 图1
氧气在催化剂表面的吸附形式[2]
Figure1.
Oxygen adsorption form on the surface of catalysts[2]
图1
氧气在催化剂表面的吸附形式[2]
Figure1.
Oxygen adsorption form on the surface of catalysts[2]
引用本文:
钟国玉, 王红娟, 余皓, 彭峰. 氧还原碳基非贵金属电催化剂研究进展[J]. 化学学报,
2017, 75(10): 943-966.
doi:
10.6023/A17040183

Citation: Zhong Guoyu, Wang Hongjuan, Yu Hao, Peng Feng. A Review of Carbon-based Non-noble Catalysts for Oxygen Reduction Reaction[J]. Acta Chimica Sinica, 2017, 75(10): 943-966. doi: 10.6023/A17040183
Citation: Zhong Guoyu, Wang Hongjuan, Yu Hao, Peng Feng. A Review of Carbon-based Non-noble Catalysts for Oxygen Reduction Reaction[J]. Acta Chimica Sinica, 2017, 75(10): 943-966. doi: 10.6023/A17040183
氧还原碳基非贵金属电催化剂研究进展
摘要:
质子交换膜燃料电池是一种直接将化学能转化为电能的能量转换装置,具有环境友好、能量密度高、转化效率高等优点,能够应用于便携能源及燃料电池电动车领域.但燃料电池阴极氧还原需要大量的铂基催化剂,铂价格昂贵、储量有限、易中毒的缺点限制了它的实际应用.因此,开发低成本、高活性、高稳定性的阴极非贵金属催化剂将能够显著推动质子交换膜燃料电池的大规模商业化应用.其中碳基非贵金属催化剂作为最有可能替代铂的氧还原催化剂,引起了广泛的研究.基于此,本文首先简单介绍了氧还原的机理;其次将碳基非贵金属催化剂分为过渡金属氮碳催化剂和非金属掺杂碳催化剂,对它们在材料制备和活性中心的研究进行了总结和讨论;最后,报道了碳基非贵金属催化剂在质子交换膜燃料电池单电池中的应用进展.
English
A Review of Carbon-based Non-noble Catalysts for Oxygen Reduction Reaction
Abstract:
Proton exchange membrane fuel cells (PEMFCs) that directly convert chemical energy into electrical energy can be applied to portable power and fuel cell electric vehicles, due to their advantages such as environment-friendliness, high power density and high convert efficiency. However, the high loading of Pt-based catalysts on the cathode oxygen reduction reaction (ORR) hinder the commercial application of PEMFCs for the high price, resource shortage and easy poisoning of Pt. Thus, developing inexpensive, high performance and durability non-noble metal cathode catalysts will promote the large-scale commercialization of PEMFCs. As the most likely alternative to Pt, carbon-based non-noble ORR catalysts have been widely studied. In this review, firstly, the electrocatalytic mechanism for ORR is simply introduced. Secondly, the carbon-based non-noble ORR catalysts are divided into transition metal-nitrogen-carbon compounds (M-N-C) and non-metal heteroatom-doped carbon catalysts; the researches of material preparations and active sites are summarized and discussed. Thirdly, the applications of carbon-based non-noble ORR catalysts in PEMFC are reviewed. Although great progress has been achieved in this area of research and development, there are still some challenges for carbon-based non-noble ORR catalysts. Firstly, the ORR electrocatalytic mechanism isn't clear, especially carbon-based non-noble catalysts. Secondly, the ORR active sites of carbon-based non-noble catalysts remain controversial, which can be mainly divided into the transition metal coordination compounds, the doped heteroatom, the filled metal and the defect sites. Thirdly, the actual activity and stability of carbon-based non-noble catalysts are still below the PEMFC target. In summary, the future research directions on carbon-based non-noble catalysts for PEMFC applications would be proposed as follows:(1) fundamentally understanding the ORR mechanisms and their relationship with catalyst active site structures and composition using both theoretical calculations and experimental approaches; (2) improving catalyst activity and stability to satisfy the practical application of PEMFC.
-
1 引言
化石能源, 如石油、天然气与煤炭的广泛投入使用推动了人类社会的快速发展, 但是化石能源的有限及其使用中引发的环境问题也是人类面临的两大难题.开发出环境友好、可再生的新能源和新能源技术已经成为当今世界最为重要的研究热点, 引起了人们的广泛关注.燃料电池是一种不经过燃烧直接将化学能转化为电能的新型能量转换装置, 与其它新能源技术相比, 具有能量转换密度高、污染小、燃料多样化、可靠性高、噪音低及便于维护等优点, 被广泛地认为是最有可能得到大规模商业化应用的新能源技术.美国的《时代周刊》曾将燃料电池技术列为21世纪的高科技之首.在我国的《国家中长期科学和技术发展规划纲要》中, 多处把燃料电池放在重要的发展方向上.而其中的质子交换膜燃料电池(PEMFC)更是具有工作温度低、能量密度高、启动速度快等优点, 有望应用于燃料电池电动汽车及便携能源领域.
目前, PEMFC的研究已经取得了很大的发展, 并陆续有示范性的质子交换膜燃料电池发电站、电动车、手机电池等面世.但是其寿命短、效率低和成本高的缺陷限制了它的大规模商业化应用. PEMFC的寿命短主要是因为铂基催化剂的溶解、铂基催化剂的毒化和Nafion膜的缓慢降解; 效率低主要是因为铂基催化剂对甲醇氧化催化性能低、Nafion膜存在甲醇渗透、阴极氧还原活性低; 成本高主要因为是Nafion膜和催化剂成本过高, 其中催化剂使用的铂系贵金属, 成本高且储量有限.从以上的分析可以看出, 铂基催化剂问题是限制PEMFC广泛应用的主要因素.其中阴极氧还原反应(ORR)的反应速率比阳极氧化反应差数个数量级, 所以需要比阳极更多的铂基催化剂用于加速氧还原反应的进行.因此, 开发低成本、高活性、高稳定性的ORR非贵金属催化剂对推动燃料电池的大规模商业化应用意义重大.在本文中, 我们先对氧还原的电催化原理进行了简单讨论, 之后较为系统地综述了近年来碳基非贵金属氧还原催化剂的研究进展.
2 阴极氧还原的电催化基本原理
2.1 氧还原反应途径
氧还原是一个多电子反应, 整个反应过程可以简单地分为四电子途径和二电子途径[1].四电子途径又分为直接四电子途径和连续四电子途径, 在酸性电解液中直接四电子途径是指反应没有中间步骤直接得到四个电子, 和四个质子生成H2O, 连续四电子途径是O2先被还原成H2O2, 然后再进一步还原为H2O.在碱性电解液中, 直接四电子途径为O2直接得到四个电子和H2O反应被还原为OH-; 连续四电子过程为O2先得到两个电子还原为HO2-, 再进一步还原为OH-.二电子途径是指在酸、碱性条件下O2被还原成H2O2或HO2-后不再得到电子, 在催化剂上解离分解为H2O或OH-和O2.在酸碱性条件下中间产物H2O2或HO2-的生成虽然有利于降低反应的活化能, 但是H2O2具有不稳定性, 会导致反应历程复杂化, 同时也会降低燃料电池的能量转换率和输出电压, 所以需要避免中间产物的生成.四电子反应具有更高的能量转化率和输出电压, 是燃料电池阴极氧还原的理想反应途径.氧气在碱性溶液和酸性溶液中还原的可能步骤及还原电位具体如下:
(a) 直接四电子途径
碱性溶液中:
酸性溶液中:
(b) H2O2反应途径
碱性溶液中:
第一步
第二步
可能是进一步地还原:
也可能是分解反应:
酸性溶液中:
第一步
第二步
可能是进一步地还原:
也可能是分解反应:
在实际的ORR反应过程中, 反应途径更为复杂, 可能多种反应都存在; 对于不同的催化剂, 其反应途径差别也很大; 甚至对于同一催化剂在酸碱条件下也可能分别是四电子或二电子途径.通常ORR反应途径, 对于过渡金属催化剂来说, 活性低的Au和Hg是二电子途径; Pt基催化剂酸碱条件下都是四电子途径; 非贵金属的碳基催化剂在碱性条件下多为四电子途径, 而在酸性条件下则是四电子途径和二电子途径都有可能.由于现有的质子交换膜技术已经能够大规模商业化, 而碱性离子膜离实际应用差距还很大, 所以酸性条件下的ORR催化更有意义.
2.2 氧还原电催化反应机理的基本认识
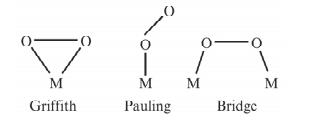
不同催化剂上氧还原的反应途径与氧气在该催化剂表面的吸附形式相关. Yeager[2]认为, 氧分子在电极上的吸附大致有三种方式, 如图 1所示.
(1) Griffiths模式
氧分子横向与催化中心作用, 氧分子中的σ轨道与中心原子中空的dzz轨道相互作用, 而中心原子中至少部分充满的dxx或dyz轨道向氧分子的π*轨道反馈, 这种较强的相互作用可以减弱O—O键, 甚至引起O2的解离吸附(双位吸附), 有利于O2的直接四电子还原反应, 在清洁Pt单晶面以及酞菁铁分子上, 氧的活化很可能按这一模式进行.
(2) Pauling模式
氧分子的一侧吸附在催化活性中心, 并通过π*轨道与中心原子中的dzz轨道相互作用, 按这种方式吸附时氧分子中只有一个原子受到较强活化, 因此有利于二电子反应, 大多数电极材料上氧的还原可能按该模式进行.
(3) Bridge(桥式)模式
如果催化活性中心原子的位置合适, 氧分子也可以同时吸附在两个中心原子上, 从而促使分子中两个氧原子同时被活化, 该模式有利于氧还原的四电子反应途径.通常在一些特殊Pt晶面以及Pt基合金上存在桥式吸附.
氧还原反应的详细反应机理和中间产物由于检测手段的限制并不清楚, 甚至是关键的第一个电子转移步骤就存在争议.以酸性条件下ORR为例, 可能的第一个电子转移步骤包括: (1) O2吸附到催化剂活性位点上后O—O键断裂生成O*(O自由基), 之后得到第一个电子; (2) O2得到一个电子吸附在催化剂活性位点, 生成超氧负离子O2-; (3) O2、一个质子和一个电子同时在催化剂活性位点上生成OOH自由基.
ORR的后续反应步骤差别也很大, 但是同样由于中间产物产生迅速、存在时间短, 反应发生在催化剂的表面微观层面, 现有的检测手段难以详细分析后续过程.目前对ORR反应机理的讨论, 通常是基于辅助实验和模拟计算进行的一系列假设[3, 4].非贵金属催化剂的ORR机理由于研究时间短, 催化中心复杂且不明确, 较Pt基贵金属更不清楚.
对于过渡金属氮碳类催化剂, 研究者们认为它们的催化机理都是类似的.以铁氮碳类催化剂(FeNC)为例[5]. FeNC催化的ORR从二价Fe开始, 在酸性条件下可能是以N4FeⅡ-OH2的形式开始. O2吸附在N4FeⅡ-OH2上同时得到一个电子和一个质子, 具体如方程式所示:
之后反应可能有两条途径.途径1, N4FeⅢ-O-OH得到一个质子和一个电子, 脱去一个水, 生成高价Fe氧自由基, 之后继续得到电子和质子, 最终又脱去一个水, 回到水合二价Fe的形式.具体步骤如下:
途径2, N4FeⅢ—O—OH再得到一个质子和一个电子生成H2O2, 而催化活性位吸附一个水后回到二价铁水合形式, 具体如下:
途径1中, O2直接得到四个质子和四个电子, 属于ORR的直接四电子途径, 在反应过程中必须生成四价Fe中间态.而途径2中O2只得到两个质子和两个电子, 属于ORR的二电子途径. Van Veen[6]认为四价Fe在FeNC催化ORR中是可能存在的, 但是必须在较高的过电位下, 这可能也解释了为什么FeNC催化剂在高过电位下为直接四电子途径, 低电位下为二电子途径.根据这一观点, 由于CoN4难以生成高价态的Co, 所以CoN4催化的ORR通常为生成H2O2的二电子途径.
Anderson和Sidik[7]认为高价态过渡金属氮碳化合物中间态难以生成, 可能还存在另外一条反应途径.生成N4FeⅡ—O—OH之后, N4FeⅡ—O—OH再得一个质子和一个电子, 但是并不解离生成H2O2, 而是O—O键断裂, 生成一个水, 之后进一步得到质子和电子, 最终O被完全还原, 具体如下面方程式:
由于该途径的中间态N4FeⅡ(—OH—OH)的结构与活性位N4FeⅡ吸附H2O2的结构是一致的, 所以理论上如果过渡金属氮碳化合物的ORR从该路径发生, 那么该催化剂应该对H2O2还原具有比较高的催化活性, 但是这与很多实验结果矛盾[8].可见, 过渡金属氮碳化合物催化ORR的机理还很模糊, 但目前有一个公认的观点是第一个电子的传递是酸性条件下ORR的速率控制步骤[6~8].
2.3 目前氧还原电催化存在的问题
目前为止, 唯一商业化的PEMFC阴阳极催化剂是碳黑负载高分散的Pt或Pt合金颗粒.以氢氧燃料电池为例, 阳极的氢气氧化非常迅速, 所以对Pt负载量的需求也比较低(<0.05 mg/cm2).所以近几十年PEMFC催化剂的研究主要集中在动力学上比较慢的阴极ORR.作为研究最多的Pt基催化剂, 对其研究已经取得了巨大进展, 通过优化Pt的负载、制备Pt基合金和优化膜电极的制备等方法已经使Pt的负载量从>1.0 mg/cm2降到了约0.4 mg/cm2, 但离实际应用还有差距[9, 10].以目前最为先进的燃料电池汽车丰田公司Mirai电动车为例, 其Pt用量高达30 g, 这与目前汽车上处理尾气的三元催化剂5 g Pt的用量还有很大差距, 只有达到或者接近这一值, PEMFC才有大规模商业化的可能.同时Pt的储量有限, 全世界储量仅有1.4万吨, 根本不足以支撑PEMFC的大规模应用.因此开出低成本、高活性、高稳定性的非贵金属ORR催化剂对推进PEMFC的大规模商业应用具有巨大的意义.
为了彻底解决PEMFC阴极贵金属催化剂的问题, 非贵金属ORR催化剂成为近年的研究热点, 也取得了巨大进展.按照催化剂材料的不同, 可以将非贵金属催化剂分为两大类[1]:一类是过渡金属氧化物、氮化物、硫化物、碳化物等; 另一类是碳基的非贵金属催化剂, 包括过渡金属氮碳类催化剂(M-N-C)和非金属掺杂碳材料.碳基的非贵金属催化剂ORR性能高, 稳定性好, 是目前最有可能替代Pt的催化剂.本文将对碳基非贵金属催化剂的研究进展进行论述.
3 过渡金属氮碳(M-N-C)催化剂
最早的关于过渡金属氮碳类催化剂的研究可以追溯到1964年, Jasinski等[11]首次发现酞菁钴显示出了一定的ORR活性.由于这类金属大环化合具有活性高、价格低廉、不存在甲醇或一氧化碳中毒, 同时可以通过调节金属配位来设计制备等优点, 研究者对其取代Pt应用于ORR催化剂寄予了厚望, 并进行了大量的研究.改变大环配体或者金属中心都可以改变催化剂的性能, N4-, N2O2, N2S2, S4等大环配体结合Cu, Co, Ni, Fe等金属都被尝试用作ORR催化剂[12, 13].在众多过渡金属大环化合物中N4-金属大环化合物, 尤其是Fe、Co的酞菁、卟啉表现出了最好的ORR催化活性.过渡金属大环化合物的性能受溶液pH的影响很大, 其酸性活性远低于碱性.即使是Fe、Co的酞菁、卟啉在酸性条件下ORR活性也远低于Pt/C[14].其次金属大环化合物的稳定性不好, 金属中心容易溶解于酸溶液中[15], 同时ORR反应过程中生成的H2O2中间体很容易氧化金属中心, 加速溶解[16].除了上面这些催化剂本身的问题, 过渡金属大环化合物, 尤其是活性最好的酞菁、卟啉, 制备过程复杂、产率低.很长一段时间, 关于过渡金属大环化合物的研究都是如何提高其催化活性和稳定性[17].以上的种种缺陷限制了金属大环类化合物类催化剂的进一步发展.
3.1 M-N-C催化剂的制备
Jahnke等[18]于1974率先报道了热处理碳载体负载的过渡金属大环化合物用于ORR催化, 之后Bagotzky[19], Wiesner和Fuhrmann[20]也进行了相似的研究.他们都认为在惰性气氛(N2, Ar等)下高温(400~1000 ℃)热处理碳载过渡金属大环化合物能得到活性和稳定性更好的催化剂.过渡金属大环化合物的热处理, 能够提高金属配合物与碳之间的结合作用[21], 同时改变了材料原来的结构, 形成了新的活性位[22].热解生成的这种新型的过渡金属氮碳类催化剂, 结构和活性组分受到热处理温度[23], 气氛[24], 碳载体[25]和前驱体影响, 且活性位以极其微小的形式(纳米簇甚至单原子)存在于催化剂内[22].所以大量的研究都是集中在研究该类催化剂的真实结构和催化活性位的本质上.
热处理过渡金属大环化合物虽然一定程度上解决了催化剂活性和稳定性的问题, 但并没有解决成本高的问题.研究者先是企图热解低成本、易合成的过渡金属大环化合物去替代酞菁、卟啉, 例如聚丙烯腈[26]、邻二氮杂菲[21]等金属大环化合物. Yeager等[26]于1989创造性地不使用金属大环化合物也制备出了具有酸性ORR活性的过渡金属氮碳类催化剂.他们将聚丙烯腈和二价的Fe盐或Co盐溶解于二甲基甲酰胺中, 将这些前体浸渍在碳载体上, 最后将混合物于800 ℃下热解得到了催化剂.这种新型的制备方法需要三种前驱体: (1) 金属前驱体; (2) 含氮前驱体; (3) 一种用来吸附或者浸渍前驱体的碳载体.根据Yeager的报道, 前驱体的混合物必须在高温条件下热解, 使N和金属元素掺入碳载体, 形成合适的催化活性中心.而之前热解金属大环化合物的方法可以看做是使用了一种同时含有氮和金属的前驱体.这种分别使用氮源和金属源的方法比起单独使用金属大环化合物, 为催化剂制备提供了更多灵活的选择, 也降低了制备的成本. Yeager认为新方法的热解过程中氮源和金属源通过自组装的方式形成了和热解金属大环化合物一样的活性中心.在他们的实验中, 聚丙烯腈高温下生成的吡啶氮与金属配位形成了C—Nx—Me活性中心.
从1989年之后, 越来越多的科研工作者投入到了使用新的氮源和金属源去制备过渡金属氮碳类催化剂, 具有代表性的工作有以下几个.
van Veen等[27]以吡咯衍生物为氮源、乙酸钴为金属源、Vulcan碳为碳载体700 ℃热解制备了Co-N-C, 制备出的催化剂具有和700 ℃热解四甲氧基苯基酞菁钴一样的ORR性能. EXAFS分析证明两者具有相同的活性中心Co-N4大环; 他们认为吡咯衍生物作为氮源, 具有和N4金属大环化合类似的结构, 有利于生成Co-N4活性中心.
Dodelet课题组[28~30]总结了一套独特的合成策略, 使用金属源和碳载体, 不加入额外的氮源, 在NH3气氛下热解制备M-N-C.研究结果表明催化剂的ORR活性受材料中微孔数量的影响, 微孔越多催化剂活性越高. NH3不仅是氮源, 在高温下它还能刻蚀碳载体增加微孔数量, 所以这种方法制备的催化剂表现出了优于直接热解金属大环化合物的性能.为了增加理想的微孔活性位, 他们设计了新的催化剂制备方法, 相关工作2009年发表在Science上[31], 如图 2.他们首先选择了含有丰富微孔的Black Pearls 2000碳黑作为碳载体, 之后用球磨的方法在微孔内填充上邻二氮菲和乙酸铁, 之后将混合物在氩气900 ℃高温热解.为了进一步提高微孔数量, 他们又对热解生成的产物在氨气气氛下进行了二次热解.最终生成的催化剂应用到H2/O2燃料电池, 催化性能已经达到商业Pt/C催化剂的水平.
同时, 美国阿拉莫斯实验室的Zelenay课题组[32]也设计出了一套催化剂的合成方法.他们将苯胺在有金属存在下聚合, 同时以高比表面积的科琴黑为碳载体高温热解制备M-N-C, 具体如图 3所示.聚苯胺与石墨碳结构上相似能够加强两者的结合, 进而有益于高温下形成活性中心.与此同时, 聚苯胺高的氮含量也有利于增加活性中心.再次, 聚苯胺的有序结构确保了氮元素均匀地分散在催化剂内.酸洗除去催化剂的金属杂质之后, 催化剂在酸性介质下的半波电位仅比Pt/C低59 mV, 单电池测试得到了最大输出功率为0.55 W/cm2, 是ORR性能最好的非贵金属催化剂之一.
M-N-C催化剂最新的研究工作主要是探讨催化活性中心、制备新类型的材料[33, 34].例如, 苏党生等[35]基于穆斯堡尔谱表征提出了Fe-N6活性中心.而魏子栋课题组[36]则对材料中存在的铁单质和碳化铁十分感兴趣.他们认为Fe-Nx是主要的活性中心, 而单质铁和碳化铁则具有辅助作用.孙世刚课题组[37]试图在Fe-N-C类催化剂中进一步掺入S元素来进一步提高Fe-N-C催化剂的活性.刘劲刚课题组[38]甚至合成了Fe, B, N, S, P五元掺杂的石墨烯, 碱性条件下该催化剂的活性已经超过了商业Pt/C.胡征等[39]通过负载MnOx在碳基材料表面诱导苯胺聚合, 从而把FeN/C活性结构高分散地制备在表面, 也是一类有特色和应用前景的制备策略.下面对M-N-C催化剂活性中心的研究进行详细的综述.
3.2 M-N-C催化剂的ORR活性中心
过渡金属大环化合物的ORR催化活性强烈地依赖于配位金属的性质. ORR发生时, O2吸附在过渡金属大环化合物中的过渡金属上.具体是O2与金属的d轨道成键, 所以O2的吸附能强烈地依赖于过渡金属d轨道的能量和电子密度.
Zagal等[40]对大量实验总结发现大环化合物中金属还原电位与ORR催化活性存在明显的正相关性, 如图 4.研究者们试图从给电子能力上给予解释.他们认为取代基的不同将会影响金属大环化合物中金属前线轨道(对金属而言即最高占据轨道)的能量, 从而影响金属的给电子能力, 进而影响电子从金属中心向O2分子的跃迁[41].简而言之, 二价金属具有更强的给电子能力, 有利于电子从活性中心向O2分子的传递; 大环化合物越容易生成二价金属(金属二价还原电位越正)则其ORR活性越高.因此有人通过调整大环化合物的取代基来控制金属的还原电位, 从而提高ORR活性[42].大环化合物中配位的过渡金属并不是都存在二价/三价的变价情况, 所以对于不同金属的大环化合物, 也有研究者们认为ORR活性和金属的d电子数有一定的相关性[43].
热解制备的M-N-C类催化剂并不能保持大环化合物结构, 最终的活性中心与热解条件和前驱体有很大关系, 所以虽然对M-N-C活性中心有大量的讨论, 但直到现在仍没有定论.在研究的早期, van Veen、Yeager和Wiesener分别提出了三种可能的活性中心.
van Veen等[44, 45]主要在500~600 ℃热解过渡金属大环化合物.在该温度下, 金属大环结构并没有完全分解, M-N4的大环化合物结构仍然完全保留, 如图 5.此时的催化剂为碳负载M-N4大环结构(M-N4/C), M-N4的大环结构为活性中心.由于热解制备的M-N-C又分为热解大环化合物和非大环化合物, 所以van Veen[27]首先验证了是否两种情况制备出的材料具有一样的活性中心.他们使用聚吡咯衍生物为氮源, 活性碳为碳载体, 乙酸钴为金属前驱体, 700 ℃高温热解.活性最高的催化剂是使用2, 5-二甲基吡咯为氮源, 钴:氮=1:10(原子个数比)的材料.在同样温度下热解活性炭负载钴卟啉获得的催化剂具有和上者一样的ORR催化活性. EXAFS的表征说明两者具有一样的Co-N4大环活性中心[46].
Yeager等[2, 26]则是在更高的热解温度(800~850 ℃)下制备M-N-C.他们认为M-N4的大环化合物在400~500 ℃开始分解.在800 ℃时, 大部分金属以金属氧化物和金属单质混合物的形式存在, 其中金属单质室温下暴露在空气中时将自发氧化.进一步的酸处理溶解其中的金属氧化物, 之后生成的金属离子与碳表面掺杂的一个或多个氮原子结合, 形成一种不同于M-N4大环的结构.最终生成M-N-C催化剂, 其中的金属-氮-碳连接的结构(M-Nx-C)被认为是真实的ORR活性中心.
Wiesener[47, 48]提出了第三种活性中心.在他看来, 与氮螯合的钴或铁在高温条件下促进螯合物的分解, 生成了氮掺杂的碳(Nx-C).掺入的氮原子被认为是真实的活性中心, 而金属元素只是中间体并不催化ORR.
以Dodelet为代表的工作者延续、发展了Yeager的观点.他们首先是用乙酸铁吸附在苝四甲酸二酐上氨气气氛下两次热解, 通过控制乙酸铁用量制备了不同铁含量的Fe-N-C催化剂[49].酸性条件下的ORR电位和铁含量的关系如图 6a所示, ORR活性先随着铁含量增多而变好, 在铁含量在2000~5000 ppm左右时达到最大, 之后略有降低, 最后保持恒定.他们首先是通过高倍XPS表征技术分别测试了不同铁含量下的铁元素和氮元素的情况, 结果如图 6b(其中铁元素的高倍XPS在铁含量2030 ppm时由于多次扫描峰信号较强).在含有铁的样品中都可以检测到Fe 2p3/2和2p1/2的峰, 这说明样品中都存在离子形式的铁; 当铁含量高于8100 ppm时, 又检测到了还原态的铁(单质铁或碳化铁).对氮的高倍XPS结果分析如图 6c, 当铁含量增加后, 吡啶氮的峰明显向高键能方向移动, 而石墨氮的峰并没有偏移; 当铁含量达到4660 ppm时, 吡啶氮的峰不再偏移, 此时吡啶氮的峰位置已经与原来相比正移了0.3 eV.他们结合这两种元素的变化情况和ORR性能的情况推断, 铁加入之后是优先与吡啶氮结合的, 吡啶氮含量有限, 之后增加铁, 铁将以单质铁或碳化铁的形式存在; 铁与吡啶氮结合的物种是主要的活性中心, 单质铁和碳化铁并没有很好的ORR催化活性.
Dodelet等[28]通过ToF-SIMES具体地揭示了铁与吡啶氮的结合形式, (1) 催化活性位包含至少两个吡啶氮, (2) 这两个吡啶氮是以类似邻二氮菲的结构与铁结合.他们在不同的温度以乙酸铁制备了Fe-N-C, 图 7显示了该催化剂不同温度下的ORR活性和ToF-SIMS测出的FeN2C4+相对丰度. ORR活性与FeN2C4+相对丰度都是随着热解温度先升高后降低, 在800 ℃左右达到最大值, 两者表现出了明显的正相关性, 所以他们认为FeN2C4+对应的物种是主要的活性中心.他们把这种活性中心定义为FeN2/C活性中心.除了FeN2C4+, ToF-SIMS也检测到了其它类型的FeNxCy+, 其中FeN4C8+被认为是Fe-N4活性中心.其它FeNxCy+都和ORR活性呈现出负的相关性.所以他们认为Fe-N4和FeN2/C两种活性中心都存在于催化剂中, 两者的相对含量与热解温度有关, 其中活性位FeN2/C的活性远高于Fe-N4活性位. Dodelet等[50]之后又将该方法制备的催化剂与Yeager等的进行了对比.他们认为两种催化剂的活性中心并不相同. Yeager提出的M-Nx-C是在催化剂酸洗过程中形成的, 而Dodelet认为的FeN2/C是在第二次热解时大量形成的, Fe-Nx-C是FeN2/C形成的前驱体.
之后Dodelet等[29, 51]又优化了Fe-N-C的制备条件, 其中选择具有大量微孔的碳材料浸渍金属盐和使用NH3气氛热解, 能够明显提高催化剂的ORR.这都揭示出微孔与Fe-N-C的活性有很大的关系.因此, 他们在FeN2/C的基础上又提出了分散在微孔中的FeN2+2/C活性中心.在FeN2+2/C结构中铁是与四个吡啶氮配位, 而FeN4结构中铁是与四个吡咯氮配位.这种结构的提出, 首先是根据之前的实验, 铁和吡啶氮直接与活性关联; 其次是这种FeN2+2/C结构确实能够稳定地存在于碳材料的微孔中[30, 52]; 最后大量的EXAFS和穆斯堡尔谱的实验都证明活性中心是金属与四个氮连接的物种[53, 54].
Dodelet等[55]也用ToF-SIMES对Co-N-C进行了表征.与Fe-N-C不同, Co-N-C中并不存在CoN2/C结构, 主要是CoN4结构.并且钴的所有结构都表现出了一样的催化活性, 并没有像Fe-N-C中有一种最好的结构形式. EXAFS的表征方法也获得了一致的结果[27, 46].
Wiesener[47]最早在1986年提出了铁只是催化形成NxC, 但是该观点一开始并未引起大家的重视.直到2004年, Maldonado和Stevenson[56]报道了酞菁铁热解制备的碳纳米纤维用于中性介质下的ORR催化, 他们认为碳纤维中的边缘缺陷和掺杂的氮是活性中心.铁在热解过程中起到催化碳纤维形成的作用, 最后以铁颗粒的形式被包裹在石墨碳内, 表面铁的含量通过XPS检测低于0.1 at%.之后, 他们又用二茂铁和吡啶化学气相沉积制备了氮掺杂的碳纤维, 并且分别在中性和碱性介质下测试其ORR催化性能.为了验证是否有表面含铁的活性中心存在, 他们对催化剂进行了CO毒化(CO极易与铁活性位形成不可逆的吸附)实验, 结果并不存在毒化现象, 他们认为这一结果验证了上面的观点.但是这种CO毒化的实验事实上可能并不能毒化FeN4的活性中心[57].值得注意的是, Stevenson的相关报道都是基于中性和碱性条件下的ORR测试, 而众所周知在该溶液条件下, 即使是不掺杂的碳材料也表现出不错的ORR催化性能.所以上面讨论中的方法并不能很好地排除金属的作用, 尤其是在更有应用意义的酸性介质中.目前大量的实验都证明铁等金属对M-N-C酸性条件下的ORR活性起了决定性作用, 而碳本身在酸性条件下却是没有活性的[58]. Popov[59, 60]和Ozkan[61]等都试图证明酸性条件下Fe-N-C类催化剂的活性中心是NxC, 并且成功的将制备的材料的ORR活性与不同形式的氮物种进行了关联.结果表明, 吡啶氮活性高但稳定性差; 石墨氮活性低但稳定性高.但是他们都缺少可靠的证据来完全排除金属的催化作用.
通常热解制备的M-N-C都含有大量的石墨层包裹的金属颗粒存在[47].一开始研究者们普遍认为这种包裹的金属相对ORR催化是不起作用的[49].但是, 最近包信和课题组[62]的研究发现碳纳米管中的金属颗粒能够降低碳纳米管表面的功函, 从而改变材料的ORR催化性能.彭峰等[63]通过简单的热解三聚氰胺和三氯化铁制备了碳化铁填充的掺氮碳纳米管.典型的样品Fe3C@NCNTs-800为100~200 nm的碳纳米管, 其表面掺入6.16 wt%氮, 而管内则以Fe3C的形式填充22.47 wt%的铁.如图 8a~b, Fe3C@NCNTs-800在酸性和碱性条件下都表现出与商业Pt/C催化剂相同的ORR催化性能, 同时优于Pt/C的耐甲醇性能和稳定性. XPS表征、毒化实验和酸洗实验揭示Fe3C@NCNTs中氮是主要的活性中心, 而管内填充铁与表面碳之间的相互作用能够促进ORR的进行, ORR催化机制如图 8c.而Li等[64]在试图制备M-N-C型催化剂时, 意外地制备出了一种石墨包裹金属颗粒的中空碳球.结果发现该材料表面的氮和铁含量都可忽略, 填充的碳化铁含量却很高, 该材料同时表现出了优异的酸性和碱性ORR催化性能.他们认为填充的碳化铁虽然不与O2接触, 但与表面的碳存在协同效应, 从而表现出ORR催化活性.
4 非金属掺杂碳催化剂
碳材料本身在碱性条件下就具有一定的ORR性能.特别是碳的本征缺陷在碱性电解液中被发现有很好的ORR活性, 已成为设计新型催化剂和探索活性结构研究中必须考虑的重要因素[65, 66].彭峰课题组[67, 68]通过实验和理论计算发现碳材料中的边缘性缺陷在碱性条件下具有不错的ORR性能, 是碳材料主要的活性位, 但酸性条件下碳材料本身并没有ORR性能.掺杂碳材料是指一些非金属元素(常见的有氮、硼、磷、硫等)取代碳材料石墨晶格中的某些sp2杂化的碳原子形成的碳材料.对碳材料的掺杂能够改变材料的电子特性, 可以得到ORR催化活性和稳定性都比较理想的非贵金属催化剂.这些材料按形貌可以分为碳纳米管、石墨烯、介孔碳等.
4.1 氮掺杂碳纳米管
早在1999年戴黎明等[69]就报道了以酞菁铁为前驱体, 石英片为载体, 在Ar和H2气氛下, 通过化学气相沉积法(CVD)800~1000 ℃制备出了掺氮的碳纳米管阵列(VA-NCNTs).但直到2009年戴黎明等[70]在Science报道VA-NCNTs作为碱性ORR催化剂表现出了比Pt/C催化剂更大的电流密度, 才揭开了掺杂碳材料在ORR领域研究的序幕.由于用该CVD法制备出的VA-NCNTs不可避免地含有金属残留, 他们首先是通过电化学氧化的方法除去金属杂质.如图 9所示, 处理前CV曲线中有铁的氧化还原峰, 处理后对应的峰消失, 而VA-NCNTs的ORR活性并没有受到影响, 甚至略有提高.对VA-NCNTs, 不掺氮的非阵列碳纳米管(NA-CCNTs)和Pt/C进行旋转环盘测试, VA-NCNTs的ORR起始电位和极限电流都远高于NA-CCNTs, 甚至略优于Pt/C.同时VA-NCNTs和Pt/C都是4e反应途径, 而NA-CCNTs则是2e反应途径.这说明氮原子的掺入不仅提高了碳纳米管的ORR催化活性, 也改变了碳纳米管的催化机理.
VA-NCNTs的优异ORR催化性能瞬间引燃了人们的研究热情, 大量关于掺氮碳纳米管的工作见诸报道[71~74].例如, 陈忠伟等[71]以乙二胺和吡啶为前驱体也制备出了碱性ORR性能可比Pt/C的NCNT.同时他们发现以吡啶为前驱体制备的NCNT具有更高的氮含量和更多的缺陷, 从而具有更高的ORR活性.进一步通过调整吡啶的用量[72], 他们发现氮含量与NCNT的ORR活性有直接的关系, 但是高的氮含量只能增加材料的ORR电流, 对ORR的起始电位和半波电位影响不大.孙学良课题组[73]对NCNT酸性和碱性介质中ORR做了详细的探讨, 他们总结如下:铁促进催化活性中心的形成, 但铁本身不是活性中心; 氮含量高ORR活性高; 酸性条件下NCNT的催化活性不高, 且没有极限电流平台. Tammeveski等[74]也发现NCNT在酸性条件下ORR活性远低于碱性条件下的活性.
绝大多数NCNT制备都使用金属催化剂, 而根据报道[75, 76]即使是痕量的金属残留也可能对ORR催化有影响, 所以很多研究者试图不使用金属制备出NCNT.例如, 戴黎明等[77]提出了不使用任何金属催化剂, 以离子溅射SiO2制备单壁NCNTs的方法, 具体制备如图 10a所示.将这种单壁NCNTs和同样条件制备的单壁CNTs进行酸性ORR测试, 如图 10b. CNTs在酸性条件下几乎没有ORR活性, 而NCNTs则表现出了一定的ORR活性. K-L方程计算得出NCNTs催化ORR的电子转移数是3.52~3.92, 说明主要是4e反应途径.连续两天的循环伏安测试发现, NCNTs的ORR活性几乎没有变化.以上结果证明氮掺杂确实能够提高碳材料酸性条件下的ORR活性.
4.2 氮掺杂石墨烯
石墨烯是一种单层的二维石墨晶体, 具有类似甚至更优于碳纳米管的性能, 研究者们很自然地也想到用掺杂石墨烯作为ORR催化剂.为了解决掺氮石墨烯难以大规模、低成本制备的问题, 大量的工作集中在掺氮石墨烯的制备上. CVD法是研究最多的原位制备掺氮石墨烯的方法.戴黎明等[78]在氨气气氛下, 以甲烷为碳源CVD法首次制备了氮掺杂的石墨烯, 和VA-NCNTs类似, 其碱性条件下的ORR性能十分优异. AFM、TEM表征证明制备出的确实是少层的石墨烯, XPS证明表面氮掺杂量为4 at%, 和VA-NCNTs类似.掺氮石墨烯碱性条件下的ORR活性如图 11, 掺氮之后石墨烯起始电位和极限电流都有极大提高, 尤其是极限电流明显高于Pt/C.与Pt/C相比, 掺氮石墨烯还表现出了优异的耐甲醇性和稳定性.
石墨烯的边缘被证明比基面更易被活化, 所以研究者们也试图用边缘功能化的方法制备掺氮石墨烯[79]. Baek等[80]用对氨基苯甲酸在磷酸/五氧化二磷溶液中将石墨边缘氨基苯甲酰化, 最后将该石墨烯高温热解制备出了掺氮石墨烯.测试显示, 掺氮石墨烯碱性条件下具有一定的催化活性, 电子转移数为3左右.但是由于制备出的掺氮石墨烯掺氮量仅有1.73 at%, 所以其ORR性能并不如戴黎明课题组制备的掺氮石墨烯.
除了原位制备掺氮石墨烯, 另一种使用较多的方法是制备好石墨烯之后通过后处理掺入氮.例如, 林跃河课题组[81]通过N等离子溅射处理石墨烯制备了氮掺杂的石墨烯. XPS测试表明, 掺氮石墨烯氮含量为8.5 at%, 其中吡啶含量26.5%, 吡咯氮含量48.5%, 石墨氮含量24%.他们认为, 高含量的吡啶氮和石墨氮被认为是掺氮石墨烯高催化活性的原因.后处理制备掺氮石墨烯的一种更简单方法是, 将石墨烯在氨气气氛或者是将石墨烯与含氮物种混合热解来得到掺氮石墨烯[82].夏兴华等[83]将氧化石墨烯和三聚氰胺混合, 然后在700~1000 ℃热处理, 制备了掺氮石墨烯.三聚氰胺在高温下先热解聚合生成石墨型氮化碳, 氧化石墨烯上的含氧官能团脱去, 为氮的掺入提供位点, 最后氮化碳分解出的氮掺入这些位点.碱性条件下CV测试证明氮的掺入提高了石墨烯的ORR起始电位和电流.刘美林等[84]使用类似的方法将三聚氰胺替换为尿素也制得了碱性条件下活性不错的掺氮石墨烯. Ruoff等[85]对比了氨气气氛热解和使用固体聚吡咯或聚苯胺热解制备掺氮石墨烯的差异.氨气气氛有利于吡啶氮和石墨氮的形成, 聚吡咯和聚苯胺则有利于吡咯氮和吡啶氮的形成.吡啶氮含量高起始电位好, 石墨氮含量高极限电流大.
4.3 其它氮掺杂碳材料
碳纳米管和石墨烯都是特殊形貌的sp2杂化石墨碳, 其它形貌的石墨碳掺入氮之后也能改变碳的电子结构, 可以用作ORR电催化[86, 87].冯新亮等[88]使用N, N-双(2, 6-二甲苯)苝-3, 4, 9, 10-四羧酸二酰亚胺为碳源, 含氮的芳香染料为氮源, SBA-15为硬模板, 不同热解温度下制备出了氮掺杂的有序介孔碳.热解温度提高, 介孔碳的氮含量降低, 但其ORR活性和4e反应途径选择性都提高.活性最高的900 ℃制备的介孔碳具有高达510 cm2/g的比表面积和2.7 wt%的氮含量, 其碱性条件的起始电位和Pt/C一致, 极限电流远高于Pt/C.他们认为高的热解温度有利于提高介孔碳的石墨化程度和石墨氮比例.使用硬模板制备的有序介孔碳虽然比表面积很高, 但是其方法过于复杂且氮掺杂量也不高.杨文等[89]考虑到这一点, 使用沸点高的离子液体和核酸碱基制备出了比表面积高达1500 cm2/g, 氮含量高达12 wt%的氮掺杂介孔碳材料, 其碱性条件的ORR活性可比Pt/C.朱珍平等[90]则更为简单地直接热解价格低廉的2, 4, 6-三硝基苯酚制备了氮掺杂的中空纳米碳颗粒.除了介孔碳, 氮掺杂的碳球也是一类研究比较多的碳材料[91].
后处理掺氮同样用在其它碳材料领域. Popov[92]在碳黑表面包覆一层含氮的聚合物, 再通过高温碳化的方式进行掺氮.对比了不同的氧化处理、含氮前驱体、热解温度等, 他们发现脂肪酸氧化的碳黑包覆聚硒脲醛800 ℃热解的样品具有最好的ORR活性, 在酸性溶液中表现出了极限电流平台.表面的吡啶氮和石墨氮, 特别是吡啶氮被认为是活性中心. Schuhmann等[93]也使用类似的方法制备掺氮碳材料, 他们主要尝试了不同的碳前驱体和聚合物前驱体, 最后发现聚吡咯包裹CNTs制备出的材料具有最好的活性.他们还发现不同的掺氮碳中, (Npyridinic+Nquaternar)/Ntotal与材料的ORR起始电位表现出了线性的正相关性, 也就是说吡啶氮和石墨氮是ORR活性中心.
4.4 其它非金属元素掺杂碳材料
除了氮掺杂以外, 其它的非金属元素如硼、磷、硫等掺杂碳材料也表现出了不错的ORR催化性能.硼是研究较早的一种掺杂元素, 胡征等[94]选择苯、三苯基硼烷和二茂铁为原料, 采用CVD法可控制备了硼含量0~2.24 at%的掺硼碳纳米管(BCNTs).碱性条件下的CV测试表明, BCNTs的ORR活性随掺硼量的增加而增加; RRDE测试证明, BCNTs催化的ORR是2e反应途径; 和NCNTs一样, BCNTs也表现出了很好的稳定性和抗中毒性能.但是BCNTs的ORR催化活性远低于Pt/C, 甚至比NCNTs也差.夏兴华等[95]则是简单地将氧化石墨和氧化硼混合热解, 制备了硼掺杂的石墨烯, 和BCNTs一样, 硼掺杂的石墨烯在碱性条件下也表现出了一定的ORR催化活性.
磷和氮是同族元素, 只差一个周期, 具有很多类似的性质, 所以研究者们也试图制备掺磷碳材料用于ORR.最具代表性的研究工作是彭峰课题组[96]通过热解甲苯和三苯基磷制备出了无任何金属杂质的掺磷石墨片.碱性条件下, 掺磷石墨的ORR起始电位和Pt/C相比还有一定的差距, 但其ORR极限电流略高于Pt/C, 同时还具有优异的抗甲醇中毒和长时间的耐久性.由于制备过程中并未使用任何金属前驱物, 所以认为掺磷石墨的高ORR活性是磷的掺入引起的, 排除了金属的作用.之后他们还制备出了掺磷的碳纳米球[97]和碳纳米管[98], ORR活性和掺磷石墨类似.
直接将硫掺入石墨的晶格比起硼、氮更加困难, 因为硫的原子半径较大[99].黄少铭课题组[100]利用氧化石墨表面丰富的官能团作为硫掺入的位点, 将氧化石墨和苯硫醚直接高温热解制备出了掺硫石墨烯.掺硫石墨烯在碱性条件也表现出了不错的ORR催化活性, 好的稳定性和耐甲醇中毒性能. Pumera等[101]则是通过CS2、SO2或H2S气氛下热剥离氧化石墨制备出掺硫的石墨烯.根据他们的报道, 热解温度是硫掺杂量的主要控制因素, 850 ℃制备的样品硫含量最高(2 at%), 其碱性条件下的ORR活性也最高.胡征课题组[102]以噻吩和苯为硫源和碳源、MgO为模板, 通过化学气相沉积法制得了硫掺杂碳纳米笼.通过控制噻吩的用量, 他们得到了不同硫掺杂量的碳材料. ORR性能测试结果表明, 硫掺入量0.84 at%时催化剂的性能最好, 硫掺入量过高时, 催化剂的ORR性能反而变差.
除了主流的氮、硼、硫、磷掺杂, 掺硒[103]、硅[104]、氟[105]等碳材料也有人研究.这些掺杂材料通常制备起来比较困难, 掺杂量很低, ORR催化活性也并不高, 所以研究较少.总的来说氮掺杂的碳材料具有远高于其它元素掺杂碳材料ORR催化活性, 尤其是在酸性条件下, 只有掺氮碳材料表现出了一定的ORR活性.但是人们在研究其它元素与氮共掺杂碳材料时却惊喜地发现, 共掺杂碳材料具有比单独氮掺杂碳材料更高的ORR活性, 近年共掺杂碳材料也成为了非金属ORR催化领域的研究热点.戴黎明课题组[106]最早用三聚氰胺和二硼酸盐, 以Ni/Fe纳米颗粒作为催化剂, 制备出了阵列硼氮共掺杂的碳纳米管用于碱性ORR催化.之后他们[107]又混合二茂铁、吡啶和三苯基磷通过CVD法制备了磷、氮共掺杂的阵列碳纳米管(PN-ACNT).碱性介质下PN-ACNT具有比单独氮或磷掺杂碳纳米管更好的ORR催化活性, 其活性甚至高于Pt/C.他们认为共掺杂碳纳米管的高活性来源于氮和其它杂元素存在协同效应.也有学者认为共掺杂碳材料不仅仅有简单的协同效应, 更多是与其构型有关.胡征课题组[108]和乔世璋课题组[109]分别以硼氮共掺杂碳管和石墨烯为研究对象, 发现不同的硼氮掺杂构型, 即硼氮处于分离或结合状态, 有着截然不同的ORR催化活性, 因此共掺杂碳材料的研究更需要关注共掺杂的构型. Woo等[110]则在含有金属催化剂的条件下热解双氰胺和磷酸制备了高比表面积的磷氮共掺杂介孔碳.值得注意的是该材料在酸性条件下也表现出了很好的ORR活性和4e反应选择性, 但该文章并未排除金属的可能作用.彭峰课题组[111]在制备好NCNTs的基础上通过硫磺高温后处理的方式制备了氮硫共掺杂碳纳米管(SN-CNTs).如图 12所示, SN-CNTs保持了NCNTs的形貌, 在酸性和碱性条件下都表现出了很好的ORR催化活性和稳定性. XPS和EELS等表征都没有检测到任何金属元素, 所以他们认为金属的作用可以排除.
4.5 掺杂碳材料催化氧还原的活性中心
掺杂能够显著提高碳材料的ORR活性被发现后, 研究者马上就对掺杂的作用机制进行了研究.其中最为典型也是活性最好的氮掺杂碳材料, 其作用机制也是研究最多的.一般而言, 氮原子掺入石墨碳的晶格后将会扰乱均匀的π-电子分布, 从而改变碳材料表面的电子状态[112].扫描隧道显微镜和理论计算证明, 氮的掺入能够提高碳材料的费米能级, 有利于电子到达导带, 使碳材料表现出一定的金属性质[112].费米能级附近态密度的增加有利于在低电压下电子的发射[113], 从而提高碳材料的电子传导能力和ORR催化能力[114]. Wiggins-Camacho等[115]运用DFT理论计算发现氮的掺入能够增加碳材料体相的电子传递能力、功函以及费米能级附近的态密度.氮的掺入量被认为是获得高ORR活性的最重要因素[116].陈忠伟等[71]制备了氮含量不同的NCNTs, 并证实了高的氮含量和多的缺陷有利于酸性ORR催化.
如图 13, 掺杂的氮可能以多种形式存在于碳材料中, 如吡啶氮、吡咯氮、石墨氮或季氮、腈、氨基等.到底是哪种形式的氮具有最好的ORR催化活性, 对优化催化剂的制备具有重要的意义.目前可以确定的是, 吡啶氮、吡咯氮和石墨氮是主要的掺杂形式, 具有最好的ORR活性, 但是三者到底哪个活性最高却存在争议.
 图13
(a) 吡啶氮, (b)吡咯氮, (c)石墨氮, (d)腈和(e)氨基
Figure13.
(a) pyridine-like N, (b) pyrrole-type N, (c) graphite-like N, (d) nitrile and (e) amidogen
图13
(a) 吡啶氮, (b)吡咯氮, (c)石墨氮, (d)腈和(e)氨基
Figure13.
(a) pyridine-like N, (b) pyrrole-type N, (c) graphite-like N, (d) nitrile and (e) amidogen
认为吡啶氮是掺氮碳材料中最主要的活性中心的相关工作是最多的[77, 78, 117~119]. Kundu等[117]在不同温度下热解乙腈和钴催化剂制备了不同氮含量和氮形式的NCNTs. 550 ℃制备的样品具有最高的吡啶氮含量, 同时也表现出了最高的ORR活性.但是该方法制备NCNTs使用了过渡金属钴催化剂, 可能有Co-N4活性中心存在.为了彻底地排除过渡金属的作用, Rao等[118]以苯乙炔、4-乙烯基吡啶、3-甲基吡咯、2-甲基-1-乙烯基咪唑、邻二氮杂苯的聚合物为前驱体, 氧化铝为硬模板制备了不同氮含量NCNTs.不同NCNTs的XPS氮分峰如图 14a, 邻二氮杂苯制备的NCNTs具有最高的总氮量(主要是石墨氮), 而2-甲基-1-乙烯基咪唑制备的NCNTs吡啶氮含量最高.如图 14b, NCNTs酸性条件下的ORR活性并不是随着总氮含量增加而增加, 而是随着吡啶氮含量的增加而增加. Wiggins-Camacho等[116]的工作发现吡啶氮在ORR催化中可能还起到了促进过氧化氢分解的作用.
最近, 也有研究者特意合成一些含有丰富特定氮掺杂形式的碳材料[85, 119]. Yasuda等[119]分别以久洛尼定和吡啶为含氮前驱体, 在石墨烯表面负载聚合物, 然后热解的方式制备掺氮石墨烯.以久洛尼定制备的掺氮石墨烯石墨氮占总氮含量高达80 at%, 而吡啶制备的掺氮石墨烯吡啶氮占总氮含量高达90 at%. ORR测试发现石墨氮为主的掺氮石墨烯催化的是2e反应途径, 而吡啶氮为主的掺氮石墨烯催化的是4e和2e混合的反应途径.这一结果证明吡啶氮对ORR的4e选择性优于石墨氮.
也有很多研究者认为石墨氮是其中活性最高的氮掺杂形式[82, 88]. Wagberg等[120]通过热处理NCNTs的方式控制NCNTs的氮掺杂形式.他们主要分析了吡咯氮(Pyrrolic-N)、吡啶氮(Pyridinic-N)、中心石墨氮(N-Qcentre)和谷位石墨氮(N-Qvalley)随着温度的变化情况以及和ORR催化活性的关系.如图 15, 他们发现随着热解温度从500 ℃升高到1000 ℃, 吡咯氮向吡啶氮转化, 而吡啶氮向石墨氮转化(其中N-Qvalley是其中主要的氮掺杂形式). ORR活性则是随着热解温度升高而变好, 这说明石墨氮特别是N-Qvalley可能是NCNTs主要的活性中心.夏兴华等[83]通过不同制备方法, 制备出不同石墨氮含量的掺氮石墨烯, 也发现石墨氮可能是主要的ORR催化活性中心.
在吡啶氮、吡咯氮和石墨氮中, 吡啶氮和吡咯氮是平面结构, 而石墨氮是三维结构.三维结构的石墨氮可能会破坏π-π共轭作用, 导致掺杂碳材料电导性和催化活性降低[121].为了提高平面的吡啶氮和吡咯氮的含量, 魏子栋课题组[122]通过蒙脱土作为硬模板制备掺氮的石墨烯.制备机理如图 16, 蒙脱土是厚度1 nm以内的层状结构, 将苯胺浸入层中聚合生成聚苯胺, 高温碳化酸洗除去蒙脱土后得到掺氮石墨烯.由于蒙脱土的限制作用, 制备出的掺氮石墨烯平面氮含量高达90.27% (56.30%吡咯氮, 33.97%吡啶氮).该方法制备出的掺氮石墨烯在酸性条件下的ORR活性极其优异, 半波电位仅比Pt/C低60 mV, 远高于之前报道过的平面氮含量低的掺氮碳材料.这说明平面氮, 特别是吡咯氮是掺氮石墨烯中主要的活性中心.
除了氮的掺杂量和掺杂形式, 掺氮碳材料的微观结构对ORR活性也有影响.陈忠伟等[123]分别用二茂铁和酞菁铁催化吡啶制备了NCNTs, 并用于ORR催化.两种方法制备的NCNTs具有接近的氮掺杂量(2.4 at%)和氮掺杂形式(石墨氮为主), 但是它们的形貌不同, 如图 17, 二茂铁制备的NCNTs管壁不光滑、竹节状明显、具有更多的缺陷.电化学性能测试表明, 二茂铁制备的NCNTs表现出了更高的ORR催化活性, 所以他们认为缺陷明显有利于ORR催化. Wong等[124]使用不同含氮前驱体(苯胺、二乙胺和乙二胺)CVD法制备了不同缺陷度的NCNTs, 也得到了同样的结论.
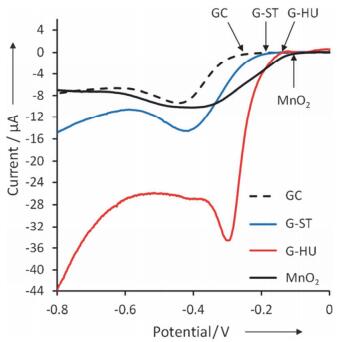
实际上绝大多数的掺杂碳材料都含有痕量甚至不低的金属.例如, 掺杂碳纳米管制备过程中普遍使用了铁、镍等催化剂[70, 125]; 基于Hummers法制备石墨烯引入了大量的金属氧化物[126]; 很多掺杂介孔碳材料也使用了金属催化剂[110].因此很多研究者对这些掺杂碳材料的非金属催化活性中心提出了质疑[75, 76]. Pumera等[76]分别选择了用Hummers法(G-HU)和Staudenmaier法(G-ST)制备的石墨烯为研究对象.原子吸收检测发现G-HU含有较多的锰残留, 而G-ST则含有较多的铁残留.如图 18, 碱性条件下的ORR的极化曲线测试发现锰含量高的G-HU具有和MnO2接近的起始还原电位, G-ST的起始还原电位却远差于前者.所以他们认为MnO2对石墨烯的ORR催化活性影响是很大的. Schuhmann等[75]则是在不含金属的掺杂碳材料上负载痕量的金属(0.05% Fe), 发现掺杂碳材料的ORR活性明显提高, ORR反应途径也由2e变为4e.戴宏杰等[127]也发现即使是痕量的铁(0.24 at%)形成Fe-N-C也会让掺杂碳材料在酸性和碱性条件下表现出很好的ORR催化活性.但也有部分研究工作通过在制备中特意避免过渡金属明确证实了无金属的碳材料至少在碱性电解液中可以表现出很好的ORR催化活性.胡征课题组[128]以MgO作为硬模板, 吡啶为碳源, 通过化学气相沉积法制备了不含过渡金属的掺氮碳纳米笼.掺氮碳纳米笼在碱性电解液中的循环伏安测试显示, 其ORR峰电流值已经大于商业Pt/C催化剂.
5 碳基非贵金属氧还原催化剂在质子交换膜燃料电池中的应用
关于碳基非贵金属氧还原催化剂的研究主要都是在半电池中进行, 而将该催化剂实际应用到质子交换膜燃料电池单电池的报道相对较少、存在的问题也更多.碳基催化剂要想取代Pt基催化剂应用到PEMFC至少需要满足以下几点[129]: (1) 在低功率运行时能够提供和Pt一样的功率(主要取决于催化剂的ORR反应速率); (2) 在实际工作情况下提供和Pt一样的功率(如电动车); (3) 在有效功率下稳定运行1000 h.对于碳基催化剂PEMFC实际应用的研究主要是在H2/O2和H2/air电池中, 下面将从以下几个部分进行简单的综述: (1) 电池组装和运行条件的优化; (2) 单电池的功率; (3) 电池的稳定性; (4) 与Pt/C催化剂的对比.
5.1 电池的组装和运行条件
PEMFC的单电池主要是由电催化剂、燃料、膜电极和双极板组成.对碳基催化剂来说, 研究较多的燃料是H2; 双极板主要采用石墨极板、金属极板和复合材料极板; 质子交换膜燃料则多采用nafion117膜; 以上部分使用的材料和应用条件与Pt/C催化剂差不多, 研究结果都比较清楚、技术也较为成熟.对于碳基催化剂在电池组装和运行条件上近期研究较多的是催化剂用量、燃料电池温度、O2的背压、膜电极厚度等. H2/air和H2/O2电池的组装、运行条件大多一致, 所以下面以H2/O2燃料电池为例.
不同文献中, 即使是同一类碳基催化剂, 在单电池中的催化剂负载量也是不同的, 负载量使用范围是0.35~10 mg/cm2[130].催化剂负载量对单电池性能有影响, 但并不是负载量越高单电池的性能就越高, 通常是有一个最合适的负载量, 大多数碳基非贵金属催化剂这一值是3.8~4.1 mg/cm2[131].在同样的负载量时, 不同的碳基催化剂的单电池性能也不同, 受到催化剂本身催化性能、微观结构以及最后得到膜电极传质情况等共同影响.由于膜电极制备是在质子交换膜上喷涂催化剂层, 催化剂负载量将影响催化剂层的厚度, 而催化剂层厚度则对传质有影响. 1 mg/cm2催化剂负载量的催化剂层厚度是20~25 μm.碳基催化剂的常用负载量3.8~4.1 mg/cm2下, 催化剂层的厚度将达到80~100 μm.而Pt/C催化剂的负载量通常比碳基催化剂低一个数量级, 对应的催化剂层的厚度为10 μm以下[132].碳基催化剂的单位质量活性远低于Pt/C, 只有使用如此高的负载量, 碳基催化剂在低功率运行时才具有和Pt/C差不多的性能.但是过厚的催化剂层严重地阻碍了燃料电池的传质, 导致非贵金属催化剂阴极在电流大于0.1 A/cm2时电池功率远低于Pt/C.只有至少提高10倍单位质量催化剂活性, 才能解决非贵金属催化剂的催化层过厚问题.
碳基催化剂组装的单电池运行温度范围是25~90 ℃. 80 ℃是最常用的H2/O2 PEMFC测试温度, 在该温度下测试结果较好, 温度较低时功率明显降低, 温度提高功率也不能明显提高[133].碳基催化剂单电池运行时O2背压则通常是150~200 kPa[59].较低的背压不一定导致单电池性能明显下降, 如碳基催化剂文献报道的最高功率之一就是在较低的背压(100~130 kPa)下测得的[37], 但背压过低(0~30 kPa)时电池功率明显下降[134].碳基催化剂使用的质子交换膜厚度范围是25~183 μm, 膜厚度越小则单电池的功率越大, 同时稳定性降低.文献报道的性能最好的碳基催化剂单电池都是在较薄的质子交换膜(25 μm)中测得的[135], 综合考虑稳定性, 大多数文献中使用的是厚度为50~100 μm[32]的质子交换膜.
5.2 单电池的功率
不同碳基催化剂的H2/O2单电池最大功率主要受其活性位类型影响.按照活性位和制备方法的差异, 我们把碳基催化剂分成四类对它们的单电池最高功率进行简单的介绍.
第一类是M-N-C催化剂, 这一类催化剂是由氮源、碳源和金属源热解制得, 研究者们认为活性中心是MNx.这类催化剂的单电池功率在不同文献报道中差别很大, 既有高达1.03 W/cm2的[135], 也有仅仅0.05 W/cm2的[136], 但大多数报道的功率是在0.05~0.65 W/cm2这一范围.以Atanassov等[137]的Fe-8CBDZ-DHT-NH3为例, 该催化剂是以甲基-苯骈咪唑-2-氨基甲酸甲酯和硝酸铁为前驱体, 二氧化硅为硬模板, N2气氛800 ℃热解, 酸洗之后NH3气氛二次热解制得. Fe-8CBDZ-DHT-NH3中铁含量为0.1~0.3 at.%, 氮含量为7.7 at.%, 热解过程中形成了FeNx活性中心, 用于单电池得到电池功率为0.56 W/cm2.碳基催化剂的单电池目前测得的最高功率也是这类催化剂得到的, 为S掺杂的Fe-N-C[37], 该S掺杂的Fe-N-C(Fe/N/C-SCN)是孙世刚课题组通过高温热解聚苯胺、磺化碳黑和硫氰化铁制得的. Fe/N/C-SCN组装电池的峰值功率达到了1.03 W/cm2.如图 19, 在电流密度低于1 A·cm-2时, Fe/N/C-SCN的性能可比商业Pt/C; 但在电流密度大于1 A·cm-2时(此时传质影响大于电化学动力学), Fe/N/C-SCN的性能远低于商业Pt/C.这是由于Pt/C催化层厚度为10 μm, Fe/N/C-SCN催化层厚度为100 μm, Fe/N/C-SCN催化层的传质远差于Pt/C.作者认为Fe/N/C-SCN的活性位和一般Fe-N-C催化剂一样, 但S元素的掺杂和高的比表面积能明显提高其单电池性能.
第二类是含有金属的氮掺杂碳材料.金属主要是Co和Fe, 在掺氮碳材料的制备过程中起了催化作用, 但并没有形成MNx的活性位, 主要活性位是NxC.这类催化剂的单电池性能总体比M-N-C催化剂的略差, 性能最好的催化剂的峰值功率为0.63 W/cm2[138], 其中含Fe的掺氮碳材料优于含Co的.典型催化剂是Popov等[139]制备的NMCC-SiO2-800-3, 他们使用乙二胺、硝酸钴、硝酸铁作为前体, 二氧化硅为硬模板, 高温Ar热解酸洗制备催化剂.该催化剂的表面氮含量为5.93 at.%, 而铁元素无法在表面检测到, 主要以碳包覆铁的形式存在, 组装单电池峰值功率为0.45 W/cm2.
第三类是不含有金属的氮掺杂碳材料.通常这些不含金属的氮掺杂碳材料在制备过程中完全不使用金属前驱体, 所以只含有NxC活性位.与含有金属的掺氮碳材料对比, 这一类催化剂的单电池功率更低.研究者们认为, 首先, 对于含有金属的掺氮碳材料, 金属催化了更多的NxC活性位形成, 其次微量的金属残余可能形成了少量的MNx活性位.典型催化剂UF-C[152]制备如下:先酸洗科琴黑除去金属杂质, 之后HNO3氧化科琴黑; 将处理后的科琴黑、脲醛和三聚氰胺混合后高温热解得到样品. XPS检测UF-C表面氮含量为2.2 wt%; ICP-MS检测样品中不含铁和钴元素; UF-C组装单电池的峰值功率仅为0.18 W/cm2.戴黎明课题组[140]制备了完全不含金属的掺氮碳纳米管/石墨烯复合材料, 用于单电池, 得到最大功率只有0.28 W/cm2.他们注意到该复合材料虽然单电池功率还不如一般的催化剂, 但单电池运行的稳定性明显优于Fe-N-C, 100 h的稳定性测试后电池功率基本没有下降.
最后一类是最新研究的金属填充掺氮碳材料, 这类催化剂的单电池功率最小.这一类催化剂制备过程中使用了大量金属前驱体, 但并没有形成MNx活性位, 主要是形成了碳包覆金属的结构, NxC是主要的活性位, 金属填充起了协同作用.典型样品如包信和课题组[62]制备的豆荚状铁填充掺氮碳纳米管, 该催化剂的单电池最大功率仅有0.05 W/cm2.
综上, 对不同碳基催化剂的H2/O2单电池最大功率、结构和组成进行简单的总结分析, 我们发现以下两点.首先, 高的比表面积和微孔数量有利于催化剂的单电池峰值功率.这一方面是由于高比表面积和微孔数量有利于传质, 另一方面是由于微孔处有利于形成FeN4活性位.其次, 氮和金属含量低时, 催化剂的单电池峰值功率一般较低; 但是在氮和金属含量足够时, 催化剂的单电池峰值功率不再受含量影响.
H2/air单电池中, 不同碳基催化剂的最大功率同样受到其活性位类型影响, 规律和H2/O2单电池一样.但碳基催化剂H2/air单电池的最大功率明显下降, 性能最好的碳基催化剂在H2/air单电池中的最大功率不到H2/O2单电池的一半[135].相对应的Pt/C催化剂在将O2改为空气作为还原气氛时, 功率密度从1.36下降到0.96 W/cm2[141].这是由于改用空气之后, 对传质的要求更高, 而非贵金属催化剂的催化层厚度过厚不利于传质.非贵金属ORR催化剂应用到H2/air PEMFC文献较少, 所以其最适宜的测试条件还没有定论, 但其测试条件和结果大部分和H2/O2类似, 因此可以根据H2/O2的结果进行一定的推测.
5.3 单电池的稳定性
除了运行功率, PEMFC的稳定性也是它的一个重要指标.美国能源部对Pt基催化剂的稳定性要求是能够在实际的电池中稳定运行至少5000 h, 而对成本较低的碳基催化剂通常把要求放低到至少1000 h[133].半电池的稳定性测试一般是通过多次循环的循环伏安法或旋转圆盘(RDE)测试.而单电池的稳定性测试则有多种方法, 首先是电势循环法, 包括启动/关闭循环测试和负载循环测试[142]; 其次是恒电位测试法, 主要是测试制定运行电位下电池输出功率或电流的变化.启动/关闭循环测试是以0.5 V·s-1的扫描速率在1.0~1.5 V(vs RHE)电势范围内进行单电池的循环测试, 这一测试主要是考察催化剂中碳载体的抗电氧化能力[143].负载循环测试是以类似脉冲周期的方式在0.6~1.0 V(vs RHE)电势范围内进行单电池的循环测试, 模拟PEMFC在运行时的情况, 主要考察催化剂在运行时的溶解、失活、中毒问题[142].碳基催化剂中碳本身属于活性中心的一部分, 很容易在高电位下发生氧化失活[144].所以这两种稳定性测试方法都不适用碳基催化剂的稳定性测试.碳基催化剂单电池的稳定性测试一般使用的是较低电位(0.4 V vs RHE左右)下的恒电位测试法.
大多数碳基催化剂在进行半电池的RDE测试时都表现出了不错的稳定性, 但在更符合实际应用的单电池测试时稳定性大多较差.以刘迪佳等[145]制备的Fe-N-C催化剂FeIM/ZIF-8为例, 如图 20a、20b, FeIM/ZIF-8催化剂经过10500圈Ar气氛或2000圈O2气氛的RDE测试后, ORR活性几乎没有变化.而图 20c显示, FeIM/ZIF-8组装的H2/O2单电池在0.5 V电位下的输出电流仅仅经过100 h就从0.2 A/cm2下降到0.05 A/cm2, 也就是输出功率下降到初始的1/3.廖世军课题组[146]和Joo课题组[147]等制备的Fe-N-C和Co-N-C催化剂也得到了类似的结果.这一结果说明, 对碳基非贵金属催化剂, 用半电池的RDE测试来确定催化剂的稳定性是不可取的, 只有用单电池测试才能得到真实的稳定性情况.下面将对碳基催化剂在H2/O2和H2/air燃料电池中的恒电位稳定性测试结果进行简单综述.
不同碳基催化剂的H2/O2单电池稳定性主要受到其活性位类型和催化剂形貌结构的影响.按照活性位和制备方法的差异, 我们把碳基催化剂分成三类对它们的H2/O2单电池稳定性进行简单介绍.
第一类, 使用含碳前驱体, 如MOF材料, 二次退火制备出的FeNC催化剂[31, 148].这一类催化剂比表面积大, 且含有大量的微孔, FeNx是主要的活性位.该类催化剂通常电池能够输出的最高功率很高, 通常可达到0.5 W/cm2以上, 但活性随着运行时间增加迅速下降.该类催化剂通常具有极高的比表面积(700~1300 m2/g), 和大量的微孔.稳定性测试的结果表明样品的微孔比例越高, 初始性能越高, 但是稳定性越差.这很可能是该类催化剂的活性位FeNx存在于微孔中, 这种活性位的初始活性很高, 但在酸性条件下并不稳定, 容易溶解失活[149].刘迪佳等[148]以金属沸石咪唑酯骨架结构材料为前驱体二次热解制备的Fe/N/CF就属于这一类催化剂.性能最好的Fe/N/CF催化剂是一种多孔的网状材料, 具有超高的比表面积(809 m2·g-1), 极高的微孔含量(99%).如图 21a为Fe/N/CF组装单电池在0.5 V的稳定性测试, 所有的样品经过100 h的测试后输出电流都明显下降, 其中性能最好的样品电流从1 A/cm2下降到0.3 A/cm2.第二类, FeNC催化剂制备方法和前一种类似, 只是不经过二次热解[150, 151]. Dodelet等[151]同样使用金属沸石咪唑酯骨架结构材料为前驱体一次热解制备了FeNC催化剂.这种方法制备的催化剂, 比表面积不如前一种, 但也较大(478 m2·g-1), 微孔含量较高(80%). 图 21b分别为第一类(蓝线)和第二类(红线)FeNC催化剂的单电池稳定性测试, 该类催化剂的性能先上升之后急速下降.结合穆斯堡尔谱的表征, 他们认为这类催化剂的活性中心和第一种一样, 只是由于没有二次热解, 暴露出的活性中心较少, 在稳定性测试时, 活性中心逐渐暴露出来, 活性先升高, 之后活性中心也不稳定, 活性下降.
第三类是使用或者没有使用Fe、Co前驱体制备的CNx催化剂[152, 153].这一类催化剂的电池功率较低, 但是稳定性高.典型的催化剂如氮掺杂的石墨烯[153]是将氧化石墨烯球磨之后与CoCl2/FeCl2混合900 ℃热解, 最后酸洗得到.作者认为该催化剂的主要活性中心是CNx, 可能含有少量的MeNx活性位.如图 21c该样品在0.4 V的单电池稳定性测试结果显示, 它初始电池功率仅为0.13 W/cm2, 经过1100 h运行也没有明显的衰退.
非贵金属催化剂的失活原因缺少深入、系统研究, 所以也没有形成统一的观点[154].目前已有的观点有:
(1) 碳载体在氧还原过程中被电氧化[155]. Meyers等[156]提出PEMFC中碳在高电位时将发生电氧化反应, C+2H2O=CO2+4H++4e-, 这一反应的进行将破坏催化剂的结构.特别是对石墨化程度较低的催化剂, 碳的电氧化很容易发生[155].
(2) 金属-氮活性位在酸性溶液中的溶解损失[26].对于金属大环化合物, 金属在酸性溶液中的溶解是其失活的主要原因[157], 高温热处理制备的FeNC催化剂虽然稳定性显著提高, 但活性位的溶解损失仍然不可避免. Ozkan等[158, 159]认为对于碳基非贵金属催化剂, 既存在CNx活性位也存在FeNx活性位, 其中FeNx活性高但不稳定, 在酸性条件下容易分解, 留下吡啶氮, 而CNx活性低但比较稳定. FeNC催化剂快速失活的原因是单电池运行过程中FeNx分解成为吡啶氮.
(3) H2O2与铁发生Fenton反应生成高氧化性物种. Fenton[160]在1894年就发现H2O2会与Fe2+形成具有高氧化活性的Fenton溶液. Gubler等[161]研究发现在PEMFC中由于O2的不完全还原生成H2O2, 很容易和铁活性中心发生Fenton反应, 具体反应为Fe2++H2O2+H+→ Fe3++·OH+H2O. Fenton反应生成的·OH是高活性的自由基, Dodelet等[162]认为·OH能够进一步腐蚀催化剂, HO·+RH(active site or carbon support)→H2O+R·R+Fe3+→Fe2++degradation products.
(4) 质子对活性位的毒化作用[154]. Popov等[163]首先发现了质子对活性位的毒化作用.他们以Fe、Co为前驱体制备出非贵金属催化剂, 认为Fe、Co只起了催化石墨化作用, CNx是该催化剂的活性中心; 稳定性试验发现, 催化剂活性随时间迅速下降.他们进一步对比了800 ℃和1000 ℃制备的样品, 发现800 ℃样品活性高, 但是稳定性低, 1000 ℃样品活性低但稳定性高.表征和模拟计算表明这是由于不同类型的N物种ORR活性不同, 吡啶氮活性最高, 石墨氮其次; 但是吡啶氮在酸性条件下并不稳定, 吡啶氮中的孤对电子将与质子结合生成NH+物种, 失去这一孤对电子之后吡啶氮不再吸附O2[60]. Dodelet课题组[164]在这一基础上提出了FeNx活性位的质子毒化理论.他们认为在FeNC催化剂中FeNx是活性位, 电池溶液中的质子首先和不活跃的N物种形成N-H自由基, FeNx活性位反应过程中形成的Fe-N4-O-O·自由基很容易和N-H吸附结合, 这种结合非常稳定, 阻止了ORR反应的进一步发生.
(5) 水在催化剂微孔中流动的传质的问题. Doudelet等[149]将失活原因和传质变化联系在了一起.他们认为非贵金属催化剂的活性中心在微孔处, 当催化剂应用到PEMFC时, 微孔处一开始没有填满水, 传质还是气相传质, 反应速率快, 当反应进一步进行, ORR生成的H2O填满催化剂的微孔处, 传质变为了液相传质, 反应速率降低.这样解释了为何微孔比例越高的催化剂活性越高, 但是稳定性越低.
5.4 与Pt/C催化剂对比
通常非贵金属催化剂是以10倍的负载量(3.8~4.1 mg/cm2)与Pt/C催化剂(0.3 mg/cm2)进行性能和稳定性对比.在该负载量下Pt催化剂的H2/O2电池峰值功率为1.36 W/cm2, H2/air电池峰值功率为0.98 W/cm2[129].即使在高倍的负载量下, 大部分碳基催化剂都还是和Pt/C有很大差距, 但最好的非贵金属催化剂, 其H2/O2电池峰值功率已经可以达到1.03 W/cm2[37], H2/air电池峰值功率可以达到0.41 W/cm2, 已经和Pt/C具有可比性.碳基催化剂在PEMFC的单电池应用中更关键的问题其实是稳定性, 活性最好的碳基催化剂100 h后单电池功率仅能保持30%左右.而Pt催化剂1100 h的稳定性测试发现, H2/O2电池功率仅从1.36 W/cm2下降到1.318 W/cm2, H2/air电池功率仅从0.92 W/cm2下降到0.898 W/cm2.对于稳定性最好的非贵金属催化剂, 1100 h的稳定性测试后电池功率只有Pt的1/10左右[153].与Pt催化剂相比, 非贵金属催化剂更难应用于燃料电池汽车领域, 因为稳定性符合要求的样品, 其体积活性远达不到美国能源部(DOE)的应用目标300 A/cm2.为了弥补催化剂活性的差距, 必须急剧地增加电池中双极板、交换膜、垫圈等的用量.即使是活性最高的非贵金属催化剂在具有和Pt一样的稳定性时, 为了达到统一的功率, 其组装的电堆成本将两倍于Pt.所以非贵金属催化剂要想替代Pt/C实际应用到PEMFC, 急需解决三个问题: (1) 提高电池功率密度; (2) 提高稳定性; (3) 降低催化剂的成本.
6 结论与展望
碳基非贵金属ORR催化剂具有较高的ORR催化活性和稳定性, 是目前最可能替代铂的催化剂之一.但即使是最好的碳基非贵金属ORR催化剂, 其ORR催化性能仍与Pt基催化剂有一定差距, 特别是在实际应用的酸性条件下.根据本文的综述, 我们认为碳基非贵金属氧还原催化剂的发展存在以下几个方面的重要问题, 值得深入研究:
(1) 氧还原催化机理不清楚.氧还原的反应机理本身极为复杂, 碳基催化剂又具有不同于铂基催化剂的催化机理, 所以现在急需通过先进的表征技术结合理论计算去探索碳基催化剂的氧化机理.
(2) 碳基非贵金属ORR催化剂的活性中心存在争议.我们将最有可能的活性中心总结在图 22中, 包括M-Nx(典型的是FeNx)、掺杂杂原子(典型的是N)、填充金属和边缘碳.这几种活性中心, 到底哪一种是碳基催化剂中主要的活性位还没有定论, 而且越来越多的文献认为多种活性位存在协同作用.所以我们认为对于碳基催化剂活性中心的研究不应拘泥于某一种活性中心, 不应受限于传统的活性中心的模式, 而应该从深层次、系统的角度, 例如材料的电子结构、表面电荷密度、功函等, 进行综合研究, 提出表面电子催化的新观点.
 图22
碳基ORR催化剂中可能的ORR活性中心
Figure22.
Possible catalytic active sites in carbon-based ORR catalysts
图22
碳基ORR催化剂中可能的ORR活性中心
Figure22.
Possible catalytic active sites in carbon-based ORR catalysts
(3) 燃料电池的实际应用问题.碳基非贵金属催化剂在不考虑用量的情况下应用到燃料电池单电池时, 已经可比商业Pt/C催化剂, 但是其应用稳定性差距还比较大; 另外, 由于催化剂用量大、电极膜层厚, 存在扩散与导电性差等问题, 实际应用中性能达不到要求.所以碳基催化剂要想取代铂基催化剂, 除了要进一步提高单位质量活性外, 还需要提高实际应用的稳定性.
我们相信经过科研工作者们更加艰苦卓绝的努力, 一定能开发出成本低、来源广、稳定性高、性能好的非贵金属ORR催化剂, 降低PEMFC成本, 推动其大规模商业化应用.
-
-
[1]
王瀛, 张丽敏, 胡天军, 化学学报, 2015, 73, 316. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344818.shtmlWang, Y.; Zhang, L.; Hu, T. Acta Chim. Sinica. 2015, 73, 316. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344818.shtml
-
[2]
Yeager, E. Electrochim. Acta 1984, 29, 1527. doi: 10.1016/0013-4686(84)85006-9
-
[3]
Li, Y.; Zhong, G.; Yu, H.; Wang, H.; Peng, F. Phys. Chem. Chem. Phys. 2015, 17, 21950. doi: 10.1039/C5CP02167G
-
[4]
陈鑫, 鄢慧君, 夏定国, 化学学报, 2017, 75, 189. doi: 10.3969/j.issn.0253-2409.2017.02.008Chen, X.; Yan, H.; Xia, D. Acta Chim. Sinica 2017, 75, 189. doi: 10.3969/j.issn.0253-2409.2017.02.008
-
[5]
Anderson, A. B.; Sidik, R. A. J. Phys. Chem. B 2004, 108, 5031. doi: 10.1021/jp037184z
-
[6]
Bouwkamp-Wijnoltz, A. L.; Visscher, W.; Van Veen, J. A. R. Electrochim. Acta 1998, 43, 3141. doi: 10.1016/S0013-4686(98)00076-0
-
[7]
Anderson, A. B.; Sidik, R. A. J. Phys. Chem. B 2004, 108, 5031. doi: 10.1021/jp037184z
-
[8]
Jaouen, F. J. Phys. Chem. C 2009, 113, 15433. doi: 10.1021/jp900838x
-
[9]
Shao, M., Electrocatalysis in Fuel Cells:A Non-and Low-Platinum Approach, Springer London, London, 2013, p. 698.
-
[10]
Wang, Y.; Balbuena, P. B. J. Chem. Theory Comput. 2005, 1, 935. doi: 10.1021/ct0500794
-
[11]
Jasinski, R. Nature 1964, 201, 1212. doi: 10.1038/2011212a0
-
[12]
Randin, J.-P. Electrochim. Acta 1974, 19, 83. doi: 10.1016/0013-4686(74)85060-7
-
[13]
Jahnke, H.; Schönborn, M.; Zimmermann, G. Physical and Chemical Applications of Dyestuffs, Springer, Verlag, 1976, p. 133.
-
[14]
Van der Putten, A.; Elzing, A.; Visscher, W.; Barendrecht, E. J. Electroanal. Chem. 1987, 221, 95. doi: 10.1016/0022-0728(87)80248-6
-
[15]
Tarasevich, M.; Radiyschkina, K.; Androuseva, S. Bioelectrochem. Bioenerg. 1977, 4, 18. doi: 10.1016/0302-4598(77)80002-0
-
[16]
Berezin, B. Russ. J. Physic Chem. 1962, 36, 258.
-
[17]
Kadish, K. M.; Smith, K. M.; Guilard, R., The Porphyrin Handbook:Inorganic, Organometallic and Coordination Chemistry, Elsevier, London, 2000, p. 15.
-
[18]
Jahnke, H. G.; Schonborn, M.; Zimmerman, G. J. Electrochem. Soc. 1974, 121, 120
-
[19]
Bagotzky, V. S.; Tarasevich, M. R.; Radyushkina, K. A.; Levina, O. A.; Andrusyova, S. I. J. Power Sources 1978, 2, 233. doi: 10.1016/0378-7753(78)85014-9
-
[20]
Fuhrmann, A.; Wiesener, K.; Iliev, I.; Gamburzev, S.; Kaisheva, A. J. Power Sources 1981, 6, 69. doi: 10.1016/0378-7753(81)80007-9
-
[21]
Shigehara, K.; Anson, F. C. J. Electroanal. Chem. 1982, 132, 107. doi: 10.1016/0022-0728(82)85010-9
-
[22]
Van der Putten, A.; Elzing, A.; Visscher, W.; Barendrecht, E. J. Electroanal. Chem. 1986, 205, 233. doi: 10.1016/0022-0728(86)90234-2
-
[23]
Lalande, G.; Cote, R.; Tamizhmani, G.; Guay, D.; Dodelet, J.; Dignard-Bailey, L.; Weng, L.; Bertrand, P. Electrochim. Acta 1995, 40, 2635. doi: 10.1016/0013-4686(95)00104-M
-
[24]
Charreteur, F.; Jaouen, F.; Dodelet, J. P. Electrochim. Acta 2009, 54, 6622. doi: 10.1016/j.electacta.2009.06.058
-
[25]
Medard, C.; Lefevre, M.; Dodelet, J.; Jaouen, F.; Lindbergh, G. Electrochim. Acta 2006, 51, 3202. doi: 10.1016/j.electacta.2005.09.012
-
[26]
Gupta, S.; Tryk, D.; Bae, I.; Aldred, W.; Yeager, E. J. Appl. Electrochem. 1989, 19, 19. doi: 10.1007/BF01039385
-
[27]
Bouwkamp-Wijnoltz, A.; Visscher, W.; Van Veen, J.; Tang, S. Electrochim. Acta 1999, 45, 379. doi: 10.1016/S0013-4686(99)00281-9
-
[28]
Lefevre, M.; Dodelet, J.; Bertrand, P. J. Phys. Chem. B 2000, 104, 11238. doi: 10.1021/jp002444n
-
[29]
Jaouen, F.; Dodelet, J. P. J. Phys. Chem. C 2007, 111, 5963. doi: 10.1021/jp068273p
-
[30]
Jaouen, F.; Lefèvre, M.; Dodelet, J. P.; Cai, M. J. Phys. Chem. B 2006, 110, 5553. doi: 10.1021/jp057135h
-
[31]
Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J. P. Science 2009, 324, 71. doi: 10.1126/science.1170051
-
[32]
Wu, G.; More, K. L.; Johnston, C. M.; Zelenay, P. Science 2011, 332, 443. doi: 10.1126/science.1200832
-
[33]
赖渊, 周德璧, 胡剑文, 崔莉莉, 化学学报, 2008, 66, 1015. doi: 10.3321/j.issn:0567-7351.2008.09.002Lai, Y.; Zhou, D. B.; Hu, J. W.; Cui, L. L. Acta Chim. Sinica 2008, 66, 1015. doi: 10.3321/j.issn:0567-7351.2008.09.002
-
[34]
郑龙珍, 陶堃, 熊乐艳, 叶丹, 韩奎, 纪忆, 化学学报, 2012, 70, 2342. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341689.shtmlZheng, L.; Tao, K.; Xiong, L.; Ye, D.; Han, K.; Ji, Y. Acta Chim. Sinica 2012, 70, 2342. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341689.shtml
-
[35]
Zhu, Y.; Zhang, B.; Liu, X.; Wang, D. W.; Su, D. S. Angew. Chem., Int. Ed. 2014, 53, 10673. doi: 10.1002/anie.201405314
-
[36]
Jiang, W. J.; Gu, L.; Li, L.; Zhang, Y.; Zhang, X.; Zhang, L. J.; Wang, J. Q.; Hu, J. S.; Wei, Z.; Wan, L. J. J. Am. Chem. Soc. 2016, 138, 3570. doi: 10.1021/jacs.6b00757
-
[37]
Wang, Y. C.; Lai, Y. J.; Song, L.; Zhou, Z. Y.; Liu, J. G.; Wang, Q.; Yang, X. D.; Chen, C.; Shi, W.; Zheng, Y. P.; Rauf, M.; Sun, S. G. Angew. Chem., Int. Ed. 2015, 54, 9907. doi: 10.1002/anie.201503159
-
[38]
Chen, W.; Sin, M.; Wei, P. J.; Zhang, Q. L.; Liu, J. G. Chin. J. Chem. 2016, 34, 878. doi: 10.1002/cjoc.201600196
-
[39]
Sun, T.; Wu, Q.; Zhuo, O.; Jiang, Y.; Bu, Y.; Yang, L.; Wang, X.; Hu, Z. Nanoscale 2016, 8, 8480. doi: 10.1039/C6NR00760K
-
[40]
Zagal, J.; Paez, M.; Tanaka, A. A.; Dos Santos, J. R.; Linkous, C. A. J. Electroanal. Chem. 1992, 339, 13. doi: 10.1016/0022-0728(92)80442-7
-
[41]
Zagal, J.; Cárdenas-Jirón, G. J. Electroanal. Chem. 2000, 489, 96. doi: 10.1016/S0022-0728(00)00209-6
-
[42]
Baker, R.; Wilkinson, D. P.; Zhang, J. Electrochim. Acta 2008, 53, 6906. doi: 10.1016/j.electacta.2008.01.055
-
[43]
Rosa, A.; Baerends, E. Inorg. Chem. 1994, 33, 584. doi: 10.1021/ic00081a029
-
[44]
Bouwkamp-Wijnoltz, A.; Visscher, W.; Van Veen, J.; Boellaard, E.; Van der Kraan, A.; Tang, S. J. Phys. Chem. B 2002, 106, 12993. doi: 10.1021/jp0266087
-
[45]
Van Veen, J.; Colijn, H.; Van Baar, J. Electrochim. Acta 1988, 33, 801. doi: 10.1016/S0013-4686(98)80010-8
-
[46]
Van Wingerden, B.; van Veen, J. R.; Mensch, C. T. J. Chem. Soc., Faraday Trans. 11988, 84, 65. doi: 10.1039/f19888400065
-
[47]
Wiesener, K. Electrochim. Acta 1986, 31, 1073. doi: 10.1016/0013-4686(86)80022-6
-
[48]
Franke, R.; Ohms, D.; Wiesener, K. J. Electroanal. Chem. 1989, 260, 63. doi: 10.1016/0022-0728(89)87099-8
-
[49]
Faubert, G.; Côté, R.; Dodelet, J.; Lefevre, M.; Bertrand, P. Electrochim. Acta 1999, 44, 2589. doi: 10.1016/S0013-4686(98)00382-X
-
[50]
Herranz, J.; Lefevre, M.; Larouche, N.; Stansfield, B.; Dodelet, J. P. J. Phys. Chem. C 2007, 111, 19033. doi: 10.1021/jp0764438
-
[51]
Charreteur, F.; Jaouen, F.; Ruggeri, S.; Dodelet, J. P. Electrochim. Acta 2008, 53, 2925. doi: 10.1016/j.electacta.2007.11.002
-
[52]
Jaouen, F.; Herranz, J.; Lefevre, M.; Dodelet, J. P.; Kramm, U. I.; Herrmann, I.; Bogdanoff, P.; Maruyama, J.; Nagaoka, T.; Garsuch, A.; Dahn, J. R.; Olson, T.; Pylypenko, S.; Atanassov, P.; Ustinov, E. A. ACS Appl. Mater. Interfaces 2009, 1, 1623. doi: 10.1021/am900219g
-
[53]
Yuasa, M.; Yamaguchi, A.; Itsuki, H.; Tanaka, K.; Yamamoto, M.; Oyaizu, K. Chem. Mater. 2005, 17, 4278. doi: 10.1021/cm050958z
-
[54]
Schulenburg, H.; Stankov, S.; Schünemann, V.; Radnik, J.; Dorbandt, I.; Fiechter, S.; Bogdanoff, P.; Tributsch, H. J. Phys. Chem. B 2003, 107, 9034. doi: 10.1021/jp030349j
-
[55]
Lefevre, M.; Dodelet, J.; Bertrand, P. J. Phys. Chem. B 2005, 109, 16718. doi: 10.1021/jp0529265
-
[56]
Maldonado, S.; Stevenson, K. J. J. Phys. Chem. B 2004, 108, 11375.
-
[57]
Birry, L.; Zagal, J. H.; Dodelet, J. P. Electrochem. Commun. 2010, 12, 628. doi: 10.1016/j.elecom.2010.02.016
-
[58]
Gara, M.; Compton, R. G. New J. Chem. 2011, 35, 2647. doi: 10.1039/c1nj20612e
-
[59]
Nallathambi, V.; Lee, J. W.; Kumaraguru, S. P.; Wu, G.; Popov, B. N. J. Power Sources 2008, 183, 34. doi: 10.1016/j.jpowsour.2008.05.020
-
[60]
Liu, G.; Li, X.; Popov, B. ECS Trans. 2009, 25, 1251.
-
[61]
von Deak, D.; Singh, D.; Biddinger, E. J.; King, J. C.; Bayram, B.; Miller, J. T.; Ozkan, U. S. J. Catal. 2012, 285, 145. doi: 10.1016/j.jcat.2011.09.027
-
[62]
Deng, D.; Yu, L.; Chen, X.; Wang, G.; Jin, L.; Pan, X.; Deng, J.; Sun, G.; Bao, X. Angew. Chem., Int. Ed. 2013, 52, 371. doi: 10.1002/anie.201204958
-
[63]
Zhong, G.; Wang, H.; Yu, H.; Peng, F. J. Power Sources 2015, 286, 495. doi: 10.1016/j.jpowsour.2015.04.021
-
[64]
Hu, Y.; Jensen, J. O.; Zhang, W.; Cleemann, L. N.; Xing, W.; Bjerrum, N. J.; Li, Q. Angew. Chem., Int. Ed. 2014, 53, 3675. doi: 10.1002/anie.v53.14
-
[65]
Jiang, Y.; Yang, L.; Sun, T.; Zhao, J.; Lyu, Z.; Zhuo, O.; Wang, X.; Wu, Q.; Ma, J.; Hu, Z. ACS Catalysis 2015, 5, 6707. doi: 10.1021/acscatal.5b01835
-
[66]
Wu, Q.; Yang, L.; Wang, X.; Hu, Z. Acc. Chem. Res. 2017, 50, 435. doi: 10.1021/acs.accounts.6b00541
-
[67]
Zhong, G.; Wang, H.; Yu, H.; Peng, F. Electrochem. Commun. 2014, 40, 5. doi: 10.1016/j.elecom.2013.12.017
-
[68]
Zhong, G.; Wang, H.; Yu, H.; Wang, H.; Peng, F. Electrochim. Acta 2016, 190, 49. doi: 10.1016/j.electacta.2015.12.216
-
[69]
Huang, S.; Dai, L.; Mau, A. W. J. Phys. Chem. B 1999, 103, 4223. doi: 10.1021/jp990342v
-
[70]
Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Science 2009, 323, 760. doi: 10.1126/science.1168049
-
[71]
Chen, Z.; Higgins, D.; Tao, H.; Hsu, R. S.; Chen, Z. J. Phys. Chem. C 2009, 113, 21008. doi: 10.1021/jp908067v
-
[72]
Chen, Z.; Higgins, D.; Chen, Z. Carbon 2010, 48, 3057. doi: 10.1016/j.carbon.2010.04.038
-
[73]
Geng, D.; Liu, H.; Chen, Y.; Li, R.; Sun, X.; Ye, S.; Knights, S. J. Power Sources 2011, 196, 1795. doi: 10.1016/j.jpowsour.2010.09.084
-
[74]
Alexeyeva, N.; Shulga, E.; Kisand, V.; Kink, I.; Tammeveski, K. J. Electroanal. Chem. 2010, 648, 169. doi: 10.1016/j.jelechem.2010.07.014
-
[75]
Masa, J.; Zhao, A.; Xia, W.; Sun, Z.; Mei, B.; Muhler, M.; Schuhmann, W. Electrochem. Commun. 2013, 34, 113. doi: 10.1016/j.elecom.2013.05.032
-
[76]
Wang, L.; Ambrosi, A.; Pumera, M. Angew. Chem., Int. Ed. 2013, 52, 13818. doi: 10.1002/anie.201309171
-
[77]
Yu, D.; Zhang, Q.; Dai, L. J. Am. Chem. Soc. 2010, 132, 15127. doi: 10.1021/ja105617z
-
[78]
Qu, L.; Liu, Y.; Baek, J. B.; Dai, L. ACS Nano 2010, 4, 1321. doi: 10.1021/nn901850u
-
[79]
Choi, E. K.; Jeon, I. Y.; Bae, S. Y.; Lee, H. J.; Shin, H. S.; Dai, L.; Baek, J. B. Chem. Commun. 2010, 46, 6320. doi: 10.1039/c0cc00753f
-
[80]
Jeon, I. Y.; Yu, D.; Bae, S. Y.; Choi, H. J.; Chang, D. W.; Dai, L.; Baek, J. B. Chem. Mater. 2011, 23, 3987. doi: 10.1021/cm201542m
-
[81]
Shao, Y.; Zhang, S.; Engelhard, M. H.; Li, G.; Shao, G.; Wang, Y.; Liu, J.; Aksay, I. A.; Lin, Y. J. Mater. Chem. 2010, 20, 7491. doi: 10.1039/c0jm00782j
-
[82]
Geng, D.; Chen, Y.; Chen, Y.; Li, Y.; Li, R.; Sun, X.; Ye, S.; Knights, S. Energy Environ. Sci. 2011, 4, 760. doi: 10.1039/c0ee00326c
-
[83]
Sheng, Z. H.; Shao, L.; Chen, J. J.; Bao, W. J.; Wang, F. B.; Xia, X. H. ACS Nano 2011, 5, 4350. doi: 10.1021/nn103584t
-
[84]
Lin, Z.; Waller, G.; Liu, Y.; Liu, M.; Wong, C. P. Adv. Energy Mater. 2012, 2, 884. doi: 10.1002/aenm.201200038
-
[85]
Lai, L.; Potts, J. R.; Zhan, D.; Wang, L.; Poh, C. K.; Tang, C.; Gong, H.; Shen, Z.; Lin, J.; Ruoff, R. S. Energy Environ. Sci. 2012, 5, 7936. doi: 10.1039/c2ee21802j
-
[86]
许智慧, 沈丽明, 吴强, 孙涛, 徐宇洋, 黎聃勤, 杜玲玉, 杨立军, 王喜章, 胡征, 化学学报, 2015, 73, 793. doi: 10.3866/PKU.WHXB201503021Xu, Z.; Shen, L.; Wu, Q.; Sun, T.; Xu, Y.; Li, D.; Du, L.; Yang, L.; Wang, X.; Hu, Z. Acta Chim. Sinica 2015, 73, 793. doi: 10.3866/PKU.WHXB201503021
-
[87]
Wu, Q.; Yang, L.; Wang, X.; Hu, Z. Acc. Chem. Res. 2017, 50, 435. doi: 10.1021/acs.accounts.6b00541
-
[88]
Liu, R.; Wu, D.; Feng, X.; Mullen, K. Angew. Chem., Int. Ed. 2010, 49, 2565. doi: 10.1002/anie.v49:14
-
[89]
Yang, W.; Fellinger, T. P.; Antonietti, M. J. Am. Chem. Soc. 2011, 133, 206. doi: 10.1021/ja108039j
-
[90]
Ma, G.; Jia, R.; Zhao, J.; Wang, Z.; Song, C.; Jia, S.; Zhu, Z. J. Phys. Chem. C 2011, 115, 25148. doi: 10.1021/jp208257r
-
[91]
Zhou, X.; Yang, Z.; Nie, H.; Yao, Z.; Zhang, L.; Huang, S. J. Power Sources 2011, 196, 9970. doi: 10.1016/j.jpowsour.2011.08.029
-
[92]
Sidik, R. A.; Anderson, A. B.; Subramanian, N. P.; Kumaraguru, S. P.; Popov, B. N. J. Phys. Chem. B 2006, 110, 1787. doi: 10.1021/jp055150g
-
[93]
Zhao, A.; Masa, J.; Muhler, M.; Schuhmann, W.; Xia, W. Electrochim. Acta 2013, 98, 139. doi: 10.1016/j.electacta.2013.03.043
-
[94]
Yang, L.; Jiang, S.; Zhao, Y.; Zhu, L.; Chen, S.; Wang, X.; Wu, Q.; Ma, J.; Ma, Y.; Hu, Z. Angew. Chem., Int. Ed. 2011, 50, 7132. doi: 10.1002/anie.v50.31
-
[95]
Sheng, Z. H.; Gao, H. L.; Bao, W. J.; Wang, F. B.; Xia, X. H. J. Mater. Chem. 2012, 22, 390. doi: 10.1039/C1JM14694G
-
[96]
Liu, Z. W.; Peng, F.; Wang, H. J.; Yu, H.; Zheng, W. X.; Yang, J. Angew. Chem., Int. Ed. 2011, 50, 3257. doi: 10.1002/anie.201006768
-
[97]
Liu, Z.; Peng, F.; Wang, H.; Yu, H.; Zheng, W.; Wei, X. J. Nat. Gas. Chem. 2012, 21, 257. doi: 10.1016/S1003-9953(11)60362-9
-
[98]
Liu, Z.; Peng, F.; Wang, H.; Yu, H.; Tan, J.; Zhu, L. Catal. Commun. 2011, 16, 35. doi: 10.1016/j.catcom.2011.08.038
-
[99]
Yang, S.; Zhi, L.; Tang, K.; Feng, X.; Maier, J.; Mullen, K. Adv. Funct. Mater. 2012, 22, 3634. doi: 10.1002/adfm.v22.17
-
[100]
Yang, Z.; Yao, Z.; Li, G.; Fang, G.; Nie, H.; Liu, Z.; Zhou, X.; Chen, X. a.; Huang, S. ACS Nano 2011, 6, 205. http://www.ncbi.nlm.nih.gov/pubmed/22201338
-
[101]
Poh, H. L.; Šimek, P.; Sofer, Z. k.; Pumera, M. ACS Nano 2013, 7, 5262. doi: 10.1021/nn401296b
-
[102]
王立伟, 冯瑞, 夏婧竹, 陈盛, 吴强, 杨立军, 王喜章, 胡征, 化学学报, 2014, 72, 1070. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344641.shtmlWang, L.; Feng, R.; Xia, J.; Chen, S.; Wu, Q.; Yang, L.; Wang, X.; Hu, Z. Acta Chim. Sinica 2014, 72, 1070. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344641.shtml
-
[103]
Jin, Z.; Nie, H.; Yang, Z.; Zhang, J.; Liu, Z.; Xu, X.; Huang, S. Nanoscale 2012, 4, 6455. doi: 10.1039/c2nr31858j
-
[104]
Liu, Z.; Fu, X.; Li, M.; Wang, F.; Wang, Q.; Kang, G.; Peng, F. J. Mater. Chem. A 2015, 3, 3289. doi: 10.1039/C4TA05937A
-
[105]
Sun, X.; Zhang, Y.; Song, P.; Pan, J.; Zhuang, L.; Xu, W.; Xing, W. ACS Catalysis 2013, 3, 1726. doi: 10.1021/cs400374k
-
[106]
Wang, S.; Iyyamperumal, E.; Roy, A.; Xue, Y.; Yu, D.; Dai, L. Angew. Chem., Int. Ed. 2011, 50, 11756. doi: 10.1002/anie.201105204
-
[107]
Yu, D.; Xue, Y.; Dai, L. J. Phys. Chem. Lett. 2012, 3, 2863. doi: 10.1021/jz3011833
-
[108]
Zhao, Y.; Yang, L.; Chen, S.; Wang, X.; Ma, Y.; Wu, Q.; Jiang, Y.; Qian, W.; Hu, Z. J. Am. Chem. Soc. 2013, 135, 1201. doi: 10.1021/ja310566z
-
[109]
Zheng, Y.; Jiao, Y.; Ge, L.; Jaroniec, M.; Qiao, S. Z. Angew. Chem., Int. Ed. 2013, 52, 3110. doi: 10.1002/anie.201209548
-
[110]
Choi, C. H.; Park, S. H.; Woo, S. I. J. Mater. Chem. 2012, 22, 12107. doi: 10.1039/c2jm31079a
-
[111]
Shi, Q.; Peng, F.; Liao, S.; Wang, H.; Yu, H.; Liu, Z.; Zhang, B.; Su, D. J. Mater. Chem. A 2013, 1, 14853. doi: 10.1039/c3ta12647a
-
[112]
Terrones, M.; Ajayan, P. M.; Banhart, F.; Blase, X.; Carroll, D. L.; Charlier, J. C.; Czerw, R.; Foley, B.; Grobert, N.; Kamalakaran, R.; Kohler-Redlich, P.; Rühle, M.; Seeger, T.; Terrones, H. Appl. Phys. A 2002, 74, 355. doi: 10.1007/s003390201278
-
[113]
Zhang, G.; Duan, W.; Gu, B. Appl. Phys. Lett. 2002, 80, 2589. doi: 10.1063/1.1469213
-
[114]
Ismagilov, Z. R.; Shalagina, A. E.; Podyacheva, O. Y.; Ischenko, A. V.; Kibis, L. S.; Boronin, A. I.; Chesalov, Y. A.; Kochubey, D. I.; Romanenko, A. I.; Anikeeva, O. B.; Buryakov, T. I.; Tkachev, E. N. Carbon 2009, 47, 1922. doi: 10.1016/j.carbon.2009.02.034
-
[115]
Wiggins-Camacho, J. D.; Stevenson, K. J. J. Phys. Chem. C 2009, 113, 19082. doi: 10.1021/jp907160v
-
[116]
Wiggins-Camacho, J. D.; Stevenson, K. J. J. Phys. Chem. C 2011, 115, 20002. doi: 10.1021/jp205336w
-
[117]
Kundu, S.; Nagaiah, T. C.; Xia, W.; Wang, Y.; Dommele, S. V.; Bitter, J. H.; Santa, M.; Grundmeier, G.; Bron, M.; Schuhmann, W.; Muhler, M. J. Phys. Chem. C 2009, 113, 14302. doi: 10.1021/jp811320d
-
[118]
Rao, C. V.; Cabrera, C. R.; Ishikawa, Y. J. Phys. Chem. Lett. 2010, 1, 2622. doi: 10.1021/jz100971v
-
[119]
Yasuda, S.; Yu, L.; Kim, J.; Murakoshi, K. Chem. Commun. 2013, 49, 9627. doi: 10.1039/c3cc45641b
-
[120]
Sharifi, T.; Hu, G.; Jia, X.; Wågberg, T. ACS Nano 2012, 6, 8904. doi: 10.1021/nn302906r
-
[121]
Casanovas, J.; Ricart, J. M.; Rubio, J.; Illas, F.; Jiménez-Mateos, J. M. J. Am. Chem. Soc. 1996, 118, 8071. doi: 10.1021/ja960338m
-
[122]
Ding, W.; Wei, Z.; Chen, S.; Qi, X.; Yang, T.; Hu, J.; Wang, D.; Wan, L. J.; Alvi, S. F.; Li, L. Angew. Chem., Int. Ed. 2013, 125, 11971 doi: 10.1002/ange.v125.45
-
[123]
Chen, Z.; Higgins, D.; Chen, Z. Electrochim. Acta 2010, 55, 4799. doi: 10.1016/j.electacta.2010.03.057
-
[124]
Wong, W. Y.; Daud, W. R. W.; Mohamad, A. B.; Kadhum, A. A. H.; Loh, K. S.; Majlan, E. H. Int. J. Hydrogen Energy 2013, 38, 9370. doi: 10.1016/j.ijhydene.2012.12.095
-
[125]
Xu, X.; Jiang, S.; Hu, Z.; Liu, S. ACS Nano 2010, 4, 4292. doi: 10.1021/nn1010057
-
[126]
Hummers, Jr, W. S.; Offeman, R. E. J. Am. Chem. Soc. 1958, 80, 1339. doi: 10.1021/ja01539a017
-
[127]
Li, Y.; Zhou, W.; Wang, H.; Xie, L.; Liang, Y.; Wei, F.; Idrobo, J. C.; Pennycook, S. J.; Dai, H. Nat. Nanotechnol. 2012, 7, 394. doi: 10.1038/nnano.2012.72
-
[128]
Chen, S.; Bi, J.; Zhao, Y.; Yang, L.; Zhang, C.; Ma, Y.; Wu, Q.; Wang, X.; Hu, Z. Adv. Mater. 2012, 24, 5593. doi: 10.1002/adma.201202424
-
[129]
Shao, M.; Chang, Q.; Dodelet, J. P.; Chenitz, R. Chem. Rev. 2016, 116, 3594 doi: 10.1021/acs.chemrev.5b00462
-
[130]
Jaouen, F., Non-Noble Metal Fuel Cell Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, p. 29.
-
[131]
Higgins, D. C.; Chen, Z. Canad. J. Chem. Eng. 2013, 91, 1881. doi: 10.1002/cjce.v91.12
-
[132]
Debe, M. K. Nature 2012, 486, 43. doi: 10.1038/nature11115
-
[133]
Larouche, N.; Chenitz, R.; Lefèvre, M.; Proietti, E.; Dodelet, J. Electrochim. Acta 2014, 115, 170. doi: 10.1016/j.electacta.2013.10.102
-
[134]
Wang, J.; Li, S.; Zhu, G.; Zhao, W.; Chen, R.; Pan, M. J. Power Sources 2013, 240, 381. doi: 10.1016/j.jpowsour.2013.03.189
-
[135]
Chokai, M.; Daidou, T.; Nabae, Y. ECS Trans. 2014, 64, 261. http://ecst.ecsdl.org/content/64/3/261.short
-
[136]
Iwazaki, T.; Obinata, R.; Sugimoto, W.; Takasu, Y. Electrochem. Commun. 2009, 11, 376. doi: 10.1016/j.elecom.2008.11.045
-
[137]
Serov, A.; Artyushkova, K.; Atanassov, P. Adv. Energy Mater. 2014, 4, 1301735. doi: 10.1002/aenm.201301735
-
[138]
Nabae, Y.; Kuang, Y.; Chokai, M.; Ichihara, T.; Isoda, A.; Hayakawa, T.; Aoki, T. J. Mater. Chem. A 2014, 2, 11561. doi: 10.1039/C4TA01828A
-
[139]
Liu, G.; Li, X.; Ganesan, P.; Popov, B. N. Electrochim. Acta 2010, 55, 2853. doi: 10.1016/j.electacta.2009.12.055
-
[140]
Shui, J.; Wang, M.; Du, F.; Dai, L. Sci. Adv. 2015, 1, e1400129 doi: 10.1126/sciadv.1400129
-
[141]
Yang, L.; Larouche, N.; Chenitz, R.; Zhang, G.; Lefèvre, M.; Dodelet, J. P. Electrochim. Acta 2015, 159, 184. doi: 10.1016/j.electacta.2015.01.201
-
[142]
Ohma, A.; Shinohara, K.; Iiyama, A.; Yoshida, T.; Daimaru, A. ECS Trans. 2011, 41, 775.
-
[143]
Reiser, C. A.; Bregoli, L.; Patterson, T. W.; Yi, J. S.; Yang, J. D.; Perry, M. L.; Jarvi, T. D. Electrochem. Solid State Lett. 2005, 8, A273. doi: 10.1149/1.1896466
-
[144]
Kramm, U. I.; Lefèvre, M.; Bogdanoff, P.; Schmeißer, D.; Dodelet, J. P. J. Phys. Chem. Lett. 2014, 5, 3750. doi: 10.1021/jz501955g
-
[145]
Zhao, D.; Shui, J. L.; Chen, C.; Chen, X.; Reprogle, B. M.; Wang, D.; Liu, D. J. Chem. Sci. 2012, 3, 3200. doi: 10.1039/c2sc20657a
-
[146]
Peng, H.; Mo, Z.; Liao, S.; Liang, H.; Yang, L.; Luo, F.; Song, H.; Zhong, Y.; Zhang, B. Sci. Rep. 2013, 3, 1765. doi: 10.1038/srep01765
-
[147]
Cheon, J. Y.; Kim, T.; Choi, Y.; Jeong, H. Y.; Kim, M. G.; Sa, Y. J.; Kim, J.; Lee, Z.; Yang, T. H.; Kwon, K.; Terasaki, O.; Park, G. G.; Adzic, R. R.; Joo, S. H. Sci. Rep. 2013, 3, 2715. doi: 10.1038/srep02715
-
[148]
Shui, J.; Chen, C.; Grabstanowicz, L.; Zhao, D.; Liu, D. J. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10629. doi: 10.1073/pnas.1507159112
-
[149]
Jaouen, F.; Lefèvre, M.; Dodelet, J. P.; Cai, M. J. Phys. Chem. B 2006, 110, 5553. doi: 10.1021/jp057135h
-
[150]
Kramm, U. I.; Lefèvre, M.; Larouche, N.; Schmeisser, D.; Dodelet, J. P. J. Am. Chem. Soc. 2014, 136, 978. doi: 10.1021/ja410076f
-
[151]
Proietti, E.; Jaouen, F.; Lefèvre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J. P. Nat. Commun. 2010, 2, 109.
-
[152]
Subramanian, N. P.; Li, X.; Nallathambi, V.; Kumaraguru, S. P.; Colon-Mercado, H.; Wu, G.; Lee, J. W.; Popov, B. N. J. Power Sources 2009, 188, 38. doi: 10.1016/j.jpowsour.2008.11.087
-
[153]
Choi, C. H.; Lim, H. K.; Chung, M. W.; Park, J. C.; Shin, H.; Kim, H.; Woo, S. I. J. Am. Chem. Soc. 2014, 136, 9070. doi: 10.1021/ja5033474
-
[154]
Banham, D.; Ye, S.; Pei, K.; Ozaki, J. i.; Kishimoto, T.; Imashiro, Y. J. Power Sources 2015, 285, 334. doi: 10.1016/j.jpowsour.2015.03.047
-
[155]
Wu, G.; Artyushkova, K.; Ferrandon, M.; Kropf, A. J.; Myers, D.; Zelenay, P. ECS Trans. 2009, 25, 1299. http://hdl.handle.net/10477/53180
-
[156]
Meyers, J. P.; Darling, R. M. J. Electrochem. Soc. 2006, 153, A1432. doi: 10.1149/1.2203811
-
[157]
Biloul, A.; Gouérec, P.; Savy, M.; Scarbeck, G.; Besse, S.; Riga, J. J. Appl. Electrochem. 1996, 26, 1139. doi: 10.1007/BF00243739
-
[158]
Mamtani, K.; Ozkan, U. S. Catal. Lett. 2015, 145, 436. doi: 10.1007/s10562-014-1434-y
-
[159]
Singh, D.; Tian, J.; Mamtani, K.; King, J.; Miller, J. T.; Ozkan, U. S. J. Catal. 2014, 317, 30. doi: 10.1016/j.jcat.2014.05.025
-
[160]
Fenton, H. J. H. J. Chem. Soc. Trans. 1894, 65, 899. doi: 10.1039/CT8946500899
-
[161]
Gubler, L.; Dockheer, S. M.; Koppenol, W. H. J. Electrochem. Soc. 2011, 158, B755. doi: 10.1149/1.3581040
-
[162]
Dodelet, J. P., N4-Macrocyclic Metal Complexes, Springer New York, New York, NY, 2006, p. 83.
-
[163]
Liu, G.; Li, X.; Lee, J. W.; Popov, B. N. Catal. Sci. Technol. 2011, 1, 207. doi: 10.1039/c0cy00053a
-
[164]
Kramm, U. I.; Herranz, J.; Larouche, N.; Arruda, T. M.; Lefevre, M.; Jaouen, F.; Bogdanoff, P.; Fiechter, S.; Abs-Wurmbach, I.; Mukerjee, S.; Dodelet, J. P. Phys. Chem. Chem. Phys. 2012, 14, 11673. doi: 10.1039/c2cp41957b
-
[1]
-









图 13 (a) 吡啶氮, (b)吡咯氮, (c)石墨氮, (d)腈和(e)氨基
Figure 13 (a) pyridine-like N, (b) pyrrole-type N, (c) graphite-like N, (d) nitrile and (e) amidogen







-



















 下载:
下载:





















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 168
- 文章访问数: 16664
- HTML全文浏览量: 2822
 下载:
下载:
