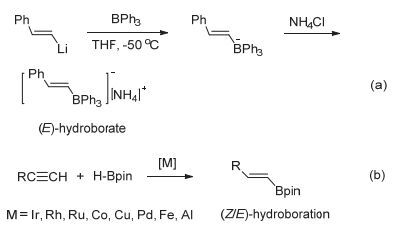
 图 1
制备硼氢化产物的方法
Figure 1.
The preparation of the hydroborate compounds
图 1
制备硼氢化产物的方法
Figure 1.
The preparation of the hydroborate compounds
引用本文:
孙国峰, 苏敏, 方洁, BorzovMaxim, 聂万丽. 有机胺盐/硼烷体系与炔烃的硼氢化加成反应机理研究[J]. 化学学报,
2017, 75(8): 824-830.
doi:
10.6023/A17040141

Citation: Sun Guofeng, Su Min, Fang Jie, Borzov Maxim, Nie Wanli. Research of the Stereoselectivity and Mechanism of the Hydroboration Reaction Between B(C6F5)3/Ammonium Chloride Systems with Terminal Alkyne[J]. Acta Chimica Sinica, 2017, 75(8): 824-830. doi: 10.6023/A17040141
Citation: Sun Guofeng, Su Min, Fang Jie, Borzov Maxim, Nie Wanli. Research of the Stereoselectivity and Mechanism of the Hydroboration Reaction Between B(C6F5)3/Ammonium Chloride Systems with Terminal Alkyne[J]. Acta Chimica Sinica, 2017, 75(8): 824-830. doi: 10.6023/A17040141
有机胺盐/硼烷体系与炔烃的硼氢化加成反应机理研究
摘要:
炔烃的立体选择性硼氢化加成反应是有机合成中重要的反应之一.在硅烷的存在下,有机胺盐酸盐/硼烷体系可与炔烃在温和的反应条件下发生计量的加成反应.该反应不仅可高立体选择性地得到Z-式构型的1,2-硼氢化胺盐加成产物,而且反应产率高,产物易于分离提纯.对有机胺盐酸盐/硼烷体系与炔烃的加成反应机理进行的研究表明,胺盐与B(C6F5)3及硅烷反应所生成的硼氢化胺盐"[R2NH2]+[H-B(C6F5)3]-",虽然被认为是受限路易斯酸碱对化学的活性中间体,但其本身并不能直接还原炔烃;炔烃必须首先被催化量的路易斯酸B(C6F5)3活化后才可与[H-B(C6F5)3]-加成.同时,胺盐氯阴离子Cl-与路易斯酸B(C6F5)3之间的弱的相互作用直接决定着产物的立体选择性,[H-B(C6F5)3]-以反式加成的方式进攻活化后的炔烃最终得到Z-式构型的硼氢化加成产物.
English
Research of the Stereoselectivity and Mechanism of the Hydroboration Reaction Between B(C6F5)3/Ammonium Chloride Systems with Terminal Alkyne
Abstract:
Stereoselective hydroboration reaction of alkynes has been considered as one of the most important organic reaction. To date a handful of metal-catalyzed systems have been demonstrated to achieve trans-hydroboration of alkynes. This paper describes the first non-metal-catalyzed systems which could stereoselectively hydroborate the terminal alkynes in a trans-configuration. The Lewis acid B(C6F5)3 and ammonium chloride have been used as the reaction substrates, and phenylsilane as the hydride source. The hydroboration reaction could be performed in a one-pot procedure by mixing of B(C6F5)3, ammonium chloride and silane together in an equivalent amount. But this one-pot reaction is not so nice since there is always mixed with the ammonium hydroborate[R2NH2]+[H-B(C6F5)3]- intermediates products. A series of ammonium hydroborates prepared from the corresponding primary, secondary, tertiary and quaternary amine hydrochlorides have been isolated, and used in the directly hydroboration with terminal alkynes. To our surprise the ammonium hydroborate[R2NH2]+[H-B(C6F5)3]- could not react with the alkynes alone. When using[R2NH2]+[H-B(C6F5)3]- to react with alkynes, trace amount of catalytic Lewis acid B(C6F5)3 is necessary to firstly activate the carbon-carbon triple bonds and form the crucial zwitterionic σ-complexes. The mechanism study has shown that different from the typical Lewis acid/Lewis base FLPs system reacted with alkynes, in this B(C6F5)3/ammonium chloride system the ammonium chloride plays an important role on the stereoselective control of the reaction. The week interaction between the Cl ion and B(C6F5)3 in the σ-complexes has not only slowed down the unfavorite 1, 1-carboboration reaction, but also stabilized the σ-complexes which has offer the chance for the nucleophilic reagent to attack the reaction center in a cis-or trans-mode. In our experiment the bulky ion[H-B(C6F5)3]-could only attach the active alkynes from the trans-side and form the Z-hydroboration product. This work demonstrates that the combination of the ammonium halides with the Lewis acid B(C6F5)3 could act as a novel "frustrated Lewis pair" to activate alkynes, and will enable the development of even more sophisticated FLP and related catalyzed reactions.
-
Key words:
- ammonium chloride
- / B(C6F5)3
- / silane
- / alkyne
- / hydroboration
-
1 引言
硼氢化反应是有机反应中重要的反应之一, 其生成的烯基硼化物可进一步转化为其他含双键的有机化合物, 故硼氢化反应在有机反应中有着重要的作用.最早制备硼氢化产物的方法报道于1991年, Baillargeon及其团队[1]将苯乙烯基锂盐与BPh3进行紫外光照辐射后与氯化铵反应, 可转化成E式的类炔烃的硼氢化铵盐加成产物(见图 1a).以过渡金属铱、铑、钯、钴、铜等络合物催化的炔烃与氢化硼烷(如HBpin, HBCat)的加成反应常得到Z式硼氢化产物(见图 1b); 而E式构型的加成产物可通过金属铝的催化来实现[2~8].
图 1
制备硼氢化产物的方法
Figure 1.
The preparation of the hydroborate compounds
近些年来针对以硼烷作为路易斯酸的非金属催化剂“受限路易斯酸碱对FLPs”化学的研究也受到了越来越多的关注[7~11].经典的受限路易斯酸碱对与炔烃主要发生两种反应: (1) 在没有路易斯碱时主要发生1, 1-碳硼烷化[12~14], 该反应所得到的加成产物Z/E构型并存; (2) FLPs与炔烃一般发生路易斯酸碱对的反式1, 2-加成反应[15].
我们课题组最近报道了一种研究受限路易斯酸碱对化学的新方法, 以有机胺盐酸盐代替常规FLPs体系中的有机胺路易斯碱, 与硼烷组成新的“路易斯酸/胺盐”反应体系.该体系在以硅烷作为还原剂时对醛酮及二氧化碳的还原均表现出独特的反应活性[16~19].发现在HCl的存在下, 该体系与炔烃的氢氯化加成产物具有较高的选择性(见图 2c)[20].并且发现当有机胺盐为碘盐时, 可高立体选择性地得到E式构型的1, 1-碳硼烷化产物(见图 2d)[21].
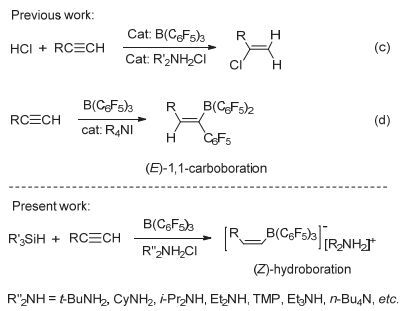 图 2
B (C6F5)3/有机胺盐体系与炔烃的反应
Figure 2.
The reaction of B (C6F5)3/ammonium systems with alkynes
图 2
B (C6F5)3/有机胺盐体系与炔烃的反应
Figure 2.
The reaction of B (C6F5)3/ammonium systems with alkynes
本文主要研究了在硅烷的存在下, 有机胺盐酸盐/硼烷体系与炔烃计量硼氢化反应.该反应在温和的反应条件下不仅可高立体选择性地得到Z-式构型的1, 2-硼氢化胺盐加成反应, 而且反应产率高, 产物易于分离提纯.本文同时也对有机胺盐酸盐/硼烷体系与炔烃的加成反应机理进行了较详细的探讨.
2 结果与讨论
2.1 硼氢化物的制备方法
其实, 硼氢化胺盐的制备方法可以有直接法和间接法两种方法, 如图 3所示.直接法是不需要分离出中间产物硼氢化胺盐4, 直接将苯硅烷、胺盐酸盐、B (C6F5)3(BCF)和炔烃计量混合反应.由于直接法通常会得到炔烃的硼氢化胺盐加成产物5与4的混合物, 这两种盐较难分离纯化.
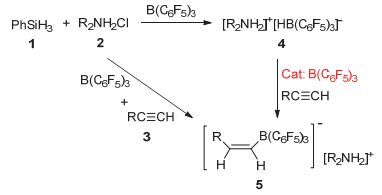 图 3
硼氢化胺盐的制备方法
Figure 3.
Preparation of hydroborate ammoniums
图 3
硼氢化胺盐的制备方法
Figure 3.
Preparation of hydroborate ammoniums
间接法制备硼氢化物胺盐5时, 需提前制备中间产物硼氢化胺盐4.中间产物4的制备及纯化较简单:向溶解有胺盐酸盐及B (C6F5)3的氯仿溶液中加入苯硅烷PhSiH3, 一般室温反应2 h后可向反应液中加入正己烷析出硼氢化胺盐4.由于硼氢化烷胺盐4通常较稳定, 加热条件下一般也不会分解.炔烃在与硼氢化烷胺盐4发生加成反应时, 必须加入催化量的B (C6F5)3来活化炔烃π键.
表 1中列出了的一系列炔烃的硼氢化胺盐加成反应都是采用的间接方法.从表中数据可以看出, 无论炔烃上带有吸电子基团还是给电子基团, 几乎所有的伯、仲、叔或季胺的硼氢化盐都可与之发生Z式的1, 2加成反应.而位阻较小的二乙胺或叔丁胺盐酸盐反应体系, 如Entries 14和15在室温或加热条件下, 加成产物可进一步转化成相应的还原产物烯烃.一般情况下, 室温反应2 h后向反应液中缓慢加入正己烷可得到适合X-单晶结构解析的晶体或纯化析出, 反应几乎计量进行.
 表 1
端基炔烃的Z式硼氢化加成反应a
Table 1.
(Z)-Hydroboration of terminal alkynesa
表 1
端基炔烃的Z式硼氢化加成反应a
Table 1.
(Z)-Hydroboration of terminal alkynesa
Entry R2NH Alkyne Product 1 CyNH2 p-F-Ph-CCH [CyNH3][F-Ph-CH=CHBCF] 2 Et3NH p-F-Ph-CCH [Et3NH][F-Ph-CH=CHBCF] 3 Et3NH Ph-CCH [Et3NH][Ph-CH=CHBCF] 4 Et3NH p-Me-Ph-CCH [Et3NH][Me-Ph-CH=CHBCF] 5 n-Bu4N p-F-Ph-CCH [n-Bu4N][F-Ph-CH=CHBCF] 6 n-Bu4N Ph-CCH [n-Bu4N][Ph-CH=CHBCF] 7 n-Bu4N p-Me-Ph-CCH [n-Bu4N][Me-Ph-CH=CHBCF] 8 TMP p-F-Ph-CCH [TMPH][F-Ph-CH=CHBCF] 9 TMP Ph-CCH [TMPH][Ph-CH=CHBCF] 10 TMP p-Me-Ph-CCH [TMPH][Me-Ph-CH=CHBCF] 11 i-Pr2NH p-F-Ph-CCH [i-Pr2NH2][F-Ph-CH=CHBCF] 12 i-Pr2NH Ph-CCH [i-Pr2NH2][Ph-CH=CHBCF] 13 i-Pr2NH p-Me-Ph-CCH [i-Pr2NH2][Me-Ph-CH=CHBCF] 14 Et2NH p-F-Ph-CCH [Et2NH2][F-Ph-CH=CHBCF]+F-Ph-CH=CH2b 15 t-BuNH2 p-F-Ph-CCH F-Ph-CH=CH2b a Reaction condition: r.t., Cat: BCF 5 mol%; b Reduction product. 2.2 有机胺盐酸盐/硼烷体系与炔烃的加成反应机理研究
在B (C6F5)3参与的炔烃加成反应中, B (C6F5)3首先会与炔烃结合形成缺电子性的σ-络合物, 如图 4所示. σ-络合物极不稳定, 在没有路易斯碱的条件下, 通常会立即发生(E/Z)-1, 1-碳硼烷化反应.当体系中有负电子性亲核试剂或路易斯碱时, 亲核试剂可以cis或trans构型与缺电子中心结合[22].一般情况下, 当亲核试剂为大位阻路易斯碱时, 通常都会形成反式1, 2加成产物, 如图中4b所示.本文在两步法制备硼氢化加成产物时, 硼氢化胺盐[R2NH2][H-BCF]是不能够直接与炔烃加成的, 必须在反应体系中加入微量的BCF作为催化剂活化炔烃形成σ-络合物, 然后胺盐阴离子[H-BCF]作为亲核基团以trans构型与σ-络合物作用给出氢负离子H-, 最后得到Z式硼氢化加成产物.
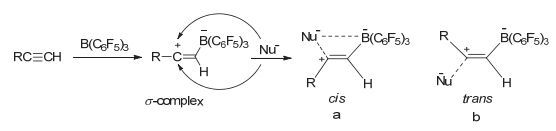 图 4
炔烃的立体选择性硼氢化加成反应机理
Figure 4.
Mechanism of the stereoselective hydroboration of alkynes
图 4
炔烃的立体选择性硼氢化加成反应机理
Figure 4.
Mechanism of the stereoselective hydroboration of alkynes
对反应机理的研究表明, 当有机胺盐/硼烷体系在与炔烃加成时, 不仅有机胺盐的卤阴离子的不同会影响到加成产物的立体结构, 而且由于卤阴离子与活性体σ-络合物的相互作用, 如图中4a所示, 在一定程度上不仅稳定了σ-络合物的存在, 同时抑制了炔烃与B (C6F5)3的1, 1-碳硼烷化副反应, 使得亲核试剂对σ-络合物的立体选择性加成成为可能.例如, 当亲核试剂是离子半径较小的Cl-离子时, 如图 2中(c)所示, 炔烃与HCl的1, 2氢氯化加成产物主要是通过cis反应机理决定加成产物的立体选择性.但是我们发现由于胺盐上质子与硼烷络合的方式会随着质子数的数量的多少而发生变化[23], 因此胺盐的用量及胺盐的种类都会对σ-络合物的稳定性产生影响.
例如当n(TMPHCl):n(BCF)比例为1:1或2:1时, 如图 5所示, 体系中游离的BCF与氯离子络合的Cl-B (C6F5)3同时存在; 而当n(TMPHCl):n(BCF)比例为3:1时, 体系中硼烷只能以Cl-B (C6F5)3形式存在, 这种络合结果与铵盐上没有质子的季铵盐体系非常类似.事实上相应的体系在与炔烃反应时, 也表现出类似的反应活性.
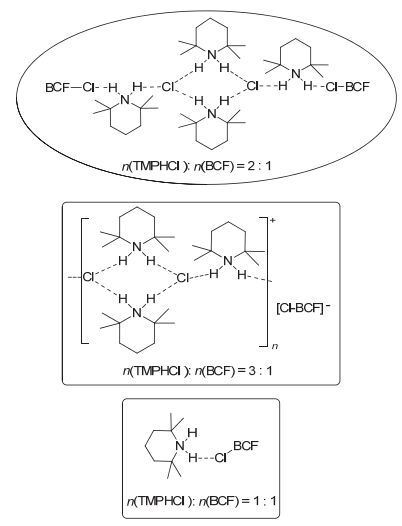 图 5
有机胺盐酸盐与B (C6F5)3的不同络合方式
Figure 5.
Different coordination modes of ammonium hydrochlorides with B (C6F5)3
图 5
有机胺盐酸盐与B (C6F5)3的不同络合方式
Figure 5.
Different coordination modes of ammonium hydrochlorides with B (C6F5)3
图 6所示为不同反应条件下相应的11B NMR峰对比图.从图中可以看出当炔烃与2, 2, 6, 6-四甲基哌啶盐酸盐(TMPHCl)反应时, 随着反应体系中n(盐酸盐)与n[B (C6F5)3]的比例从1:1增加到3:1时, 由于络合效应的影响, 体系中自由的B (C6F5)3相应减少, 对应于δ -12 (σ-络合物)处的单峰明显减弱; 而Cl-B (C6F5)3却不断增多, 对应于δ -5~8处的较宽单峰明显增强; 当n(TMPHCl)与n[B (C6F5)3]的比例为3:1时, 与季铵盐如氯代四正丁铵体系类似, 仅在δ -8处出现Cl-B (C6F5)3较宽的单峰, 观察不到σ-络合物的生成.同时我们也观察到增加炔烃的比例不会影响σ-络合物的稳定性.
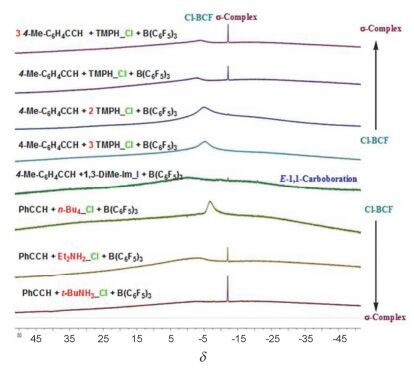 图 6
R2NH2Cl/B (C6F5)3体系与炔烃反应的11B核磁共振谱图
Figure 6.
11B NMR of R2NH2Cl/B (C6F5)3 reaction with alkynes
图 6
R2NH2Cl/B (C6F5)3体系与炔烃反应的11B核磁共振谱图
Figure 6.
11B NMR of R2NH2Cl/B (C6F5)3 reaction with alkynes
当胺盐与硼烷的比例为1:1时, 不同种类的伯、仲、叔、季胺盐酸盐反应体系也会对σ-络合物的稳定性产生影响.如图 6中所示, 质子数较多的伯胺盐酸盐反应体系在δ -12处出现较强的σ-络合物信号峰; 而仲胺盐酸盐反应体系可同时观察到Cl-B (C6F5)3及σ-络合物的相应信号峰.
当亲核试剂是离子半径较大的I-离子时, 如图 2中(d)所示, 由于碘负离子主要是通过trans构型与σ-络合物作用, 同时又由于碘负离子是很好的离去基团, 因此主要得到高立体选择性的(E)-1, 1-碳硼烷化产物[24].在图 5所示的1, 3-二甲基咪唑碘盐/B (C6F5)3体系与炔烃的反应中, 其硼谱上仅能观察到微弱的σ-络合物生成.
3 结论
以有机胺盐酸盐代替常规FLPs体系中的烷基胺路易斯碱, 与硼烷组成新的“路易斯酸/胺盐”反应体系, 在一定程度上扩展了FLPs化学的研究方法.而有机胺盐/硼烷体系在与炔烃加成反应时, 有机胺盐阴离子与活性体σ-络合物的相互作用, 不仅直接影响到炔烃加成产物的立体结构, 而且通过改变阴离子的种类, 可进一步扩展炔烃的加成反应的类型.我们实验室正积极地开展着有关这方面的研究工作.
4 实验部分
本实验的所有操作都是在无水无氧条件下进行的, 所有反应溶剂及氘代试剂均在氮气保护下经过干燥除水处理[25].
4.1 硼氢化烷胺盐的制备
参照文献[19]以Et3NHHBCF的制备为例, 分别将13.7 mg Et3NHCl、55 mg BCF和10.8 mg PhSiH3加入10 mL CH2Cl2中反应1 h后, 向该体系加入正己烷, 在反应过程中大部分的硼氢化铵盐会沉淀析出, 通过无水无氧方法过滤、CH2Cl2洗涤3次、真空抽滤干燥过夜可得到较纯净硼氢化铵盐.
4.2 (Z)-[TMPH][Ph-CH=CHBCF]的制备
在核磁管中加入6.50 mg (0.01 mmol) TMPHHBCF溶于CDCl3中, 再加入1.0 mg (0.01 mmol)苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅橙色, 后又变为浅色.所分离得到的硼氢化产物产率为: 64.84%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.39 (s, 12H, 2, 6-CH3), 1.73~1.65 (m, 4H, 3, 5-CH2), 1.84~1.75 (m, 2H, 4-CH2), 4.90 (br, 2H, NH2), 6.66 (d, J=15.20 Hz, 1H, =C-H), 6.82 (d, J=15.20 Hz, 1H, =C-H), 6.97~6.91 (m, 1H, p-C6H5), 7.06~6.98 (m, 4H, C6H5); 11B NMR (128 MHz, 298 K, CDCl3) δ: -15.9 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.0 (d, J=20.99 Hz, o-C6F5), -162.70 (t, J=20.99 Hz, p-C6F5), -166.43 (t, J=20.99 Hz, m-C6F5); 13C {1H}NMR (100 MHz, 298 K, CDCl3) δ: 15.70, 27.97, 35.33, 60.59, 125.27, 127.04, 127.98, 132.90, 136.50, 137.60, 140.81, 148.27.
4.3 (Z)-[TMPH][Me-Ph-CH=CHBCF]的制备
在核磁管中加入13.1 mg (0.02 mmol) TMPHHBCF溶于CDCl3中, 再加入2.4 mg (0.02 mmol)对甲基苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色, 一段时间后颜色稍加深.所分离得到的硼氢化产物产率为: 57.14%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.36 (s, 12H, 2, 6-CH3), 1.73~1.63 (m, 4H, 3, 5-CH2), 1.83~1.73 (m, 2H, 4-CH2), 2.19 (s, 3H, CH3-C6H4), 4.69 (bs), 6.61 (d, J=14.6 Hz, 1H, =C-H), 6.75 (d, J=14.6Hz, 1H, =C-H), 6.83 (d, J=7.6 Hz, 2H, m-C6H4), 6.93 (d, J=7.6 Hz, 2H, o-C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.05 (d, J=20.9 Hz, o-C6F5), -162.81 (t, J=20.9 Hz, p-C6F5), -166.47 (t, J=20.9 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 8.2, 15.6, 20.89, 27.85, 35.1, 60.7, 126.13, 127.97, 132.75, 135.05, 136.26, 137.95, 138.13, 148.27.
4.4 (Z)-[TMPH][F-Ph-CH=CHBCF]的制备
在核磁管中加入13.1 mg (0.02 mmol) TMPHHBCF溶于CDCl3中, 再加入2.7 mg对氟苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅橙色, 又迅速变为浅黄色.所分离得到的硼氢化产物产率为: 59.77%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.42 (s, 12H, 2, 6-CH3), 1.75~1.68 (m, 4H, 3, 5-CH2), 1.86~1.78 (m, 2H, 4-CH2), 4.80 (t, J=46.1 Hz, 2H, NH2), 6.61 (d, J=14.6 Hz, 1H, =C-H), 6.68 (t, J=8.7 Hz, 2H), 6.75 (d, J=14.6 Hz, 1H, =C-H), 6.96 (dd, J=7.3, 8.7 Hz, 2H); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -118.01 (s, F-C6H4), -131.94 (d, J=20.77 Hz, o-C6F5), -162.56 (t, J=19.0 Hz, p-C6F5), -166.31 (t, J=19.0 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 15.6, 28.1, 35.1, 61.1, 113.41, 113.58, 127.76, 129.37, 129.45, 132.09, 136.32, 137.13, 137.89, 141.18, 159.53, 161.97.
4.5 (Z)-[n-Bu4N][Ph-CH=CHBCF]的制备
在核磁管中加入7.5 mg (0.01 mmol) n-Bu4NHHBCF溶于CDCl3中, 再加入1.2 mg (0.01 mmol)苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 66.67%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 0.968 (t, J=7.30 Hz, 12H, CH3), 1.34 (sxt, J=7.30 Hz, 8H, CH2), 1.59~1.46 (m, 8H, CH2), 3.05~2.93 (m, 8H, CH2), 6.67 (t, J=15.10 Hz, 1H, =C-H), 6.75 (d, J=15.10 Hz, 1H, =C-H), 7.01~6.85 (m, 5H, C6H5); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.3 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -131.70 (d, J=21.36 Hz, o-C6F5), -163.89 (t, J=21.36 Hz, p-C6F5), -167.35 (t, J=21.36 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 13.35, 19.50, 23.60, 58.80, 124.65, 126.5, 127.95, 132.92, 136.33, 137.67, 141.64, 148.40.
4.6 (Z)-[n-Bu4N][Me-Ph-CH=CHBCF]的制备
在核磁管中加入7.5 mg (0.01 mmol) n-Bu4NHHBCF溶于CDCl3中, 再加入1.3 mg (0.01 mmol)对甲基苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为棕红色, 一段时间后, 溶液颜色变浅.所分离得到的硼氢化产物产率为: 62.50%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 0.96 (t, J=7.30 Hz, 12H, CH3), 1.34 (sxt, J=7.32 Hz, 8H, CH2), 1.57~1.46 (m, 8H, CH2), 2.18 (s, 3H, CH3C6H4), 3.03~2.92 (m, 8H, CH2), 6.63 (d, J=14.90 Hz, 1H, =C-H), 6.69 (d, J=14.90 Hz, 1H, =C-H), 6.76 (d, J=7.9 Hz, 2H, C6H4), 6.86 (d, J=7.9 Hz, 2H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H} NMR (376 MHz, 298 K, CDCl3) δ: -131.69 (d, J=18.60 Hz, o-C6F5), -164.0 (t, J=20.44 Hz, p-C6F5), -167.41 (t, J=19.14 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 13.4, 19.5, 21.0, 23.6, 58.80, 127.19, 127.86, 132.87, 134.13, 136.14, 137.9, 138.77, 148.0.
4.7 (Z)-[n-Bu4N][F-Ph-CH=CHBCF]的制备
在核磁管中加入7.5 mg (0.01 mmol) n-Bu4NHHBCF溶于CDCl3中, 再加入1.3 mg (0.01 mmol)对氟苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液无明显颜色变化.所分离得到的硼氢化产物产率为: 72.45%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.35 (sxt, J=7.4 Hz, 8H, CH2), 1.60~1.50 (m, 8H, CH2), 3.04~2.96 (m, 8H, CH2), 6.65 (t, J=8.75 Hz, 2H, C6H4), 6.73 (d, J=14.35 Hz, 1H, =C-H), 6.95 (dd, J=8.4, 5.8 Hz, 2H); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -119.1 (s, F-C6H4), -131.71 (d, J=20.28 Hz, o-C6F5), -163.72 (t, J=20.46 Hz, p-C6F5), -167.22 (t, J=20.04 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 13.7, 19.5, 23.6, 59.0, 77.0.
4.8 (Z)-[i-Pr2NH2][Ph-CH=CHBCF]的制备
在核磁管中加入6.1 mg (0.01 mmol) i-Pr2NH2HBCF溶于CDCl3中, 再加入1.2 mg (0.01 mmol)苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 74.70%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.31 (d, J=6.5 Hz, 12H, CH3), 3.49 (sep, J=6.5 Hz, 1H, CH3CH), 5.27 (br, 2H, NH2), 6.62 (d, J=13.47 Hz, 1H, =C-H), 6.83 (d, J=14.87 Hz, 1H, =C-H), 7.00~6.94 (m, 1H, C6H4), 7.07~7.00 (m, 4H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.43 (d, J=20.77 Hz, o-C6F5), -162.39 (t, J=20.77 Hz, p-C6F5), -166.31 (t, J=20.77 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 20.2, 50.7, 125.66, 127.30, 128.04, 132.88, 135.15, 137.88, 140.32, 148.23.
4.9 (Z)-[i-Pr2NH2][Me-Ph-CH=CHBCF]的制备
在核磁管中加入6.1 mg (0.01 mmol) i-Pr2NH2HBCF溶于CDCl3中, 再加入1.2 mg (0.01 mmol)对甲基苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为棕红色, 一段时间后, 颜色变浅.所分离得到的硼氢化产物产率为: 63.86%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.30 (d, J=5.91 Hz, 12H, CH3CH), 2.20 (s, 3H, CH3), 3.47 (sep, J=5.91 Hz, 2H, CH3CH), 5.48 (bs, 2H, NH2), 6.58 (d, J=14.72 Hz, 1H, =C-H), 6.77 (d, J=14.92 Hz, 1H, =C-H), 6.85 (t, J=7.89 Hz, 2H, C6H4), 6.95 (d, J=7.89 Hz, 2H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.47 (d, J=20.60 Hz, o-C6F5), -162.50 (t, J=20.60 Hz, p-C6F5), -166.35 (t, J=20.60 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 19.7, 21.0, 50.5, 127.94, 128.02, 132.68, 135.40, 137.39.
4.10 (Z)-[i-Pr2NH2][F-Ph-CH=CHBCF]的制备
在核磁管中加入6.2 mg (0.01 mmol) i-Pr2NH2HBCF溶于CDCl3中, 再加入1.2 mg (0.01 mmol)对氟苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 59.77%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.36 (d, J=6.27 Hz, 12H, CH3), 3.58 (sep, J=6.27 Hz, 2H, CH3CH), 6.59 (d, J=14.82 Hz, 1H, =C-H), 6.70 (t, J=8.78 Hz, 2H, C6H4), 6.75 (d, J=14.82 Hz, 1H, =C-H), 6.96 (dd, J=7.94 Hz, 2H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -117.36 (s, F-C6H4), -132.3 (d, J=20.40 Hz, o-C6F5), -162.18 (t, J=20.48 Hz, p-C6F5), -166.16 (t, J=20.40 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 19.5, 50.5, 113.46/113.67, 100.7, 127.7, 129.44/129.34, 132.12.
4.11 (Z)-[Et3NH][Ph-CH=CHBCF]的制备
在核磁管中加入6.1 mg (0.01 mmol) Et3NHHBCF溶于CDCl3中, 再加入1.0 mg (0.01 mmol)苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 57.30%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.29 (t, J=7.30 Hz, 9H, CH2CH3), 3.15 (q, J=7.25 Hz, 6H, CH2CH3), 6.62 (d, J=14.68 Hz, 1H, =C-H), 6.80 (d, J=14.68 Hz, 1H, =C-H), 7.11~6.93 (m, 5H, C6H5); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.84 (d, J=20.46 Hz, 2F, o-C6F5), -162.51 (t, J=20.44 Hz, 2F, p-C6F5), -166.57 (t, J=20.46 Hz, 1F, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 8.7, 47.7, 125.53, 127.15, 127.88, 132.7, 140.5, 136.50, 137.90, 148.10.
4.12 (Z)-[Et3NH][Me-Ph-CH=CHBCF]的制备
在核磁管中加入6.1 mg (0.01 mmol) Et3NHHBCF溶于CDCl3中, 再加入1.5 mg (0.01 mmol)对甲基苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为棕红色, 一段时间后, 溶液颜色变浅.所分离得到的硼氢化产物产率为: 59.09%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.28 (t, J=7.3 Hz, 9H, CH2CH3), 2.20 (s, 3H, CH3C6H4), 2.44 (bs), 3.17~3.07 (m, 6H, CH2CH3), 6.58 (d, J=14.7 Hz, 1H, =C-H), 6.73 (d, J=14.7 Hz, 1H, =C-H), 6.84 (d, J=7.8 Hz, 2H, C6H4), 6.94 (d, J=7.8 Hz, 2H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -132.9 (d, J=20.06 Hz, o-C6F5), -162.6 (t, J=20.4 Hz, p-C6F5), -166.6 (t, J=20.0 Hz, m-C6F5); 13C {H}NMR (101 MHz, 298 K, CDCl3) δ: 8.5, 21.0, 23.6, 47.7, 127.7, 127.85, 135.34, 137.47.
4.13 (Z)-[Et3NH][F-Ph-CH=CHBCF]的制备
在核磁管中加入6.1 mg (0.01 mmol) Et3NHHBCF溶于CDCl3中, 再加入1.2 mg (0.01 mmol)对氟苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 57.64%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.29 (t, J=7.3 Hz, 9H, CH2CH3), 3.20~3.08 (m, 6H, CH2CH3), 6.69 (t, J=8.5 Hz, 2H, F-C6H4), 6.59 (d, J=14.6 Hz, 1H, =C-H), 6.73 (d, J=14.6 Hz, 1H, =C-H), 6.98 (dd, J=7.2, 8.5 Hz, 2H, F-C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -117.65 (s, F-C6H4), -132.75 (d, J=20.5 Hz, o-C6F5), -162.42 (t, J=20.5 Hz, p-C6F5), -166.51 (t, J=20.5 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 8.7, 47.9, 100.74, 113.18/113.70, 127.17, 129.34/129.44, 131.96, 136.41, 136.85, 140.10, 148.22, 159.63, 161.97.
4.14 (Z)-[Cy2NH2][F-Ph-CH=CHBCF]的制备
在核磁管中加入7.3 mg (0.01 mmol) Cy2NH2HBCF溶于CDCl3中, 再加入1.4 mg (0.01 mmol)对氟苯乙炔, 后又补加0.8 mg对氟苯乙炔, 最后加入催化剂BCF 1.0 mg (0.002 mmol).溶液由无色变为浅黄色.所分离得到的硼氢化产物产率为: 62.89%; 1H NMR (400 MHz, 298 K, CDCl3) δ: 1.14 (t, J=12.85 Hz, 2H), 1.25 (t, J=16.20 Hz, 6H), 1.41 (q, J=11.68 Hz, 4H), 1.69 (d, J=11.68 Hz, 2H), 1.82 (d, J=14.0 Hz, 2H), 2.02 (d, J=10.51 Hz, 4H), 3.00~3.16 (m, 2H), 5.74 (br, 2H, NH2), 6.68 (t, J=8.70 Hz, 2H, F-C6H4), 6.65~6.75 (m, 2H, =C-H), 7.00~6.94 (m, 2H, C6H4); 11B NMR (128 MHz, 298 K, CDCl3) δ: -16.0 (s); 19F {1H}NMR (376 MHz, 298 K, CDCl3) δ: -118.24 (s, F-C6H4), -132.05 (d, J=20.0 Hz, o-C6F5), -162.94 (t, J=20.91 Hz, p-C6F5), -166.57 (t, J=20.0 Hz, m-C6F5); 13C {1H}NMR (101 MHz, 298 K, CDCl3) δ: 24.42, 24.66, 29.87, 54.78, 113.35, 129.42.
-
-
[1]
Kropp, M. A.; Baillargeon, M.; Park, K. M.; Ahamidapaty, K.; Schuster, G. B. J. Am. Chem. Soc. 1991, 113, 2155. doi: 10.1021/ja00006a038
-
[2]
Ohmura, T.; Yamamoto, Y.; Miyaura, N. J. Am. Chem. Soc. 2000, 122, 4990. doi: 10.1021/ja0002823
-
[3]
Gunanathan, C.; Hoelscher, M.; Pan, F.; Leitner, W. J. Am. Chem. Soc. 2012, 134, 14349. doi: 10.1021/ja307233p
-
[4]
Obligacion, J. V.; Neely, J. M.; Yazdani, A. N.; Pappas, I.; Chirik, P. J. J. Am. Chem. Soc. 2015, 137, 5855. doi: 10.1021/jacs.5b00936
-
[5]
(a) Jang, W. J. ; Lee, W. L. ; Moon, J. H. ; Lee, J. Y. ; Yun, J. Org. Lett. 2016, 18, 1390. (b) Liu, Y. -Y. ; Zhang, W. -B. Chin. J. Org. Chem. 2016, 36(10), 2249. (刘媛媛, 张万斌, 有机化学, 2016, 36(10), 2249. )
-
[6]
Xu, S.-M.; Zhang, Y.-Z.; Li, B.; Liu, S. H.-Y. J. Am. Chem. Soc. 2016, 138(44), 14566. doi: 10.1021/jacs.6b09759
-
[7]
(a) Chen, J. ; Xi, T. ; Lu, Z. Org. Lett. 2014, 16, 6452. (b) He, X. -W. ; Hu, X. -Q. ; Tao, J. -J. ; Han, G. ; Shang, Y. -J. Chin. J. Org. Chem. 2016, 36(7), 1465. (何心伟, 胡小倩, 陶佳佳, 韩光, 商永嘉, 有机化学, 2016, 36(7), 1465. )
-
[8]
Bismuto, A.; Thomas, S. P.; Cowley, M. J. Angew. Chem., Int. Ed. 2016, 55, 15356. doi: 10.1002/anie.v55.49
-
[9]
Welch, G. C.; San Juan, R. R.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124. doi: 10.1126/science.1134230
-
[10]
Welch, G. C.; Stephan, D. W. J. Am. Chem. Soc. 2007, 129, 1880. doi: 10.1021/ja067961j
-
[11]
Chen, D.-J.; Wang, Y.; Klankermayer, J. Angew. Chem., Int. Ed. 2010, 49, 9475. doi: 10.1002/anie.201004525
-
[12]
Stephan, D. W.; Erker, G. Angew. Chem., Int. Ed. 2010, 49, 46. doi: 10.1002/anie.200903708
-
[13]
Stephan, D. W. Acc. Chem. Res. 2015, 48, 306. doi: 10.1021/ar500375j
-
[14]
Chen, C.; Kehr, G..; Fröhlich, R.; Erker, G. J. Am. Chem. Soc. 2010, 132, 13594. doi: 10.1021/ja106365j
-
[15]
Chen, C.; Voss, T.; Fröhlich, R.; Kehr, G.; Erker, G. Org. Lett. 2011, 13, 62. doi: 10.1021/ol102544x
-
[16]
Jiang, C.; Blacque, O.; Berke, H. Organometallics 2010, 29, 125. doi: 10.1021/om9008636
-
[17]
Reddy, J. S.; Xu, B.-H.; Mahdi, T.; Fröhlich, R.; Kehr, G.; Stephan, D. W.; Erker, G. Organometallics 2012, 31, 5638. doi: 10.1021/om3006068
-
[18]
Nie, W.-L.; Klare, H. F. T.; Oestreich, M.; FrÖhlich, R.; Kehr, G.; Erker, G. Z. Naturforsch. 2012, 67b, 987.
-
[19]
徐莹莹, 李钊, Borzov, Maxim, 聂万丽, 化学进展, 2012, 24(8), 1526. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmXu, Y.-Y.; Li, Z.; Borzov, M.; Nie, W.-L. Chem. Prog. 2012, 24(8), 1526. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[20]
田冲, 姜亚, Borzov, Maxim, 聂万丽, 化学学报, 2015, 73, 1203 http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345179.shtmlTian, C.; Jiang, Y.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2015, 73, 1203. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345179.shtml
-
[21]
胡茜, 田冲, Borzov, Maxim, 聂万丽, 化学学报, 2015, 73, 1025. doi: 10.3866/PKU.WHXB201504141Hu, X.; Tian, C.; Jiang, Y.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2015, 73, 1025. doi: 10.3866/PKU.WHXB201504141
-
[22]
温志国, 田冲, Borzov, Maxim, 聂万丽, 化学学报, 2016, 74, 498. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345525.shtmlWen, Z.-G.; Tian, C.; Jiang, Y.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2016, 74, 498 http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345525.shtml
-
[23]
E-Stereospecific 1, 1-carboboration of terminal arylalkynes with[I-B(C6F5)3], 待发.
-
[24]
Nie, W.-L.; Sun, G.-F.; Tian, C.; Borzov, M. Naturforsch. 2016, 71(10) b, 1029.
-
[25]
Detailed experimental procedures, characterization and X-ray data are available from the Supporting Information. (一些有代表性的氯代硼胺盐及炔烃硼氢化胺盐的合成、结构及X-射线单晶结构详见支持信息. )
-
[1]
-

图 2 B (C6F5)3/有机胺盐体系与炔烃的反应
Figure 2 The reaction of B (C6F5)3/ammonium systems with alkynes

图 4 炔烃的立体选择性硼氢化加成反应机理
Figure 4 Mechanism of the stereoselective hydroboration of alkynes

图 5 有机胺盐酸盐与B (C6F5)3的不同络合方式
Figure 5 Different coordination modes of ammonium hydrochlorides with B (C6F5)3

图 6 R2NH2Cl/B (C6F5)3体系与炔烃反应的11B核磁共振谱图
Figure 6 11B NMR of R2NH2Cl/B (C6F5)3 reaction with alkynes
表 1 端基炔烃的Z式硼氢化加成反应a
Table 1. (Z)-Hydroboration of terminal alkynesa
Entry R2NH Alkyne Product 1 CyNH2 p-F-Ph-CCH [CyNH3][F-Ph-CH=CHBCF] 2 Et3NH p-F-Ph-CCH [Et3NH][F-Ph-CH=CHBCF] 3 Et3NH Ph-CCH [Et3NH][Ph-CH=CHBCF] 4 Et3NH p-Me-Ph-CCH [Et3NH][Me-Ph-CH=CHBCF] 5 n-Bu4N p-F-Ph-CCH [n-Bu4N][F-Ph-CH=CHBCF] 6 n-Bu4N Ph-CCH [n-Bu4N][Ph-CH=CHBCF] 7 n-Bu4N p-Me-Ph-CCH [n-Bu4N][Me-Ph-CH=CHBCF] 8 TMP p-F-Ph-CCH [TMPH][F-Ph-CH=CHBCF] 9 TMP Ph-CCH [TMPH][Ph-CH=CHBCF] 10 TMP p-Me-Ph-CCH [TMPH][Me-Ph-CH=CHBCF] 11 i-Pr2NH p-F-Ph-CCH [i-Pr2NH2][F-Ph-CH=CHBCF] 12 i-Pr2NH Ph-CCH [i-Pr2NH2][Ph-CH=CHBCF] 13 i-Pr2NH p-Me-Ph-CCH [i-Pr2NH2][Me-Ph-CH=CHBCF] 14 Et2NH p-F-Ph-CCH [Et2NH2][F-Ph-CH=CHBCF]+F-Ph-CH=CH2b 15 t-BuNH2 p-F-Ph-CCH F-Ph-CH=CH2b a Reaction condition: r.t., Cat: BCF 5 mol%; b Reduction product.  下载: 导出CSV
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 11
- 文章访问数: 3871
- HTML全文浏览量: 823
 下载:
下载:
