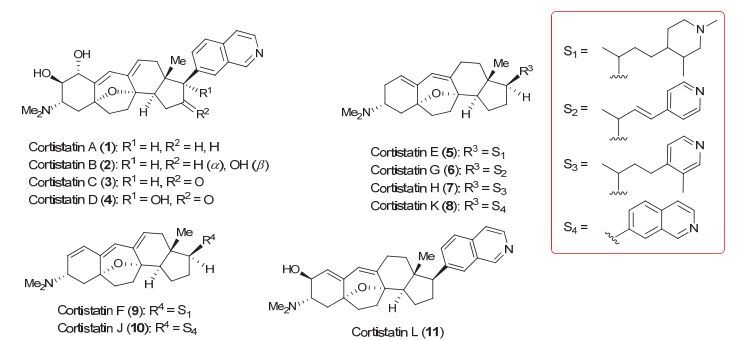
 图 1
Cortistatin类型天然产物
Figure 1.
Cortistatin family of natural products
图 1
Cortistatin类型天然产物
Figure 1.
Cortistatin family of natural products
引用本文:
顾月青, 袁浩, 傅俊凯, 龚建贤, 杨震. Cortistatin类型天然产物的不对称形式全合成:金催化串联Semi-Pinacol重排反应策略[J]. 化学学报,
2017, 75(8): 798-807.
doi:
10.6023/A17040190

Citation: Gu Yueqing, Yuan Hao, Fu Junkai, Gong Jianxian, Yang Zhen. Asymmetric Formal Synthesis of Cortistatins via a Gold-Catalyzed Semi-Pinacol Rearrangement Strategy[J]. Acta Chimica Sinica, 2017, 75(8): 798-807. doi: 10.6023/A17040190
Citation: Gu Yueqing, Yuan Hao, Fu Junkai, Gong Jianxian, Yang Zhen. Asymmetric Formal Synthesis of Cortistatins via a Gold-Catalyzed Semi-Pinacol Rearrangement Strategy[J]. Acta Chimica Sinica, 2017, 75(8): 798-807. doi: 10.6023/A17040190
Cortistatin类型天然产物的不对称形式全合成:金催化串联Semi-Pinacol重排反应策略
摘要:
本工作详细报道了Cortistatin类型天然产物不对称形式全合成的研究路线.以近期作者发展的金催化串联semi-pinacol重排反应构建[3,2,1]七元氧桥环结构的方法学为基础,进一步将其应用于复杂体系,高效构建了Cortistatin类型天然产物独特的七元氧桥环核心骨架,从而完成了该类型天然产物的不对称形式全合成.
-
关键词:
- 金催化
- / 氧桥环
- / 形式全合成
- / semi-pinacol重排
- / Cortistatins
English
Asymmetric Formal Synthesis of Cortistatins via a Gold-Catalyzed Semi-Pinacol Rearrangement Strategy
Abstract:
Over the past decade, Gold complexes have emerged as efficient and mild catalysts for the transformation of substrates possessing alkyne functionality into a range of useful scaffolds. These powerful methods have enabled the development of novel approaches for the total synthesis of biologically active natural products by gold catalysis. In this case, we found that the intramolecular nucleophilic addition of a hydroxyl group to a carbon-carbon triple bond, which activated by a gold catalyst, followed by further useful transformation has proven to be an excellent method for rapid construction of structural diversity of molecular scaffolds. The cortistatins are a family of 11 steroidal alkaloids which exhibit significant biological activities. The intriguing biological properties and their low natural abundance have elevated cortistatins to be a typical target for both partial and total synthesis. Up to now, more than a dozen research groups have published approaches directed toward the synthesis of cortistatins, including one semi-synthesis, five total syntheses and five formal syntheses, as well as a number of synthetic studies about the pentacyclic core and some illuminating model studies. One of the biggest challenges for the synthesis of cortistatins is how to construct the unprecedented oxabicyclo [3.2.1]octane ring system which lies within a complex tetracarbocyclic skeleton. In our previous work, we have developed a gold-catalyzed semi-pinacol rearrangement strategy to diastereoselective synthesis of the oxabicyclo [3.2.1]octane ring system. The wide substrate scope as well as the high diastereoselectivity have made us to apply this method into the asymmetric formal synthesis of Cortistatins. Herein, full details about our efforts towards the formal synthesis of cortistatins were described by employing our developed gold-catalyzed cascade reaction to oxabicyclo[3.2.1]octane ring systems. This route is featured with a novel gold-catalyzed cascade reaction involving intramolecular nucleophilic addition of hydroxyl group to the carbon-carbon triple bond, followed by an oxonium ion initiated semi-pinacol-type 1, 2-migration to construct the key oxabicyclo [3.2.1]octane skeleton.
-
1 引言
Cortistatin类型天然产物是由Kobayashi小组[1]从印度尼西亚的一种名为“Corticium simplex”的海绵体中分离得到的一类甾体生物碱.该类型天然分子具有非常独特的化学结构(图 1), 含有一个[6, 7, 6, 5]四环系的中心碳环骨架, 以及多个连续手性中心和季碳中心, 此外中心碳环骨架中还含有一个氧桥结构, 从而形成一个特殊的[3, 2, 1]七元氧桥环结构.除了独特的化学结构之外, Cortistatin类型天然产物还具有十分显著的生物活性[2].例如Cortistatin A对人类的脐静脉血管内皮细胞(human umbilical vein endothelial cells, HUVECs)显示出非常强的抑制作用, 其IC50达到1.8 nmol•L-1的浓度水平.该分子可以抑制由血管内皮生长因子(VEGF)或碱性纤维原细胞生长因子(bFGF)诱导的脐静脉血管内皮细胞的迁移和微管形成, 且选择性指数是其他常见纤维细胞和肿瘤细胞的3300倍.这些数据表明Cortistatin A对于血管新生具有非常高的选择性抑制作用, 该类型天然分子具备开发新一代高选择性抗癌药物的潜力.
图 1
Cortistatin类型天然产物
Figure 1.
Cortistatin family of natural products
Cortistatin类型天然产物突出的生物学活性以及新颖的骨架结构, 吸引了世界范围内众多合成化学家的目光[3].到目前为止, 已经有六例全合成[4]和五例形式全合成[5]工作相继报道, 同时还有许多关于该类天然产物核心骨架结构的研究报道[6].该类型天然分子合成的最大难点和挑战之一在于如何有效构建七元氧桥环骨架结构.前人已经通过许多的策略来实现该氧桥环骨架的构建以及该类天然产物的全合成, 如Baran教授小组[4a]利用二碘化钐促进的扩环反应构建了七元环核心骨架, Nicolaou教授小组[4c]发展了一个非常高效的分子内Oxy-Michael/Aldol/Dehydration串联反应一步完成核心骨架的构建, Shair教授小组[4e]运用新颖的分子内串联Aza-Prins反应和分子内成醚反应一步实现了高度官能团化的六元环和七元环氧桥的立体选择性构建, Myers教授小组[4f]运用分子内RCM以及去芳构化反应实现了该家族分子的多样性全合成, 此外Hirama教授小组[4g]的自由基环化反应以及Funk教授小组[4h]的分子内[4+3]环加成反应都能有效构建七元氧桥核心骨架. 2011年, 我们[5d]报道了以分子内Diels-Alder (DA)反应, 氧化去芳构化以及氧杂Michael加成作为关键反应构建该类分子的七元氧桥骨架结构, 并完成了Cortistatin类型天然产物的形式全合成.考虑到分子内DA反应的收率不高以及氧化去芳构化选择性较低的问题, 发展一种更加高效的方法来构建核心七元氧桥环骨架结构, 从而完成Cortistatin类型天然产物的合成依然非常重要.本文详细阐述了以近期发展的金催化串联反应构建七元氧桥环结构为核心步骤的Cortistatin类型天然产物的形式全合成[7].
2 结果与讨论
2.1 金催化串联反应构建七元氧桥环骨架结构
金催化剂作为一种温和且高效的催化剂, 能够有效地活化炔烃官能团并将其转化为一系列有用的骨架结构.由于金催化反应通常具备温和的反应条件以及突出的官能团兼容性, 在过去的几十年之中, 以金催化剂活化π键为起始步, 继而引发一系列串联反应来构建复杂骨架的方法受到化学家们的广泛关注[8].
基于先前对金催化串联反应研究的认识[9], 我们认为利用羟基官能团分子内亲核进攻被金催化剂活化的碳碳三键, 继而进一步引发串联反应是构建复杂骨架结构非常有效的方法之一[10].基于此, 我们设想在金催化剂的作用下, 炔基二醇化合物A中的羟基以分子内的方式对被金催化剂活化的炔烃结构进行亲核加成得到烯基醚中间体B, 随后中间体B快速异构化形成一个具有高度张力的氧鎓离子中间体C[11], 继而引发semi-pinacol类型的1, 2-碳迁移反应[12], 便可得到七元氧桥环结构D (图 2).
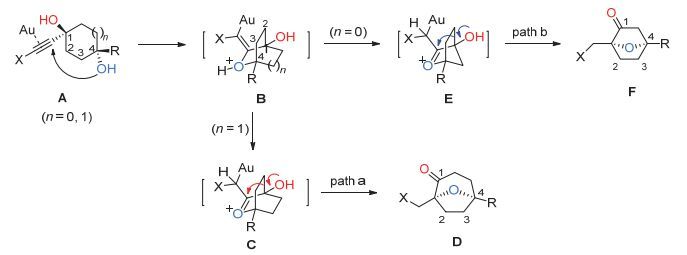 图 2
金催化串联semi-pinacol重排反应
Figure 2.
Gold-catalyzed tandem semi-pinacol rearrangement reactions
图 2
金催化串联semi-pinacol重排反应
Figure 2.
Gold-catalyzed tandem semi-pinacol rearrangement reactions
通过系统的条件筛选, 我们最终确定的最优条件是以Ph3PAuNTf2作为金催化剂, 以二氯甲烷作为溶剂, 底物在室温或70 ℃的条件下反应2 h (图 3).对于炔烃末端连有不同取代基的[6, 6]以及[6, 5]并环结构的底物, 在最优条件下, 均能很好地发生串联反应并以较好的收率得到相应的产物13a~13h.单环二醇前体同样适用于该金催化串联反应, 从而得到相应的七元氧桥环产物 13i~13m.此外, 如果以1, 3-环戊二醇为前体, 通过金催化串联反应, 可以得到六元氧桥环产物13n~13r.该类型六元氧桥环产物的形成, 可能是经历了图 2中path b的反应途径[13].
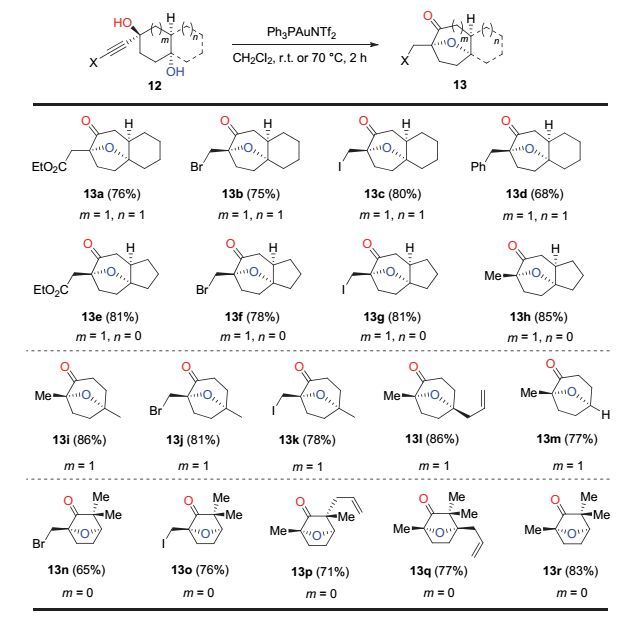 图 3
金催化串联semi-pinacol重排反应的底物适用范围
Figure 3.
Scope of the gold-catalyzed tandem semi-pinacol rearrangement reactions
图 3
金催化串联semi-pinacol重排反应的底物适用范围
Figure 3.
Scope of the gold-catalyzed tandem semi-pinacol rearrangement reactions
2.2 模型研究
上述的金催化串联反应方法学提供了一种高效的合成七元氧桥环骨架结构的方法, 为完成Cortistatin类型天然分子的合成提供了一种新的合成策略.具体来说, 对于完成Cortistatin类型天然产物的全合成可以分为三个部分, 即A环上官能团的引入, D环上异喹啉片段的引入以及该类分子核心四环骨架结构的构建.由于前两部分都可以在合成后期进行引入, 因此, 完成该类天然产物合成的关键在于如何高效地构建其核心四环骨架.为了探索上述金催化串联反应在Cortistatin类型天然产物全合成中应用的可行性, 我们首先对该类型天然分子中ABC环的合成进行了模型研究(图 4).
 图 4
Cortistatin类型天然产物ABC环系的模型研究
Figure 4.
Model study to the ABC rings of Cortistatins
图 4
Cortistatin类型天然产物ABC环系的模型研究
Figure 4.
Model study to the ABC rings of Cortistatins
从金催化方法学得到的产物13a出发, 首先将该化合物的羰基用乙二醇进行保护得到缩酮化合物14, 接着利用二异丁基氢化铝将酯基还原成相应的醛基得到化合物15.化合物15通过Wittig反应以90%的收率转化成烯烃16.该烯烃化合物可与异丙烯基硼酸频哪醇酯发生烯烃复分解反应得到17, 随后将硼酸酯结构进一步氧化得到甲基酮化合物18[14].酮化合物18在酸性条件下脱除乙二醇保护基得到二酮化合物19.最后在CH3ONa/MeOH的碱性条件下二酮化合物19进行分子内羟醛缩合反应得到ABC三环骨架化合物20, 进而完成了该模型的研究(图 5).
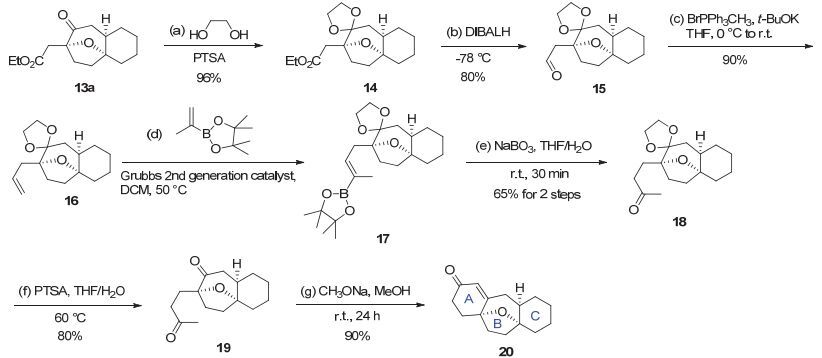 图 5
第一代模型研究
Figure 5.
First generation of model study
图 5
第一代模型研究
Figure 5.
First generation of model study
但是考虑到上述路线较为冗长, 且合成中包含有多步保护/脱保护以及氧化/还原过程, 因此我们对该模型研究进行了改进.在研究过程中, 溴代产物13b引起了我们的关注, 该化合物可以通过我们发展的金催化串联反应从底物12b以75%的收率得到.随后通过自由基方式的烯丙基化反应得到烯烃化合物21[15], 接着经过Wacker氧化以两步70%的收率得到二酮中间体19, 最后在CH3ONa/MeOH的条件下进行分子内羟醛缩合反应得到ABC三环骨架化合物20.改进后的模型研究路线从金催化前体12b出发, 仅需4步就能以47%的总收率得到模型化合物20 (图 6).
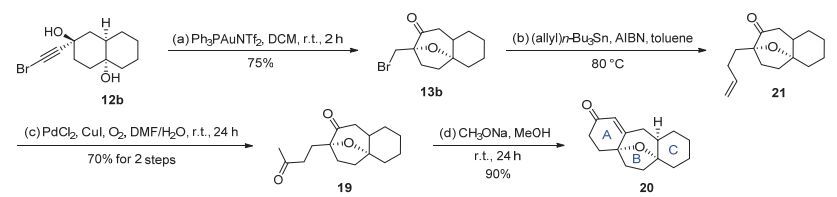 图 6
第二代模型研究
Figure 6.
Second generation of model study
图 6
第二代模型研究
Figure 6.
Second generation of model study
2.3 Cortistatin类型天然产物的形式全合成研究
基于第二代模型研究的结果, 我们开始了Cortistatin类型天然产物的合成研究.受Myers教授小组[4f]对于Cortistatin A, L, J, K四个天然产物的多样性全合成研究的启发, 我们的逆合成路线如图 7所示.
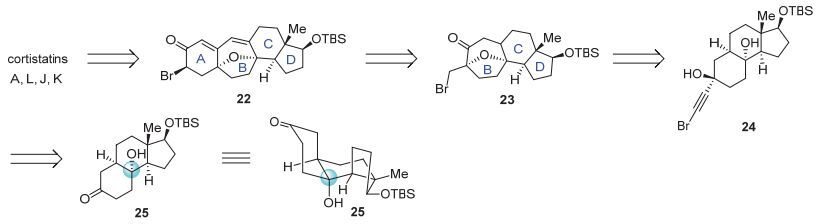 图 7
Cortistatin A, L, J, K的逆合成分析
Figure 7.
Retrosynthetic analysis of cortistatin A, L, J, K
图 7
Cortistatin A, L, J, K的逆合成分析
Figure 7.
Retrosynthetic analysis of cortistatin A, L, J, K
Cortistatin A, L, J, K可以逆推至共同高级中间体22, 高级中间体22中的A环可以参照第二代模型研究的方法由溴代物23经多步转化得到, 而溴代物23中的七元氧桥环即可通过我们发展的金催化串联反应从前体化合物24得到.前体24则可以由酮中间体25进行亲核加成获得.因此, 合成的首要任务是构建热力学上不利的[6, 6]顺式并环化合物25.
在合成化合物25的过程中, 我们注意到, Shair教授在Cortistatin A的全合成中使用的中间体33与化合物25非常相近, 其中唯一的差别是将化合物25中的三级羟基通过MEM保护基进行了保护.由于我们在金催化方法学研究过程中发现, 经过MEM保护的三级羟基同样能够像没被保护的裸羟基一样发生金催化串联反应得到相同的七元氧桥环产物[7].因此, 我们根据Shair教授的路线从Hajos-parrish酮的衍生物26出发经过7步化学转化得到了中间体33[4e].但是其中从烯醇硅醚29转化成α-羟基酮30的过程中, 环氧化中间体很容易分解, 导致该反应重复性较差, 产率极不稳定, 往往较低.因此, 我们对该步反应进行了条件优化, 最终利用OsO4/NMO[16]作为氧化剂可以将连续三步总收率提高至74% (图 8).
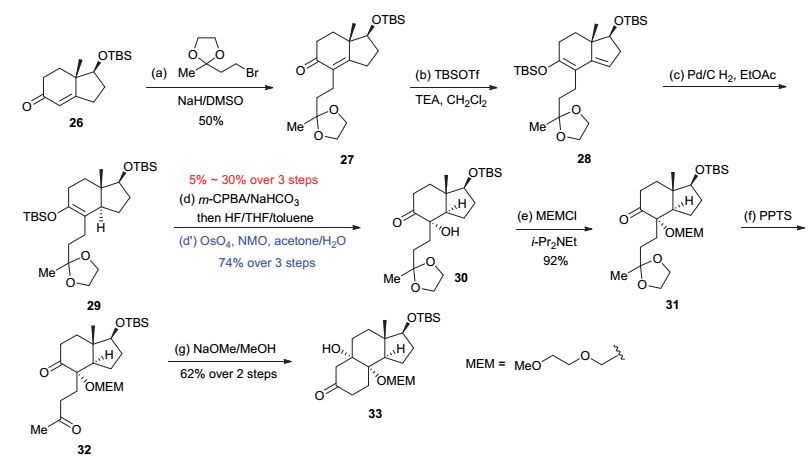 图 8
中间体33的优化合成路线
Figure 8.
A modified route towards the synthesis of intermediate 33
图 8
中间体33的优化合成路线
Figure 8.
A modified route towards the synthesis of intermediate 33
得到化合物33之后, 我们开始尝试制备金催化串联反应的关键前体.由于化合物33中存在裸露的三级羟基, 其能够阻止亲核试剂对其结构上的酮进行加成.于是, 我们将该化合物在二氯亚砜和吡啶的条件下发生羟基消除反应得到中间体34, 接着对酮进行亲核加成反应得到金催化反应前体35.然而, 该前体在金催化串联反应条件下并没有得到我们预期的七元氧桥环产物38, 而是分别以25%和30%的收率得到了消除副产物36以及取代副产物37 (图 9).
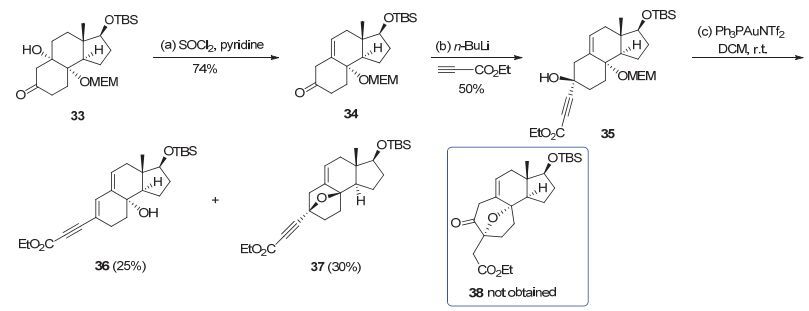 图 9
通过金催化串联反应合成七元氧桥环的第一次尝试
Figure 9.
First attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
图 9
通过金催化串联反应合成七元氧桥环的第一次尝试
Figure 9.
First attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
化合物36和37的形成表明烯烃官能团的存在能够诱导炔丙基的三级醇发生消除反应形成共轭体系, 同时也有利于OMEM基团离去形成烯丙基正离子中间体, 进而影响金催化串联反应的发生.因此, 我们尝试移除烯烃官能团.然而对中间体34的直接氢化除了回收原料之外没有得到任何的氢化产物.于是我们首先使用硼氢化钠将34还原成相应的醇39, 随后尝试双键的氢化, 结果发现在60个大气压力的氢化条件下该中间体能以20%的低收率得到氢化产物40.接着氢化产物40通过DMP氧化以及分子间亲核加成反应得到金催化关键反应前体42.然而, 该前体在金催化串联反应条件下依然没能得到产物43, 反应底物也在金催化剂的作用下逐渐分解(图 10).
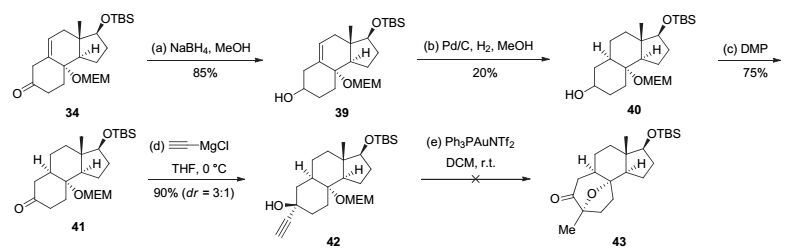 图 10
通过金催化串联反应合成七元氧桥环的第二次尝试
Figure 10.
Second attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
图 10
通过金催化串联反应合成七元氧桥环的第二次尝试
Figure 10.
Second attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
以上失败的结果使我们对于金催化前体的构象要求有了更进一步的认识.炔烃官能团和OMEM基团需要充分接近才能发生分子内亲核进攻反应.因此, R基团与OMEM基团保持顺式构型, 并且R基团不能是氢原子对于金催化串联反应具有十分重要的影响(图 11).
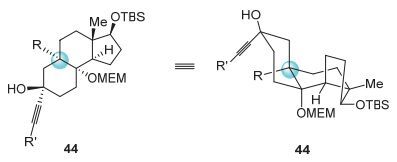 图 11
金催化串联反应前体的结构分析
Figure 11.
Structural analysis of the key precursor for the gold-catalyzed tandem reaction
图 11
金催化串联反应前体的结构分析
Figure 11.
Structural analysis of the key precursor for the gold-catalyzed tandem reaction
因此, 我们首先尝试将中间体33中的叔醇官能团用TMS保护基进行保护.将化合物33与TMSOTf和三乙胺进行反应, 在该反应条件下, 化合物33中的酮羰基也发生反应形成了烯醇硅醚, 因此我们将得到的粗混合物进一步与PPTS反应释放出酮官能团.然而, 我们并没有得到预期的产物而是以78%的收率得到了二醇被保护的中间体45.这可能是TMSOTf作为路易斯酸促进了MEM基团的部分离去, 得到的氧鎓中间体被叔醇所捕获, 从而形成化合物45.接着将45与炔基格氏试剂反应得到关键前体46.然而, 该前体在金催化剂的作用下不发生任何反应, 甚至将温度提升至70 ℃也只能回收原料(图 12).
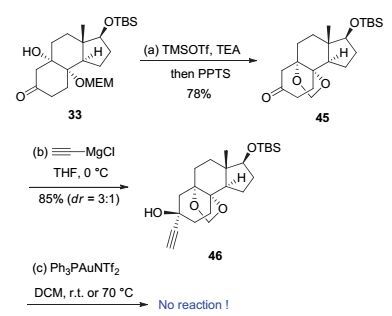 图 12
通过金催化串联反应合成七元氧桥环的第三次尝试
Figure 12.
Third attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
图 12
通过金催化串联反应合成七元氧桥环的第三次尝试
Figure 12.
Third attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
使用硅保护基保护叔醇失败后, 我们尝试选择其它保护基.我们认为甲基保护基可能是个不错的选择, 首先甲基体积较小, 容易对叔醇进行保护; 其次甲基的化学性质不活泼, 不会对后续的金催化反应产生影响.但是利用不同的碱与碘甲烷对33中叔醇结构进行直接的甲基保护均没有得到相应的产物, 在低温下反应不发生, 而将温度升高后反应底物迅速分解.因此, 为了得到甲基保护的金催化反应前体, 我们首先将化合物33还原成相应的醇47, 接着选择性地将二级醇进行乙酰基保护得到中间体48.在氢化钠和碘甲烷的条件下将叔醇用甲基保护得到化合物49, 然后进行选择性羟基脱保护和氧化得到化合物51, 接着将其与炔基格氏试剂反应得到关键反应前体52.幸运的是, 该前体在金催化剂的作用下能够以83%的收率得到七元氧桥环产物53.化合物53的结构随后通过X射线单晶衍射得到进一步确认[17](图 13).
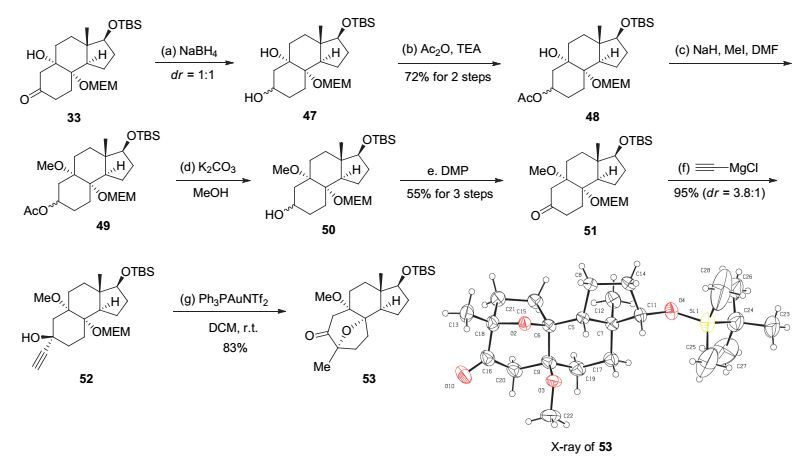 图 13
通过金催化串联反应合成七元氧桥环
Figure 13.
Synthesis of the 8-oxabicyclo[3.2.1]octane in cortistatins via Au-catalyzed tandem reaction
图 13
通过金催化串联反应合成七元氧桥环
Figure 13.
Synthesis of the 8-oxabicyclo[3.2.1]octane in cortistatins via Au-catalyzed tandem reaction
实现了关键反应之后, 我们对金催化的前体化合物52的合成进行了优化.首先将中间体33保护成相应的缩酮化合物54, 接着在氢化钾和碘甲烷的条件下将叔醇进行甲基保护得到化合物55.由于MEM基团对酸性条件比较敏感, 因此利用酸性条件脱除缩酮保护基没有成功, 反而导致底物分解.经过一系列条件筛选之后, 我们发现使用CAN在碱性条件下能够顺利脱除缩酮保护基, 得到酮化合物51[18].随后酮51与炔基格氏试剂反应即可得到炔基化合物52 (图 14).
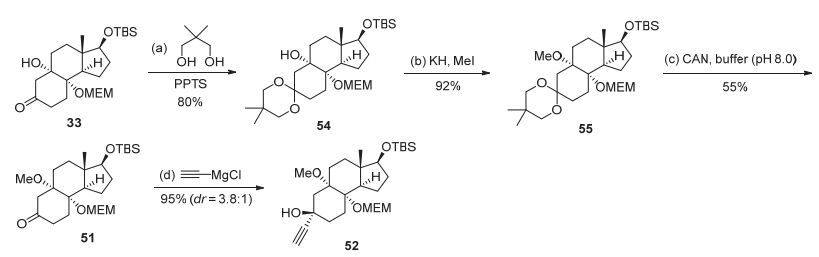 图 14
中间体52的优化合成路线
Figure 14.
An optimized route for the key precursor52
图 14
中间体52的优化合成路线
Figure 14.
An optimized route for the key precursor52
得到炔基化合物52之后, 后续的步骤即可参照模型研究中的反应条件得以实现.将端炔52与NBS和催化量的硝酸银反应得到金催化反应前体56[19], 该前体在金催化剂的作用下能够以81%的收率得到相应的七元氧桥产物57.接着通过自由基烯丙基化反应顺利得到烯烃58, 随后利用Wacker氧化以90%的收率得到二酮中间体59, 该中间体在CH3ONa/MeOH的碱性条件下发生串联aldol condensation/β-elimination反应得到四环系核心骨架60[20].最后通过羰基α位溴代反应得到高级中间体22, 根据Myers教授[4f]的工作, 中间体22可以通过后续的转化, 实现Cortistatin A, L, J, K的全合成(图 15).
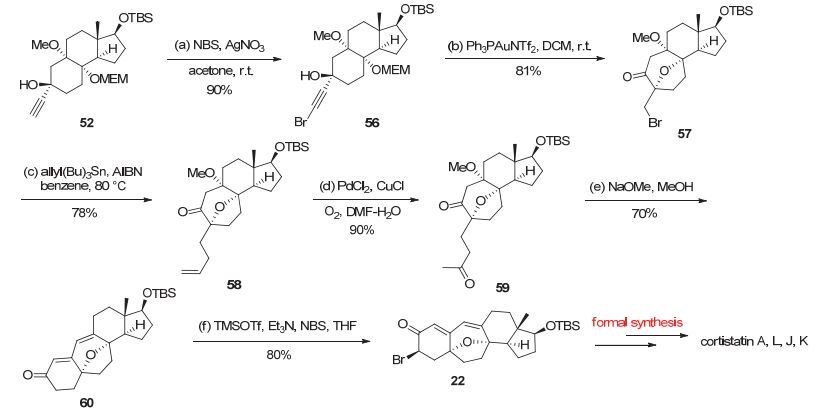 图 15
Cortistatin A, L, J, K的形式全合成
Figure 15.
Formal synthesis of cortistatin A, L, J, K
图 15
Cortistatin A, L, J, K的形式全合成
Figure 15.
Formal synthesis of cortistatin A, L, J, K
3 结论
在本工作中, 我们详细报道了天然产物Cortistatin A, L, J, K的形式全合成研究工作.整个合成路线以我们小组近期发展的金催化串联semi-pinacol重排反应为关键步骤, 高效地构建Cortistatin类型天然产物中七元氧桥环核心骨架结构.利用该金催化串联反应实现其它含有氧桥结构天然产物分子全合成的研究工作目前仍在进行之中.
-
-
[1]
Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. J. Am. Chem. Soc. 2006, 128, 3148. doi: 10.1021/ja057404h
-
[2]
(a) Aoki, S.; Watanabe, Y.; Tanabe, D.; Arai, M.; Suna, H.; Miyamoto, K.; Tsujibo, H.; Tsujikawa, K.; Yamamoto, H.; Kobayashi, M. Bioorg. Med.Chem. 2007, 15, 6758. (b) Watanabe, Y.; Aoki, S.; Tanabe, D.; Setiawan, A.; Kobayashi, M. Tetrahedron 2007, 63, 4074. (c) Aoki, S.; Watanabe, Y.; Tanabe, D.; Setiawan, A.; Arai, M.; Kobayashi, M. Tetrahedron Lett. 2007, 48, 4485.
-
[3]
For reviews on the synthesis of the cortistatins, see: (a) Nising, C. F.; Brase, S. Angew. Chem. Int. Ed. 2008, 47, 9389. Angew. Chem. 2008, 120, 9529. (b) Narayan, A. R. H.; Simmons, E. M.; Sarpong, R. Eur. J. Org. Chem.2010, 3553. (c) Chen, D. Y. K.; Tseng, C. C Org. Biomol. Chem.2010, 8, 2900.
-
[4]
(a) Shenvi, R. A.; Guerrero, C. A.; Shi, J.; Li, C. C.; Baran, P. S.J. Am. Chem. Soc. 2008, 130, 7241. (b) Shi, J.; Manolikakes, G.; Yeh, C. H.; Guerrero, C. A.; Shenvi, R. A.; Shigehisa, H.; Baran, P. S. J. Am. Chem. Soc. 2011, 133, 8014. (c) Nicolaou, K. C.; Sun, Y. P.; Peng, X. S.; Polet, D.; Chen, D. Y. Angew.Chem. Int. Ed. 2008, 47, 7310. Angew.Chem. 2008, 120, 7420. (d) Nicolaou, K. C.; Peng, X. S.; Sun, Y. P.; Polet, D.; Zou, B.; Lim, C. S. Chen, D. Y. J. Am. Chem.Soc. 2009, 131, 10587. (e) Lee, H. M.; Nieto-Oberhuber, C.; Shair, M. D. J. Am. Chem. Soc. 2008, 130, 16864. (f) Flyer, A. N.; Si, C.; Myers, A. G. Nat. Chem. 2010, 2, 886. (g) Yamashita, S.; Iso, K.; Kitajima, K.; Himuro, M.; Hirama, M.J. Org. Chem. 2011, 76, 2408. (h) Nilson, M. G.; Funk, R. L. J. Am. Chem. Soc. 2011, 133, 12451.
-
[5]
(a) Yamashita, S.; Iso, K.; Hirama, M. Org. Lett. 2008, 10, 3413. (b) Yamashita, S.; Kitajima, K.; Iso, K.; Hirama, M. Tetrahedron Lett. 2009, 50, 3277. (c) Simmons, E. M.; Hardin-Narayan, A. R.; Guo, X.; Sarpong, R. Tetrahedron 2010, 66, 4696. (d) Fang, L.; Chen, Y.; Huang, J.; Liu, L.; Quan, J.; Li, C. C.; Yang, Z. J. Org.Chem. 2011, 76, 2479. (e) Kuang, L. P.; Liu, L. L.; Chiu, P.Chem. Eur. J. 2015, 21, 14287.
-
[6]
(a) Dai, M.; Danishefsky, S. J. Tetrahedron Lett. 2008, 49, 6610. (b) Dai, M.; Wang, Z.; Danishefsky, S. J. Tetrahedron Lett.2008, 49, 6613. (c) Kurti, L.; Czako, B.; Corey, E. J. Org.Lett. 2008, 10, 5247. (d) Simmons, E. M.; Hardin, A. R.; Guo, X.; Sarpong, R. Angew. Chem. Int. Ed. 2008, 47, 6650. Angew. Chem. 2008, 120, 6752. (e) Kotoku, N.; Sumii, Y.; Hayashi, T.; Kobayashi, M. Tetrahedron Lett. 2008, 49, 7078. (f) Craft, D. T.; Gung, B. W. Tetrahedron Lett. 2008, 49, 5931. (g) Magnus, P.; Littich, R. Org. Lett. 2009, 11, 3938. (h) Yu, F.; Li, G.; Gao, P.; Gong, H.; Liu, Y.; Wu, Y.; Cheng, B.; Zhai, H.Org. Lett. 2010, 12, 5135. (i) Frie, J. L.; Jeffrey, C. S.; Sorensen, E. J. Org. Lett. 2009, 11, 5394. (j) Baumgartner, C.; Ma, S.; Liu, Q.; Stoltz, B. M. Org. Biomol. Chem.2010, 8, 2915. (k) Liu, L. L.; Chiu, P. Chem. Commun.2011, 47, 3416. (l) Kotoku, N.; Sumii, Y.; Kobayashi, M. Org.Lett. 2011, 13, 3514. (m) Wang, Z.; Dai, M. J.; Park, P. K.; Danishefsky, S. J. Tetrahedron 2011, 67, 10249. (n) Aquino, C.; Greszler, S. N.; Micalizio, G. C. Org. Lett. 2016, 18, 2624.
-
[7]
Fu, J.; Gu, Y.; Yuan, H.; Luo, T.; Li, S.; Lan, Y.; Gong, J.; Yang, Z. Nat. Commun. 2015, 6, 8617.
-
[8]
For selected reviews, see: (a) Hashmi, A. S. K. Chem. Rev.2007, 107, 3180. (b) Friend, C. M.; Hashmi, A. S. K. Acc. Chem.Res. 2014, 47, 729. (c) Zhang, L. Acc. Chem.Res. 2014, 47, 877. (d) Wang, Y. M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889. (e) Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028. (f) Dorel, R.; Echavarren, A. M. J. Org. Chem. 2015, 80, 7321. (g) Hopkinson, M. N.; Tlahuext-Aca, A.; Glorius, F. Acc. Chem.Res. 2016, 49, 2261.
-
[9]
(a) Shi, H.; Fang, L.; Tan, C.; Shi, L.; Zhang, W.; Li, C. C.; Luo, T.; Yang, Z. J. Am. Chem. Soc. 2011, 133, 14944. (b) Shan, Z.; Liu, J.; Xu, L.; Tang, Y.; Chen, J.; Yang, Z. Org. Lett. 2012, 14, 3712. (c) Yue, G.; Zhang, Y.; Fang, L.; Li, C.; Luo, T.; Yang, Z. Angew. Chem. Int. Ed. 2014, 53, 1837. Angew. Chem. 2014, 126, 1868. (d) Shi, H.; Tan, C.; Zhang, W.; Zhang, Z.; Long, R.; Luo, T.; Yang, Z. Org. Lett. 2015, 17, 2342.
-
[10]
For selected examples, see: (a) Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J. P. J. Am. Chem. Soc. 2005, 127, 9976. (b) Hashmi, A. S. K.; Bührle, M.; Wçlfle, M.; Rudolph, M.; Wieteck, M.; Rominger, F.; Frey, W. Chem. Eur. J. 2010, 16, 9846. (c) Bihelovic. F.; Saicic, R. N. Angew. Chem. Int.Ed. 2012, 51, 5687. Angew. Chem. 2012, 124, 5785. (d) Noey, E. L.; Luo, Y.; Zhang, L.; Houk, K. N. J. Am. Chem.Soc. 2012, 134, 1078. (e) Zeng, X. Chem. Rev.2013, 113, 6864. (f) Li, D. Y.; Chen, H. J.; Liu, P. N. Angew.Chem. Int. Ed. 2016, 55, 373. Angew. Chem.2016, 128, 381.
-
[11]
(a) Barluenga, J.; Diéguez, A.; Fernández, A.; Rodríguez, F.; Fañanás, F. J. Angew. Chem. Int. Ed. 2006, 45, 2091. Angew. Chem. 2006, 118, 2145. (b) Barluenga, J.; Fernández, A.; Diéguez, A.; Rodríguez, F.; Fañanás, F. J. Chem. Eur.J. 2009, 15, 11660. (c) Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226. (d) Nagaraju, C.; Prasad, K. R. Angew. Chem. Int. Ed. 2014, 53, 10997;Angew. Chem. 2014, 126, 11177.
-
[12]
(a) Kirsch, S. F.; Binder, J. T.; Liébert, C.; Menz, H. Angew. Chem. Int. Ed.2006, 45, 5878. Angew. Chem. 2006, 118, 6010. (b) Crone, B.; Kirsch, S. F. Chem. Eur. J. 2008, 14, 3514. (c) Song, Z. L.; Fan, C. A.; Tu, Y. Q. Chem. Rev. 2011, 111, 7523. (d) Zhang, X. M.; Tu, Y. Q.; Zhang, F. M.; Chen, Z. H.; Wang, S. H. Chem. Soc.Rev. 2017, 46, 2272.
-
[13]
Gu, Y.; Zhang, P.; Fu, J.; Liu, S.; Lan, Y.; Gong, J.; Yang, Z. Adv. Synth. Catal. 2016, 358, 1392. doi: 10.1002/adsc.201600218
-
[14]
(a) Morrill, C.; Funk, T. W.; Grubbs, R. H. Tetrahedron Lett.2004, 45, 7733. (b) Hemelaere, R.; Carreaux, F.; Carboni, B. J.Org. Chem. 2013, 78, 6786.
-
[15]
Keck, G. E.; Yates, J. B. J. Am. Chem. Soc. 1982, 104, 5829. doi: 10.1021/ja00385a066
-
[16]
Kotoku, N.; Sumii, Y.; Hayashi, T.; Kobayashi, M. Heterocycles 2011, 83, 1535. doi: 10.3987/COM-11-12195
-
[17]
The X-ray crystallography data for compound 53, see SI of ref. 7.
-
[18]
Marḱo, I. E.; Ates, A.; Gautier, A.; Leroy, B.; Plancher, J. M.; Quesnel, Y.; Vanherck, J. C. Angew. Chem. Int. Ed. 1999, 38, 3207. Angew. Chem. 1999, 111, 3411.
-
[19]
Ghosh, N.; Nayak, S.; Prabagar, B.; Sahoo, A. K. J. Org. Chem. 2014, 79, 2453 doi: 10.1021/jo4027319
-
[20]
Tan, D. S.; Dudley, G. B.; Danishefsky, S. Angew. Chem.Int. Ed. 2002, 41, 2185. Angew. Chem.2002, 114, 2289..
-
[1]
-

图 2 金催化串联semi-pinacol重排反应
Figure 2 Gold-catalyzed tandem semi-pinacol rearrangement reactions

图 3 金催化串联semi-pinacol重排反应的底物适用范围
Figure 3 Scope of the gold-catalyzed tandem semi-pinacol rearrangement reactions

图 5 第一代模型研究
Figure 5 First generation of model study
Reagents and Conditions: a) Ethylene glycol (15.0 equiv.), PTSA (0.1 equiv.), Benzene (0.1 mol•L-1), refulx, 12 h, 96%; b) DIBAL-H (1.3 equiv.), DCM (0.1 mol•L-1), -78 ℃, 2 h, 80%; c) BrPPh3CH3 (4.0 equiv.), t-BuOK (4.0 equiv.), THF (0.2 mol•L-1), 0 ℃, 12 h, 90%; d) 4, 4, 5, 5-tetramethyl-2-(prop-1-en-2-yl)-1, 3, 2-dioxaboroane (8.0 equiv.), Grubbs 2nd catalyst (0.2 equiv.), DCM (0.05 mol•L-1), 50 ℃, 6 h; e) NaBO3•4H2O (6.0 equiv.), THF/H2O (V/V, 1:1, 0.05 mol•L-1), r.t., 0.5 h, 65% for two steps; f) PTSA (0.4 equiv.), THF/H2O (V/V, 1:1, 0.04 mol•L-1), 60 ℃, 12 h, 80%; g) MeONa (4.0 equiv.), MeOH (0.04 mol•L-1), r.t., 24 h, 90%.

图 6 第二代模型研究
Figure 6 Second generation of model study
Reagents and Conditions: a) Ph3PAuCl (0.025 equiv.), AgNTf2 (0.025 equiv.), DCM (0.2 mol•L-1), r.t., 2 h, 75%; b) allyltributyltin (3.0 equiv.), AIBN (0.5 equiv.), toluene (0.05 mol•L-1), 80 ℃, 7 h; c) PdCl2 (0.5 equiv.), CuCl (2.0 equiv.), O2 (balloon pressure), DMF/H2O (V/V, 7:1, 0.05 mol•L-1), r.t., 12 h, 70% for two steps. d) MeONa (4.0 equiv.), MeOH (0.04 mol•L-1), r.t., 24 h, 90%.

图 7 Cortistatin A, L, J, K的逆合成分析
Figure 7 Retrosynthetic analysis of cortistatin A, L, J, K

图 8 中间体33的优化合成路线
Figure 8 A modified route towards the synthesis of intermediate 33
Reagents and Conditions: a) NaH (1.05 equiv.), 2-(2-bromoethyl)-2-methyl-1, 3-dioxolane (1.1 equiv.), DMSO (1.0 mol•L-1), r.t., 15 h, 50%; b) TBSOTf (1.2 equiv.), 2, 6-lutidine (2 equiv.), CH2Cl2 (0.3 mol•L-1), 0 ℃, 2 h; c) H2 (balloon pressure), Pd/C (0.08 equiv.), EtOAc (0.3 mol•L-1), r.t., 12 h; d) m-CPBA (1.1 equiv.), NaHCO3 (10 equiv.), toluene (0.6 mol•L-1), -10 ℃, 30 min, then HF (10 equiv.), THF/toluene (V/V, 1:1, 0.4 mol•L-1), 0 ℃, 30 min (5%~30% over three steps); d') NMO (2.0 equiv.), OsO4 (0.2 equiv.), acetone/water (V/V, 8:1, 0.1 mol•L-1), 50 ℃, 18 h (74% over three steps); e) MEMCl (2.0 equiv.), i-Pr2NEt (4.0 equiv.), 1, 2-dichloroethane (0.2 mol•L-1), 80 ℃, 18 h, 92%; f) PPTS (0.1 equiv.), acetone/water (V/V, 4:1, 0.2 mol•L-1), 60 ℃, 4 h; g) NaOMe (5.0 equiv.), MeOH (0.2 mol•L-1), 70 ℃, 1 h (62% over two steps).

图 9 通过金催化串联反应合成七元氧桥环的第一次尝试
Figure 9 First attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
Reagents and Conditions: a) pyridine (6.0 equiv.), SOCl2 (3.0 equiv.), DCM (0.05 mol•L-1), -10 ℃, 0.5 h, 74%; b) n-BuLi (1.5 equiv.), ethyl propiolate (1.5 equiv.), THF (0.03 mol•L-1), -78 ℃, 3 h, 50%; c) Ph3PAuCl (0.025 equiv.), AgNTf2 (0.025 equiv.), DCM (0.2 mol•L-1), r.t., 2 h, 25% for 36, 30% for 37.

图 10 通过金催化串联反应合成七元氧桥环的第二次尝试
Figure 10 Second attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
Reagents and Conditions: a) NaBH4 (1.0 equiv.), EtOH (0.05 mol•L-1), -78 ℃, 3 h, 85%; b) palladium on carbon (0.1 equiv.), H2 (6.0 MPa), MeOH (0.3 mol•L-1), r.t., 18 h, 20%; c) DMP (1.1 equiv.), NaHCO3 (1.1 equiv.), DCM (0.01 mol•L-1), r.t., 2 h, 75%; d) ethynylmagnesium chloride (2.0 equiv.), THF (0.01 mol•L-1), 0 ℃ to r.t., 3 h, 90%, dr=3:1.

图 11 金催化串联反应前体的结构分析
Figure 11 Structural analysis of the key precursor for the gold-catalyzed tandem reaction

图 12 通过金催化串联反应合成七元氧桥环的第三次尝试
Figure 12 Third attempt to synthesize the 8-oxabicyclo[3.2.1]octane of cortistatins via Au-catalyzed tandem reaction
Reagents and Conditions: a) triethylamine (6.0 equiv.), TMSOTf (3.0 equiv.), DCM (0.03 mol•L-1), 0 ℃ to r.t., 3 h, then PPTS (0.2 equiv.), DCM (0.03 mol•L-1), r.t., 2 h, 78%; b) ethynylmagnesium choride (2.0 equiv.), THF (0.01 mol•L-1), 0 ℃ to r.t., 3 h, 85%, dr=3:1; c) Ph3PAuCl (0.025 equiv.), AgNTf2 (0.025 equiv.), DCM (0.2 mol•L-1), r.t. or 70 ℃, 2 h.

图 13 通过金催化串联反应合成七元氧桥环
Figure 13 Synthesis of the 8-oxabicyclo[3.2.1]octane in cortistatins via Au-catalyzed tandem reaction
Reagents and Conditions: a) NaBH4 (2.0 equiv.), EtOH (0.1 mol•L-1), -78 ℃, 3 h, dr=1:1; b) TEA (10.0 equiv.), Ac2O (5.0 equiv.), DMAP (0.1 equiv.), DCM (0.1 mol•L-1), 40 ℃, 16 h, 72% for two steps; c) NaH (5.0 equiv.), MeI (10.0 equiv.), DMF (0.1 mol•L-1), 0 ℃ to r.t., 4 h; d) K2CO3 (1.5 equiv.), MeOH (0.1 mol•L-1), 40 ℃, 12 h; e) DMP (2.0 equiv.), DCM (0.1 mol•L-1), 0 ℃ to r.t., 2 h, 55% for three steps; f) ethynylmagnesium chloride (3.0 equiv.), THF (0.5 mol•L-1), 0 ℃ to r.t., 4 h, 95%, dr=3.8:1; g) Ph3PAuCl (0.025 equiv.), AgNTf2 (0.025 equiv.), DCM (0.02 mol•L-1), r.t., 1 h.

图 14 中间体52的优化合成路线
Figure 14 An optimized route for the key precursor52
Reagents and Conditions: a) 2, 2-dimethylpropane-1, 3-diol (10.0 equiv.), PPTS (0.1 equiv.), benzene (0.1 mol•L-1), 50 ℃, 4 h, 80%; b) KH (4.0 equiv.), MeI (8.0 equiv.), 0 ℃ to r.t., 92%; c) CAN (0.05 equiv.), borate buffer (pH=8.0), 60 ℃, 5 h, 55%; d) ethynylmagnesium chloride (3.0 equiv.), THF (0.5 mol•L-1), 0 ℃ to r.t., 4 h, 95%, dr=3.8:1.

图 15 Cortistatin A, L, J, K的形式全合成
Figure 15 Formal synthesis of cortistatin A, L, J, K
Reagents and Conditions: a) NBS (1.3 equiv.), AgNO3 (0.05 equiv.), acetone (0.08 mol•L-1), r.t., 0.5 h, 90%; b) Ph3PAuCl (0.025 equiv.), AgNTf2 (0.025 equiv.), DCM (0.02 mol•L-1), r.t., 1 h, 81%; c) allyltributyltin (10.0 equiv.), AIBN (0.5 equiv.), benzne (0.05 mol•L-1), 80 ℃, 18 h, 78%; d) PdCl2 (0.2 equiv.), CuCl (1.5 equiv.), O2 (balloon pressure), DMF/H2O (V/V, 7:1, 0.1 mol•L-1), r.t., 12 h, 90%; e) NaOMe (2.0 equiv.), MeOH (0.03 mol•L-1), r.t., 10 h, 70%; f) triethylamine (10.0 equiv.), trimethylsilyl trifluoromethanesulfonate (6.0 equiv.), N-bromosuccinimide (3.0 equiv.), THF (0.02 mol•L-1), 0 ℃, 1 h, 80%.
-


 下载:
下载:














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 4016
- HTML全文浏览量: 408
 下载:
下载:
