S. ; Liu, H. Angew. Chem. , Int. Ed. 2007, 46, 7636. (e) Wang, H. J. ; Zhao, Y. ; Wang, C. ; Fu, Y. ; Guo, Q. X. Acta Chim. Sinica 2009, 67, 893 (in Chinese). (王华静, 赵岩, 王晨, 傅尧, 郭庆祥, 化学学报, 2009, 67, 893. )
-
[4]
(a) Xu, C. P.; Arancon, R. A. D.; Labidi, J.; Luque, R. Chem.Soc. Rev. 2014, 43, 7485. (b) Upton, B. M.; Kasko, A. M. Chem. Rev. 2016, 116, 2275. (c) Li, C. Z.; Zhao, X. C.; Wang, A. Q.; Huber, G. W.; Zhang, T. Chem. Rev. 2015, 115, 11559. (d) Deng, W. P.; Zhang, H. X.; Xue, L. Q.; Zhang, Q. H.; Wang, Y. Chin. J. Catal. 2015, 36, 1440.
-
[5]
(a) Zaheer, M. ; Kempe, R. ACS Catal. 2015, 5, 1675. (b) Zeng, W. P. ; Li, X. H. ; Du, J. ; Li, J. M. ; Zhang, P. ; Hu, C. W. ; Meng, X. G. Acta Chim. Sinica 2010, 68, 27 (in Chinese). (曾伟鹏, 李小红, 杜娟, 李建梅, 张平, 胡常伟, 孟祥光, 化学学报, 2010, 68, 27. )
-
[6]
(a) Sergeev, A. G.; Hartwig, J. F. Science 2011, 332, 439. (b) Sergeev, A. G.; Webb, J. D.; Hartwig, J. F. J.Am. Chem. Soc. 2012, 134, 20226.
-
[7]
(a) Zhou, C. H. ; Xia, X. ; Lin, C. X. ; Tong, D. S. ; Berltramini, J. Chem. Soc. Rev. 2011, 40, 5588. (b) Wang, W. L. ; Wang, J. Y. ; Dong, X. Z. ; Chen, P. Chem. Bull. 2016, 79, 731 (in Chinese). (王唯黎, 王景芸, 董晓哲, 陈平, 化学通报, 2016, 79, 731. )
-
[8]
Shiramizu, M.; Toste, F. D. Angew. Chem., Int. Ed. 2012, 51, 8082. doi: 10.1002/anie.v51.32
-
[9]
Zakzeski, J.; Bruijnincx, P. C. A.; Jongerius, A. L.; Weckhuysen, B. M. Chem. Rev. 2010, 110, 3552. doi: 10.1021/cr900354u
-
[10]
Alonso, D. M.; Wettstein, S.; G. Dumesic, J. A. Chem. Soc. Rev. 2012, 41, 8075. doi: 10.1039/c2cs35188a
-
[11]
(a) Zhang, H. F. ; Yang, J. Y. ; Wu, J. X. ; Mao, H. F. ; Sun, X. L. Chin. J. Org. Chem. 2016, 36, 1266 (in Chinese). (张海峰, 杨军艳, 吴建新, 毛海舫, 孙小玲, 有机化学, 2016, 36, 1266. ) (b) Ouyang, X. P. ; Yang, Y. ; Zhu, G. D. ; Qiu, X. Q. Chin. Chem. Lett. 2015, 26, 980.
-
[12]
(a) He, J.; Zhao, C.; Lercher, J. A. J. Am. Chem.Soc. 2012, 134, 20768. (b) Song, Q.; Wang, F.; Cai, J. Y.; Wang, Y. H.; Zhang, J. J.; Yu, W. Q.; Xu, J. Energy Environ. Sci.2013, 6, 994; (c) Wang, X.; Rinaldi, R. ChemSusChem 2012, 5, 1455. (d) Sturgeon, M. R.; O'Brien, M. R.; Ciesielski, P. N.; Katahira, R.; Kruger, J. S.; Chmely, S. C.; Hamlin, J.; Lawrence, K.; Hunsinger, G. B.; Foust, T. D.; Baldwin, R. M.; Biddy, M. J.; Beckham, G. T. Green Chem. 2014, 16, 824. (e) Song, Q.; Cai, J. Y.; Zhang, J. J.; Yu, W. Q.; Wang, F.; Xu, J. Chin. J. Catal. 2013, 34, 651.
-
[13]
Ren, Y. L.; Yan, M. J.; Wang, J. J.; Zhang, Z. C.; Yao, K. S. Angew. Chem., Int. Ed. 2013, 52, 12674. doi: 10.1002/anie.201305342
-
[14]
Harm, R. G.; Markovits, I.-I. E.; Drees, M.; Mult, H. C.; Herrmann, W. A.; Cokoja, M.; Kuhn, F.-E. ChemSusChem 2014, 7, 429. doi: 10.1002/cssc.201300918
-
[15]
Nichols, J. M.; Bishop, L. M.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2010, 132, 12554. doi: 10.1021/ja106101f
-
[16]
(a) Zhou, X. Y. ; Mitra, J. ; Rauchfuss, T. B. ChemSusChem 2014, 7, 1623. (b) Yan, L. ; Pang, H. ; Huang, Y. B. ; Fu, Y. Acta Chim. Sinica 2014, 72, 1005 (in Chinese). (严龙, 庞欢, 黄耀兵, 傅尧, 化学学报, 2014, 72, 1005. )
-
[17]
(a) Guo, H. W.; Zhang, B.; Li, C. Z.; Peng, C.; Dai, T.; Xie, H. B.; Wang, A. Q.; Zhang, T. ChemSusChem 2016, 9, 3220. (b) Heather, J.; Parker, H. J.; Chuck, C. J.; Woodman, T.; Jones, M. D. Catal. Today 2016, 269, 40. (c) Xiao, Y. W.; Xiu, Y. L. Chin. J. Chem. 2011, 22, 733.
-
[18]
(a) Hanson, S. K.; Baker, R. T.; Gordon, J. C.; Scott, B. L.; Thorn, D. L. Inorg. Chem. 2010, 49, 5611. (b) Hanson, S. K.; Wu, R.; Silks, L. A. P. Angew. Chem., Int. Ed. 2012, 51, 3410. (c) Zhang, G.; Scott, B. L.; Wu, R. L.; Silks, L. A. P.; Hanson, S. K. Inorg. Chem. 2012, 51, 7354. (d) Sedai, B.; Urrutia, C. D.; Baker, R. T.; Wu, R. L.; Silks, L. A. P.; Hanson, S. K. ACS. Catal.2013, 3, 3111. (e) Diaz-Urrutia, C.; Sedai, B.; Leckett, K. C.; Baker, R. T.; Hanson, S. ACS Sustanable Chem. Eng. 2016, 4, 6244.
-
[19]
(a) Ma, Y. Y.; Du, Z. T.; Liu, J. X.; Xia, F.; Xu, J. Green Chem.2015, 17, 4968. (b) Ma, Y. Y.; Du, Z. T.; Xia, F.; Ma, J. P.; Gao, J.; Xu, J. RSC Adv. 2016, 6, 110229.
-
[20]
(a) Gazi, S.; Ng, W. K. H.; Ganguly, R.; Moeljadi, A. M. P.; Soo, H. S. Chem. Sci. 2015, 6, 7130. (b) Mottweiler, J.; Puche, M.; Rauber, C.; Schmidt, T.; Concepcion, P.; Corma, A.; Bolm, C. ChemSusChem 2015, 8, 1206.
-
[21]
(a) Mottweiler, J.; Rinesch, T.; Besson, C.; Buendia, J.; Bolm, C. Green Chem. 2015, 17, 5001. (b) Stein, T V.; Hartog, T. D.; Buendia, J.; Stoychev, S.; Mottweiler, J.; Bolm, C.; Klankermayer, J.; Leitner, W. Angew. Chem., Int.Ed. 2015, 54, 5859. (c) Deng, W. P.; Zhang, H. X.; Wu, X. J.; Li, R. S.; Zhang, Q. H.; Wang, Y. Green Chem. 2015, 17, 5009. (d) Mitchell, L. J.; Moody, C. J. J. Org. Chem. 2014, 79, 11091.
-
[22]
(a) Rahimi, A.; Ulbrich, A.; Coon, J. J.; Stahl, S. S. Nature 2015, 515, 249. (b) Rahimi, A.; Azarpira, A.; Kim, H.; Ralph, J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 6415.
-
[23]
(a) Patil, N. D.; Yan, N. Catal. Commun. 2016, 84, 155. (b) Yao, S. G.; Meier, M. S.; Pace Ⅲ, R. B.; Crocker, M. RSC Adv.2016, 6, 104742. (c) Mobley, J. K.; Yao, S. G.; Crocker, M.; Meier, M. RSC Adv. 2015, 5, 105136. (d) Nguyen, J. D.; Matsuura, B. S.; Stepemson, C. R. J. J. Am. Chem. Soc. 2014, 136, 1218. (e) Luo, J.; Zhang, J. J. Org. Chem. 2016, 81, 9131. (f) Karakas, D. M.; Bosque, I.; Stephenson, C. R. J. Org.Lett. 2016, 18, 5166.
-
[24]
(a) Wang, M.; Lu, J. M.; Zhang, X. C.; Li, L. H.; Li, Hong. J.; Luo, N. C.; Wang, F. ACS Catal. 2016, 6, 6086. (b) Wang, M.; Li, L. H.; Lu, J. M.; Li, H. J.; Zhang, X. C.; Liu, H. F.; Luo, N. C.; Wang, F. Green Chem. 2017, 19, 702. (c) Liu, H. F.; Wang, M.; Li, H. J.; Luo, N. C.; Xu, S. T.; Wang, F. J. Catal. 2017, 346, 170.
-
[25]
(a) Yang, Y. Y.; Fan, H. L.; Song, J. L.; Meng, Q. L.; Zhou, H. C.; Wu, L. Q.; Yang, G. Y.; Han, B. X. Chem. Coummn. 2015, 51, 4028. (b) Paitil, N. D.; Yan, N. Tetrahedron Lett. 2016, 57, 3024.
-
[26]
(a) Luo, N. C.; Wang, M.; Li, H.; Zhang, J.; Liu, H.; Wang, F. ACS Catal. 2016, 6, 7716. (b) Zhang, J.; Liu, Y.; Chiba, S.; Loh, T.-P. Chem. Commun. 2013, 49, 11439.
-
[27]
Dakkach, M.; Atlamsani, A.; Sebti, S. C. R. Chim. 2012, 15, 482. doi: 10.1016/j.crci.2012.03.003
-
[28]
(a) Wang, W. H.; Niu, M.; Hou, Y. C.; Wu, W. Z.; Liu, Z. Y.; Liu, Q. Y.; Ren, S. H.; Marsh, K. N. Green Chem. 2014, 16, 2614. (b) Niu, M.; Hou, Y.-C.; Ren, S. H.; Wang, W. H.; Zheng, Q. T.; Wu, W. Z. Green Chem. 2015, 17, 335. (c) Niu, M.; Hou, Y. C.; Ren, S. H.; Wu, W. Z.; Marsh, K. N. Green Chem. 2015, 17, 453.
-
[29]
(a) Custodis, V. B. F. ; Karakoulia, S. A. ; Triantafyllidis, K. S. ; van Bokhoven, J. A. ChemSusChem 2016, 9, 1134. (b) Shu, R. Y. ; Xu, Y. ; Zhang, Q. ; Ma, L. L. ; Wang, T. J. CIESC J. 2016, 67, 4523 (in Chinese). (舒日洋, 徐莹, 张琦, 马隆龙, 王铁军, 化工学报, 2016, 67, 4523. )
-
[30]
(a) Shuai, L.; Amiri, M. T.; Questell-Santiago, Y. M.; Heroguel, F.; Li, Y. D.; Kim, H.; Meilan, R.; Chapple, C.; Ralph, J.; Luterbacher, J. Science 2016, 354, 329. (b) Wang, L.; Meng, Y. Z.; Wang, S. J.; Shang, X. Y.; Li, L.; Hay, A. S. Macromolecules 2004, 37, 3151.
-
[31]
Huang, X. Q.; Li, X. Y.; Zou, M. C.; Song, S.; Tang, C. H.; Yuan, Y. Z.; Jiao, N. J. Am. Chem. Soc. 2014, 136, 14858. doi: 10.1021/ja5073004
-
[32]
Jiang, Y. Y.; Yan, L.; Zhang, Q.; Fu, Y. ACS Catal. 2016, 6, 4399. doi: 10.1021/acscatal.6b00239
-
[1]
-

图 2 反应2 h后的反应混合物1H NMR谱图
Figure 2 1H NMR spectrum of the mixtures after reaction for 2 h
表 1 反应催化剂的筛选a
Table 1. Catalysts screening of the model reaction
Entry Catalyst Conversionb/% Yieldb/% 1b 1c 1 NH4VO3 35.7 17.2 24.0 2 VOSO4 21.3 13.1 10.4 3 VOPO4 10.0 8.0 8.2 4 VO2 4.0 3.1 2.7 5 V2O5 <5 — — aReaction condition: 1a (0.5 mmol), 30 mol% catalysts, added into DMSO (2 mL), keep in 100 ℃ for 8 h at 101 kPa O2 atmosphere; bConversions and yields were confirmed by GC.  下载: 导出CSV
下载: 导出CSV
表 2 溶剂对反应结果的影响a
Table 2. Solvent influence for the reaction
Entry Solventb (V:V) Conversionc/% Yieldc/% 1b 1c 1 DMSO 35.7 17.2 24.0 2 HOAc 93.0 62.6 55.3 3 CH3CN — — — 4 H2O — — — 5 DMSO/HOAc (1:1) 100 80.5 68.5 6 H2O/HOAc (1:1) 81.4 57.9 63.2 7 CH3CN/HOAc (1:1) 67.6 36.2 36.2 8 DMSO/HOAc (1:3) 100 73.8 67.2 9d DMSO/HOAc (3:1) 96.4 82.1 88.1 10 DMSO/H3PO4 (1:1) 100 76.1 55.0 11e DMSO/HOAc (3:1) 61.2 34.1 33.5 12f DMSO/HOAc (3:1) 100 — 48.1 13g DMSO/HOAc (3:1) 100 — 41.4 a0.5 mmol of 1a, 30 mol% NH4VO3 were added into the reaction under 101 kPa O2 atmosphere and kept 100 ℃ for 8 h; bThe ratio of acids and organic solvents is based on volume; cConversions and yields were confirmed by GC; d The isolated yeilds of 1b and 1c are 75.2% and 71.4%, respectively; e0.5 mmol 2-phenoxy-1-phenylethanol was used as substrate; f The initial O2 pressure is 0.5 MPa; g The initial O2 pressure is 1 MPa.
下载: 导出CSV
表 3 不同氧化剂对反应的影响a
Table 3. The study of different oxidants
Entry Oxidant Atmosphere Conversion/% Yieldb/% 1b 1c 1 VO2 (0.3 equiv.) O2 71.4 55.1 53.8 2 VO2 (4.0 equiv.) Ar — — — 3 V2O5 (4.0 equiv.) Ar 100 69.4 68.9 aReaction condition: 1a 0.5 mmol, 30 mol% catalysts, added into 2 mL of DMSO/HOAc (V:V=3:1), keep in 100 ℃ for 8 h at 101 kPa O2 atmosphere; bConversions and yields were confirmed by GC.
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 3205
- HTML全文浏览量: 190
 下载:
下载:
