引用本文:
张涌灵, 王敏, 曹鹏, 廖建. 铜催化苯乙烯不对称硼胺化反应[J]. 化学学报,
2017, 75(8): 794-797.
doi:
10.6023/A17040144

Citation: Zhang Yongling, Wang Min, Cao Peng, Liao Jian. Copper-Catalyzed Enantioselective Aminoboration of Styrenes with Chiral Sulfoxide Phosphine Ligand[J]. Acta Chimica Sinica, 2017, 75(8): 794-797. doi: 10.6023/A17040144
Citation: Zhang Yongling, Wang Min, Cao Peng, Liao Jian. Copper-Catalyzed Enantioselective Aminoboration of Styrenes with Chiral Sulfoxide Phosphine Ligand[J]. Acta Chimica Sinica, 2017, 75(8): 794-797. doi: 10.6023/A17040144
铜催化苯乙烯不对称硼胺化反应
English
Copper-Catalyzed Enantioselective Aminoboration of Styrenes with Chiral Sulfoxide Phosphine Ligand
Abstract:
To date, copper catalysis has become an attractive approach to access multifunctional alkylborons through borylative coupling processes, many important protocols such as carboboration, stannylboration and aminoboration were developed. Among these methods, however, there is no report involving enantioselective aminoboration of simple styrene substrates, which can generate a class of useful chiral compounds. In this work, an enantioselective Cu-catalyzed aminoboration of styrenes by using a chiral sulfoxide-phosphine (SOP) ligand was developed, chiral β-aminoalkylboranes were obtained in satisfied yields and ee values, and these products can be readily converted to a class of valuable β-hydroxylalkylamines. A general procedure for the aminoboration of styrenes is as following:in glove box, CuCl (0.02 mmol), chiral sulfoxide phosphine L1 (0.022 mmol) and 2.0 mL of dried tetrahydrofuran were added into a flame-dried tube, the resolved solution was stirred for 30 min at room temperature, then bis(pinacolato)diboron (B2pin2) (0.3 mmol), t-BuOLi (0.6 mmol) and styrene (0.2 mmol) were added. The tube was taken out of the glove box and cooled to 0℃. Electrophilic amination reagent, O-benzoyl-N, N-dibenzylhydroxylamine (2a, 0.3 mmol), was dissolved in 1.0 mL of ethyl acetate and added to the mixture, the resolved mixture was stirred at 0℃ for 24 h. The crude product was filtered through a celite pad, concentrated and oxidized by NaBO3·4H2O. The mixture was extracted three times with ethyl acetate, concentrated and purified with silica gel chromatography to give the desired β-hydroxylalkylamines, the enantioselective excess of products were determined by chiral HPLC analysis. Broad substrate scope which related to steric and electronic effect were compatible in this catalysis under the standard conditions. To demonstrate the utility of this method, a gram scale experiment was performed and the desired product was obtained in 92% isolated yield and 90% ee. The benzyl group of products can be readily removed via a Pd/C-catalyzed hydrogenation process and the corresponding product with a free amino group in excellent yield (95%).
-
1 引言
有机硼化合物是一类非常重要的有机合成砌块, 广泛被应用于各类碳—碳键(C—C)和碳—杂键(C—X)形成反应中[1], 因此, 对于这类化合物的合成方法学研究一直是有机化学领域的热点之一.过渡金属(Pd/Pt[2], Cu[3], Co/Fe[4], Ni[5])催化的对碳碳不饱和键的硼化加成反应能够方便、有效地构建烷基硼或烯基硼化合物, 其中, 铜催化因其低毒及相对廉价的优点得到了较为广泛的关注, 通过铜催化硼化偶联反应[6](硼碳化, 硼锡化和硼胺化等)可以很好地构建多官能化烷基硼化合物, 在有机合成中具有广泛的应用前景. α-氨基硼酯类化合物被广泛应用在药物、天然产物和活性分子的合成中[7].铜催化的烯烃1, 2-硼胺化反应[8]是构建该类骨架较为理想的方法之一. 2013年, Miura课题组[8a]报道了首例以联硼酸频哪醇酯(pinB-Bpin)和O-苯甲酰基-N, N-二苄基羟胺试剂(Bn2NOBz)分别作为亲核硼源和亲电氮源, 铜催化实现了苯乙烯类化合物的硼胺化反应.同时对不对称硼胺化进行了初步尝试, 以(S, S)-Me-Duphos作为手性配体, 实现了反式-β-甲基苯乙烯的硼胺化, 得到较好的对映选择性(80%~86% ee)和中等到较好的收率(51%~83%).随后, Miura和Shi课题组分别报道了系列特定结构烯烃如双环烯烃[8e]、烯基硅烷[8f]、烯基硼酯[8g]和含环丙烷烯烃[8b, 8h]等的不对称硼胺化反应.到目前为止, 针对大量商业可得的简单苯乙烯的不对称硼胺化反应尚没有报道.我们课题组近期的研究兴趣集中在通过我们自己设计的手性亚砜配体去实现简单烯烃的不对称官能化, 特别是不对称双官能化.最近, 我们实现了手性亚砜膦配体(SOP)/铜络合物催化苯乙烯的硼碳化反应和硼锡化反应, 获得了优秀的收率和对映选择性[9].在这些前期研究工作基础上, 我们在本工作中对铜催化苯乙烯的不对称硼胺化反应进行了研究, 以手性亚砜膦配体(SOP)/铜络合物作为催化剂, 实现了简单苯乙烯的不对称1, 2-硼胺化反应, 获得了较高的收率和最高达95%的对映选择性.该方法为手性氨基硼化合物提供了一条高效的合成路径.
2 结果与讨论
我们选取苯乙烯(1a)、亲电性胺试剂(2a)和联硼酸频哪醇酯(pinB-Bpin)作为反应底物, 用手性亚砜膦配体(SOP) L1和CuCl原位络合生成活性催化剂, 以叔丁醇锂(LiOBu-t)为碱, 四氢呋喃(THF)为溶剂, 设定反应温度为0 ℃, 对反应进行了初步尝试(表 1, Entry 1).反应24 h后, 以较高的收率(85%)和很好的对映选择性(89% ee)得到烯烃的硼胺化产物.但由于硼胺化产物在硅胶柱层析分离纯化过程中不稳定, 容易发生分解, 因此在实际后处理过程中, 我们将得到的硼胺化产物直接氧化为稳定的β-羟胺化合物3a, 并对其进行表征分析.通过测定3a的比旋光值, 与已知文献进行比对[10], 确定了产物3a的绝对构型为S.为了进一步提高反应的对映选择性, 我们对反应条件进行了优化.
 表 1
反应条件优化a
Table 1.
Reaction conditions screening
表 1
反应条件优化a
Table 1.
Reaction conditions screening

Entry L Base Solvent (V:V) Yieldb/% eec/% 1 L1 t-BuOLi THF 85 89 2 L2 t-BuOLi THF 79 -30 3 L3 t-BuOLi THF 60 50 4 L4 t-BuOLi THF 64 50 5 L5 t-BuOLi THF 49 67 6 L1 t-BuOLi CH3CN 48 88 7 L1 t-BuOLi MTBE 57 82 8 L1 t-BuOLi Tol trace ndf 9d L1 t-BuOLi EA 40 94 10d L1 t-BuOLi THF/EA (1:2) 50 94 11d L1 t-BuOLi THF/EA (1:1) 56 92 12d L1 t-BuOLi THF/EA (2:1) 83 (76) e 93 13d L1 t-BuOK THF/EA (2:1) trace ndf 14d L1 KOH THF/EA (2:1) 71 95.5 15d L1 LiOMe THF/EA (2:1) 56 96 a Conducted with 1a (0.2 mmol), B2(pin)2 (0.3 mmol), Bn2NOBz (0.3 mmol), CuCl (10 mol%), ligand (11 mol%), base (0.6 mmol) in solvent (2.0 mL) at 0 ℃ for 24 h. Then, the resulting mixture was filtered through celite, concentrated, and oxidized by NaBO3•4H2O (8 equiv.) in THF/H2O (V:V=1:1, 4 mL) for 5 h at 20 ℃.b Determined by 1H NMR spectroscopy. c Determined by chiral HPLC. dSolvent is 3.0 mL. e Value in parentheses is isolated yield of 3a. fnd=not detected. 我们首先考察了手性配体(表 1, Entries 2~5), 包括其它我们研究小组所发展的SOP配体(详细信息见SI)和几种商业性双膦配体.与手性亚砜膦配体L1相比, (S, S)-Me-Duphos (L2), (R)-BINAP (L3), (S, S)-BDPP (L4)以及(R)-SEGPHOS (L5)不管是在反应活性还是对映选择性上, 都没有表现出更好的结果(表 1, Entries 2~5).随后, 我们考察了反应溶剂对反应的影响(表 1, Entries 6~12).以乙腈作为溶剂, 反应的对映选择性基本相当, 但是活性却明显降低(表 1, Entry 6);甲基叔丁基醚(MTBE)给出的收率和对映选择性均较低(表 1, Entry 7);该反应在甲苯中无法得到目标产物(表 1, Entry 8);当使用乙酸乙酯(EA)作为溶剂时, 反应的对映选择性得到明显提高(94% ee), 但是收率较低, 这主要是由亲电性胺试剂2a在乙酸乙酯中溶解性较差所致(表 1, Entry 9).值得高兴的是, 我们综合考虑了四氢呋喃的高收率和乙酸乙酯的高对映选择性, 选取了混合溶剂(THF/EA, V:V=2/1), 可同时实现产物3a的高收率和高对映选择性(表 1, Entry 12).最后我们考察了碱对反应的影响(表 1, Entries 13~15).由于亲电性胺试剂2a的转酯化作用, 叔丁醇钾只能得到痕量的产物(表 1, Entry 13);氢氧化钾和甲醇锂均可得到相当的对映选择性, 但是收率较低(表 1, Entry 14, 15).此外我们对亲电性胺试剂苯甲酰基取代基以及氮上不同取代基也进行了系统考察, 但并没有获得更好的结果(见SI).
综合考虑上述实验结果, 我们确定了反应的最优条件:以10 mol% CuCl为催化剂前体, 11 mol% L1为手性配体, 以四氢呋喃/乙酸乙酯(V:V=2:1) 为溶剂, 叔丁醇锂作为碱, 0 ℃条件下反应24 h.
在最优条件下, 我们对铜催化苯乙烯不对称硼胺化反应底物普适性进行了考察(表 2).针对苯环上取代基电子效应和位阻效应, 各种取代苯乙烯底物被引入到反应体系中, 均可以很好地与亲电性胺试剂2a和硼试剂(B2pin2)发生反应, 目标产物以较好的收率和对映选择性获得(产率55%~78%, 60%~95% ee).卤素取代(Cl, Br, F)苯乙烯在该反应中耐受, 可取得中等结果.当苯环取代基为供电子基团时, 相对于缺电子烯烃, 可获得较高的对映选择性.对位强吸电子基取代苯乙烯(3q)因发生强烈的质子化(即硼氢化)竞争副反应, 导致较低的收率和对映选择性.比较有趣的是, 缺电子烯烃和富电子烯烃在位阻方面表现出了不一样的趋势.例如, 间位(3f)和对位(3k)甲基取代的富电子苯乙烯可以获得比邻位(3b)甲基苯乙烯更好的对映选择性.然而卤素(Cl, Br)取代的缺电子苯乙烯却表现出了相反的结果:邻位取代(3c, 3d)的苯乙烯比间位(3g, 3h)和对位(3m, 3n)取代的苯乙烯表现出了更好的对映选择性.相同位置取代基的位阻增大有利于对映选择性, 但是反应活性会有一定程度的降低.当烯烃底物是2-乙烯基噻吩时, 反应同样可以得到很好的对映选择性, 但是收率却很低(3u, 29%, 89% ee), 可能的原因是底物中硫原子与铜有配位作用导致催化剂的活性降低.此外, 非芳香性基团取代烯烃如降冰片二烯也能顺利得到预期产物, 有较好的收率和对映选择性(3v, 75%, 80% ee), 其活性可能来源于其特殊二烯结构和骨架具有较强的张力, 其它非芳香烯烃如环己烯、脂肪链取代的端烯(3w~3y)等在该反应体系中则无活性.由于位阻原因, β-取代苯乙烯(3z)在该体系同样无反应活性, 本工作研究结果表明, 亚砜膦配体与双膦配体如DuPhos在底物适用范围上具有互补性[8a].
表 2
硼胺化底物范围考察a
Table 2.
Substrate scope of aminoboration

为了体现该反应的实用性, 我们对放大实验进行了尝试.在标准反应条件下, 将模板反应规模扩大到克级(1a, 1.04 g), 获得了比小规模反应更好分离收率(92% vs 76%)和基本保持的对映选择性(90% ee).同时, 在温和的反应条件下, 通过钯/碳催化氢化, 可高效地脱除产物的氮苄基保护基(95%), 得到(S)-(+)-2-苯甘氨醇.
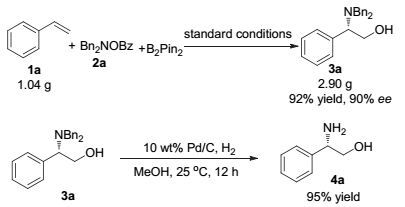 图式1
放大实验和3a脱保护反应
图式1.
Scaled-up experiment and deprotection of 3a
图式1
放大实验和3a脱保护反应
图式1.
Scaled-up experiment and deprotection of 3a
3 结论
我们以铜/手性亚砜膦络合物作为催化剂, 通过对反应条件的优化, 实现了简单苯乙烯底物的不对称1, 2-硼胺化反应, 取得了较高的收率和最高达95%的对映选择性.通过温和的氧化, 可将产物的硼基团转化为羟基, 为手性β-羟胺类化合物的合成提供了一种有效的方法.
-
-
[1]
(a) Pelter, A.; Smith, K.; Brown, H. C. Borane Reagents, Academic Press, London, 1988; (b) Miyaura, N.; Suzuki, A. Chem. Rev.1995, 95, 2457; (c) Davison, M.; Hughes, A. K.; Marder, T. B.; Wade, K. Contemporary Boron Chemistry, RSC, Cambridge, U. K., 2000; (d) Boronic Acids, 2nd ed.; Ed.: Hall, D. G., Wiley-VCH, Weinheim, Germany, 2011.
-
[2]
(a) Ishiyama, T.; Matsuda, N.; Miyaura, N.; Suzuki, A. J. Am.Chem. Soc. 1993, 115, 11018; (b) Ishiyama, T.; Matsuda, N.; Murata, M.; Ozawa, F.; Suzuki, A.; Miyaura, N. Organometallics 1996, 15, 713; (c) Lesley, G.; Nguyen, P.; Taylor, N. J.; Marder, T. B. Organometallics 1996, 15, 5137; (d) Ishiyama, T.; Yamamoto, M.; Miyaura, N. Chem.Commun. 1996, 2073; (e) Ishiyama, T.; Yamamoto, M.; Miyaura, N. Chem.Commun. 1997, 689; (f) Thomas, R. L.; Souza, F. E. S.; Marder, T. B. J. Chem. Soc., Dalton Trans. 2001, 1650; (g) Yang, F.-Y.; Cheng, C.-H. J. Am. Chem. Soc. 2001, 123, 761; (h) Burks, H. E.; Kliman, L. T.; Morken, J. P. J. Am.Chem. Soc. 2009, 131, 9134; (i) Kliman, L. T.; Mlynarski, S. N.; Morken, J. P. J. Am. Chem. Soc. 2009, 131, 13210; (j) Iwadate, N.; Suginome, M. J. Am. Chem. Soc. 2010, 132, 2548; (k) Coombs, J. R.; Haeffner, F.; Kliman, L. T.; Morken, J. P. J. Am. Chem. Soc. 2013, 135, 11222; (l) Coombs, J. R.; Zhang, L.; Morken, J. P. J. Am.Chem. Soc. 2014, 136, 16140; For review, see: Ishiyama, T.; Ishida, K.; Miyaura, N. Tetrahedron 2001, 57, 9813 and references therein.
-
[3]
Selected examples, see: (a) Ito, H. ; Yamanaka, H. ; Tateiwa, J. -i. ; Hosomi, A. Tetrahedron Lett. 2000, 41, 6821; (b) Zhu, W. ; Ma, D. Org. Lett. 2006, 8, 261; (c) Beenen, M. A. ; An, C. ; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6910; (d) Lee, J. E. ; Yun, J. Angew. Chem. , Int. Ed. 2008, 47, 145; (e) Lipshutz, B. H. ; Boskovic, Z. V. ; Aue, D. H. Angew. Chem. , Int. Ed. 2008, 47, 10183; (f) Chen, I. H. ; Yin, L. ; Itano, W. ; Kanai, M. ; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 11664; (g) Kleeberg, C. ; Dang, L. ; Lin, Z. ; Marder, T. B. Angew. Chem. , Int. Ed. 2009, 48, 5350; (h) Lillo, V. ; Prieto, A. ; Bonet, A. ; Díaz-Requejo, M. M. ; Ramírez, J. s. ; Pérez, P. J. ; Fernández, E. Organometallics 2009, 28, 659; (i) Noh, D. ; Chea, H. ; Ju, J. ; Yun, J. Angew. Chem. , Int. Ed. 2009, 48, 6062; (j) O'Brien, J. M. ; Lee, K. S. ; Hoveyda, A. H. J. Am. Chem. Soc. 2010, 132, 10630; (k) Lee, J. C. ; McDonald, R. ; Hall, D. G. Nat. Chem. 2011, 3, 894; (l) Moure, A. L. ; Arrayas, R. G. ; Carretero, J. C. Chem. Commun. 2011, 47, 6701; (m) Solé, C. ; Whiting, A. ; Gulyás, H. ; Fernández, E. Adv. Synth. Catal. 2011, 353, 376; (n) Burns, A. R. ; Solana Gonzalez, J. ; Lam, H. W. Angew. Chem. , Int. Ed. 2012, 51, 10827; (o) Ito, H. ; Kubota, K. Org. Lett. 2012, 14, 890; (p) Yang, C. T. ; Zhang, Z. Q. ; Tajuddin, H. ; Wu, C. C. ; Liang, J. ; Liu, J. H. ; Fu, Y. ; Czyzewska, M. ; Steel, P. G. ; Marder, T. B. ; Liu, L. Angew. Chem. , Int. Ed. . 2012, 51, 528; (q) Feng, X. ; Jeon, H. ; Yun, J. Angew. Chem. , Int. Ed. 2013, 52, 3989; (r) Semba, K. ; Nakao, Y. J. Am. Chem. Soc. 2014, 136, 7567; (s) Smith, K. B. ; Logan, K. M. ; You, W. ; Brown, M. K. Chem. Eur. J. 2014, 20, 12032; (t) Logan, K. M. ; Smith, K. B. ; Brown, M. K. Angew. Chem. , Int. Ed. 2015, 54, 5228; (u) Su, W. ; Gong, T. J. ; Lu, X. ; Xu, M. Y. ; Yu, C. G. ; Xu, Z. Y. ; Yu, H. Z. ; Xiao, B. ; Fu, Y. Angew. Chem. , Int. Ed. 2015, 54, 12957; (v) Liu, Q. ; Tian, B. ; Tian, P. ; Tong, X. ; Lin, G. -Q. Chin. J. Org. Chem. 2015, 35, 1; (刘强, 田兵, 田平, 童晓峰, 林国强, 有机化学, 2015, 35, 1. ) (w) Semba, K. ; Ohtagaki, Y. ; Nakao, Y. Org. Lett. 2016, 18, 3956; (x) Smith, J. J. ; Best, D. ; Lam, H. W. Chem. Commun. 2016, 52, 3770; (y) Logan, K. M. ; Brown, M. K. Angew. Chem. , Int. Ed. 2017, 56, 851.
-
[4]
Selected examples, see: (a) Adams, C. J.; Baber, R. A.; Batsanov, A. S.; Bramham, G.; Charmant, J. P.; Haddow, M. F.; Howard, J. A.; Lam, W. H.; Lin, Z.; Marder, T. B.; Norman, N. C.; Orpen, A. G. Dalton Trans. 2006, 1370; (b) Obligacion, J. V.; Chirik, P. J. Org. Lett.2013, 15, 2680; (c) Obligacion, J. V.; Chirik, P. J. J. Am.Chem. Soc. 2013, 135, 19107. (d) Zhang, L.; Peng, D.; Leng, X.; Huang, Z. Angew. Chem., Int. Ed. 2013, 52, 3676; (e) Cao, Y.; Zhang, Y.; Zhang, L.; Zhang, D.; Leng, X.; Huang, Z. Org. Chem. Front.2014, 1, 1101; (f) Chen, J.; Xi, T.; Lu, Z.Org. Lett. 2014, 16, 6452; (g) Chen, J.; Xi, T.; Ren, X.; Cheng, B.; Guo, J.; Lu, Z. Org. Chem. Front. 2014, 1, 1306; (h) Zhang, L.; Zuo, Z.; Leng, X.; Huang, Z. Angew. Chem., Int. Ed. 2014, 53, 2696; (i) Zhang, L.; Zuo, Z.; Wan, X.; Huang, Z. J. Am. Chem. Soc. 2014, 136, 15501; (j) Guo, J.; Chen, J.; Lu, Z. Chem. Commun. 2015, 51, 5725; (k) Zhang, L.; Huang, Z. J. Am. Chem. Soc. 2015, 137, 15600; (l) Zhang, H.; Lu, Z. ACS Catal. 2016, 6, 6596; (m) Zuo, Z.; Huang, Z. Org. Chem. Front. 2016, 3, 434; (n) Zuo, Z.; Yang, J.; Huang, Z. Angew. Chem., Int.Ed. 2016, 55, 10839; (o) Xi, T.; Lu, Z. ACS Catal. 2017, 7, 1181.
-
[5]
(a) Suginome, M.; Matsuda, T.; Yoshimoto, T.; Ito, Y. Org. Lett.1999, 1, 1567; (b) Suginome, M.; Shirakura, M.; Yamamoto, A. J.Am. Chem. Soc. 2006, 128, 14438; (c) Ely, R. J.; Morken, J. P. J. Am. Chem. Soc. 2010, 132, 2534.
-
[6]
Reviews: (a) Semba, K. ; Fujihara, T. ; Terao, J. ; Tsuji, Y. Tetrahedron 2015, 71, 2183. (b) Liu, Y. ; Zhang, W. Chin. J. Org. Chem. 2016, 36, 2249. (刘媛媛, 张万斌, 有机化学, 2016, 36, 2249. )
-
[7]
(a) Bloch, R. Chem. Rev. 1998, 98, 1407; (b) Ramadhar, T. R.; Batey, R. A. Synthesis 2011, 1321; (c) Yus, M.; González-Gómez, J. C.; Foubelo, F. Chem. Rev. 2013, 113, 5595.
-
[8]
(a) Matsuda, N.; Hirano, K.; Satoh, T.; Miura, M. J. Am.Chem. Soc. 2013, 135, 4934; (b) Sakae, R.; Matsuda, N.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 1228; (c) Parra, A.; Amenos, L.; Guisan-Ceinos, M.; Lopez, A.; Garcia Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833; (d) Sakae, R.; Hirano, K.; Miura, M. J. Am. Chem. Soc.2015, 137, 6460; (e) Sakae, R.; Hirano, K.; Satoh, T.; Miura, M. Angew.Chem., Int. Ed. 2015, 54, 613; (f) Kato, K.; Hirano, K.; Miura, M. Angew. Chem., Int. Ed. 2016, 55, 14400; (g) Nishikawa, D.; Hirano, K.; Miura, M. Org. Lett.2016, 18, 4856; (h) Shi, M. Chem. Commun. 2016, 52, 5273.
-
[9]
(a) Chen, B.; Cao, P.; Yin, X.; Liao, Y.; Jiang, L.; Ye, J.; Wang, M.; Liao, J. ACS Catal. 2017, 7, 2425; (b) Jia, T.; Cao, P.; Wang, B.; Lou, Y.; Yin, X.; Wang, M.; Liao, J. J. Am.Chem. Soc. 2015, 137, 13760; (c) Jia, T.; Cao, P.; Wang, D.; Lou, Y.; Liao, J. Chem. Eur. J.2015, 21, 4918.
-
[10]
Metro, T. X.; Appenzeller, J.; Pardo, D. G.; Cossy, J. Org. Lett. 2006, 8, 3509. doi: 10.1021/ol061133d
-
[1]
-

图式1 放大实验和3a脱保护反应
Scheme 1 Scaled-up experiment and deprotection of 3a
Reaction conditions: 10 wt% Pd/C wetted with ca. 55% water (0.04 mmol), (S)-3a (0.4 mmol) in MeOH (3.0 mL) under hydrogen by balloon. The suspension was stirred at 25 ℃ for 12 h.
表 1 反应条件优化a
Table 1. Reaction conditions screening
Entry L Base Solvent (V:V) Yieldb/% eec/% 1 L1 t-BuOLi THF 85 89 2 L2 t-BuOLi THF 79 -30 3 L3 t-BuOLi THF 60 50 4 L4 t-BuOLi THF 64 50 5 L5 t-BuOLi THF 49 67 6 L1 t-BuOLi CH3CN 48 88 7 L1 t-BuOLi MTBE 57 82 8 L1 t-BuOLi Tol trace ndf 9d L1 t-BuOLi EA 40 94 10d L1 t-BuOLi THF/EA (1:2) 50 94 11d L1 t-BuOLi THF/EA (1:1) 56 92 12d L1 t-BuOLi THF/EA (2:1) 83 (76) e 93 13d L1 t-BuOK THF/EA (2:1) trace ndf 14d L1 KOH THF/EA (2:1) 71 95.5 15d L1 LiOMe THF/EA (2:1) 56 96 a Conducted with 1a (0.2 mmol), B2(pin)2 (0.3 mmol), Bn2NOBz (0.3 mmol), CuCl (10 mol%), ligand (11 mol%), base (0.6 mmol) in solvent (2.0 mL) at 0 ℃ for 24 h. Then, the resulting mixture was filtered through celite, concentrated, and oxidized by NaBO3•4H2O (8 equiv.) in THF/H2O (V:V=1:1, 4 mL) for 5 h at 20 ℃.b Determined by 1H NMR spectroscopy. c Determined by chiral HPLC. dSolvent is 3.0 mL. e Value in parentheses is isolated yield of 3a. fnd=not detected.  下载: 导出CSV
下载: 导出CSV
-


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 24
- 文章访问数: 2292
- HTML全文浏览量: 345
 下载:
下载:
